

Abstract
Mycoplasma hyopneumoniae, the agent of porcine enzootic pneumonia (EP), is able to persist in the lung tissue and evade destruction by the host for several weeks. To understand the mechanism of pathogen survival, phagocytic uptake of M. hyopneumoniae by primary porcine alveolar macrophages was investigated. Intracellular location and survival of the pathogen were explored using gentamicin survival assays, flow cytometry and confocal microscopy of M. hyopneumoniae 232 labelled with green fluorescent protein (GFP). Following 1 h and 16 h of co-incubation, few viable M. hyopneumoniae were recovered from inside macrophages. Flow cytometric analysis of macrophages incubated with M. hyopneumoniae expressing GFP indicated that the mycoplasmas became associated with macrophages, but were shown to be extracellular when actin-dependent phagocytosis was blocked with cytochalasin D. Confocal microscopy detected GFP-labelled M. hyopneumoniae inside macrophages and the numbers increased modestly with time of incubation. Neither the addition of porcine serum complement or convalescent serum from EP-recovered pigs was able to enhance engulfment of M. hyopneumoniae. This investigation suggests that M. hyopneumoniae evades significant uptake by porcine alveolar macrophages and this may be a mechanism of immune escape by M. hyopneumoniae in the porcine respiratory tract.
Introduction
The porcine respiratory pathogen Mycoplasma hyopneumoniae is considered to be the primary agent of porcine enzootic pneumonia [1]. Enzootic pneumonia, a chronic disease, is a source of great economic loss worldwide because M. hyopneumoniae-infected animals exhibit reduced average daily weight gain and feed conversion [2, 3]. It has been shown that M. hyopneumoniae infection increases susceptibility of the pig to secondary infections which have further economic and welfare implications [4, 5]. At the time of slaughter, lung pathology arising from M. hyopneumoniae infection can be seen as regions of well-demarcated, dark purple, atelectatic tissue located in the anterior of the cranial and middle lung lobes, and apical anterior portion of the caudal lobe [6]. Microscopic inspection of lung lesions typically reveals loss of cilia from the respiratory ciliated epithelium, accumulation of lymphocytes and plasmocytes in peribronchial, peribronchiolar and perivascular areas, hyperplasia of bronchus associated lymphoid tissue, thickening of alveolar septae and exudate in bronchi and bronchiole lumens containing neutrophils and macrophages [7]. M. hyopneumoniae-induced lung lesions indicate the porcine host is producing an immune response to infection, but this response is not removing the pathogen quickly.
Management and control of enzootic pneumonia is achieved with good animal husbandry, disease surveillance and vaccination [8]. Many pig-producing countries practise vaccination against M. hyopneumoniae using adjuvanted inactivated whole-cell preparations, but live-attenuated vaccines are available in China and Mexico [8–10]. At present, all commercial M. hyopneumoniae vaccines offer reduced clinical and pathological signs of disease; however, they all fall short of providing complete protection from M. hyopneumoniae infection and the associated lung pathology. Hence, there is room to improve upon current M. hyopneumoniae vaccines [11].
In order to design and produce more effective vaccines against M. hyopneumoniae, better understanding of M. hyopneumoniae pathogenicity is required. A recent review by Maes et al. [10] recognised knowledge surrounding M. hyopneumoniae pathogenicity remains scarce but progress has been made. At present, lipoproteins at the surface of M. hyopneumoniae cells are recognised as adhesion proteins that facilitate attachment and colonisation of the ciliated epithelium [12, 13]. Some of these adhesion proteins are also thought to function as moonlighting proteins. For example, aminopeptidases at the cell-surface of M. hyopneumoniae bind to a number of different host molecules, and are also able to bind and cleave plasminogen to plasmin [14, 15]. The significance M. hyopneumoniae-directed plasmin activation in the in vivo environment and its role in disease pathogenesis remains to be determined. Speculative mechanisms of M. hyopneumoniae virulence include hydrogen peroxide production, modulation and evasion of host immunity [10]. More research is required to reveal pathogenicity mechanisms employed by M. hyopneumoniae to infect and colonise the porcine host.
During early infection and colonisation, M. hyopneumoniae is likely to encounter resident pulmonary phagocytes such as macrophages. Respiratory macrophages are an important first line of defence against invading microorganisms. They provide protection by engulfing and destroying susceptible organisms, they can also release chemical mediators to trigger an inflammatory immune response [16]. With this in mind, it is not clear from previous experimental data what role macrophages play in defence or propagation of M. hyopneumoniae infection. A number of pathogenic Mycoplasma spp. have demonstrated the ability to evade phagocytic uptake by host macrophages. For example, M. pulmonis, M. bovis, M. dispar, M. pneumoniae and M. ovipneumoniae all appear to resist uptake by host macrophages in the absence of specific opsonins. Upon addition of specific antisera these pathogenic mycoplasmas are readily engulfed [17–22]. It is possible that M. hyopneumoniae resists phagocytic uptake by respiratory macrophages, and this may in-part explain why M. hyopneumoniae is not quickly cleared by the respiratory immune system.
In this study, evasion of host immunity by M. hyopneumoniae was examined by investigating the interactions between M. hyopneumoniae and primary porcine alveolar macrophages. Experiments were designed to ascertain whether M. hyopneumoniae is phagocytosed by alveolar macrophages, and whether components of normal and convalescent porcine serum facilitate phagocytosis of M. hyopneumoniae.
Materials and methods
Bacterial strains and culture conditions
M. hyopneumoniae strain 232 was routinely cultured static at 37 °C in modified liquid Friis medium, and in a humidified atmosphere of 5% CO2 at 37 °C on modified Friis agar [1, 23–25].
Escherichia coli DH5α (Thermo Fisher Scientific, Massachusetts, USA), a K-12 derivative, was routinely cultured in LB broth [1% (w/v) tryptone (Oxoid, Hampshire, UK), 1% (w/v) NaCl and 0.5% (w/v) yeast extract (Oxoid)] at 37 °C with agitation at 210 rpm, and on LB agar [LB broth supplemented with 1.5% (w/v) bacteriological agar no. 1 (Oxoid)].
Isolation and culture of primary porcine alveolar macrophages
Two conventional large white specific-pathogen free pigs were sourced from a herd which did not demonstrate viral-, enzootic pneumonia-, nor pleuropneumonia-like lung pathology at abattoir surveillance. The animals were euthanised by sedation with a ketamine and xylazine mixture, followed by overdose of sodium pentobarbital via ear vein injection. Primary alveolar macrophages were collected as done by Cullen, Rycroft [26]. Briefly, ice-cold sterile phosphate buffered saline (PBS) supplemented with 10 µg/mL gentamicin sulphate (Sigma Aldrich, Dorest, UK) was poured into the lungs, followed by gentle massage for 4 s and then the lavage fluid was collected into a sterile ice-cold glass bottle. The lungs from each animal appeared normal and did not present any macroscopic lesions suggestive of infection with respiratory pathogens such as M. hyopneumoniae, Actinobacillus pleuropneumoniae or porcine reproductive and respiratory syndrome virus. The lavage fluid was centrifuged at 200 × g for 10 min at room temperature. The cell pellet was resuspended in RPMI 1640 1 × medium (GIBCO, Thermo Fisher Scientific), supplemented with 10% heat-treated foetal bovine serum (FBS) (GIBCO). Viability counts were performed by trypan blue exclusion. If the number of viable cells was ≥ 90%, the cells were frozen on the day of procurement in FBS supplemented with 10% dimethyl sulfoxide (DMSO). These cells were identified as macrophages based on cellular morphology under the microscope and positive staining with anti-porcine CD-163 mouse antibody [27]. Two batches of alveolar macrophages were prepared, A and B, each batch originated from a different pig and were collected 1 week apart.
The alveolar macrophages were routinely cultured in RPMI 1640 Glutamax supplemented with HEPES (GIBCO) and 10% heat-treated FBS (referred to as tissue culture medium). The macrophages were maintained in vitro in 24-well plates in a humidified atmosphere of 5% CO2 at 37 °C for no more than 32 h.
Plasmid construction
Polymerase chain reaction (PCR) was performed using Phusion High Fidelity DNA polymerase (NEB, Hertfordshire, UK) according to the manufacturers instructions. DNA oligonucleotides used in PCR amplification reactions are listed in Table 1. The plasmid pMHGFP-P97 was constructed by amplifying M. hyopneumoniae strain 232 P97 promoter sequence from pMHC9-1 [28], using primers P97F and P97R, and was subsequently cloned into plasmid pMHGFP2. Plasmid pMHGFP2, constructed from pMHO-1 [29], contained the green fluorescent protein gene, gfpmut2, under control of the spiralin promoter sequence of Spiroplasma citri [28]; gfpmut2 was amplified from the pKENmut2 vector [30] using primers gfpF and gfpR. The spiralin promoter sequence was digested from pMHGFP2, and replaced with the P97 promoter sequence at restriction sites MluI and BamHI to give rise to plasmid pMHGFP-P97.
Table 1.
DNA oligonucleotides
| Name | 3′ sequence 5′ |
|---|---|
| gfpF | TTACTAGTTGTGTGGAATTCGAGCTCGG |
| gfpR | TTGCATGCCTGCAGGTCTGG |
| P97F | GTCACGCGTCTTTTAATTATTAGTCTTCC |
| P97R | GTCGGATCCTTACTCATATTTTAAACCTC |
Underlined nucleotides indicate restriction enzyme sequence
Bacterial transformation
E. coli DH5α were transformed with ligation reactions by heat shock transformation. Briefly, ice-cold competent E. coli DH5α (100 μL) were incubated on ice for 30 min with ligation reactions (4 µL). E. coli were heat-shock transformed at 42 °C for 45 s and then cooled on ice for 2 min, followed by recovery in SOC medium (2% (w/v) tryptone (Oxoid), 0.5% (w/v) yeast extract (Oxoid), 0.05% (w/v) NaCl, 12.5 mM KCl, 10 mM MgCl2 and 20 mM glucose) (900 μL) for 1 h at 37 °C and 210 rpm. Transformed E. coli (100 μL) were spread on LB agar supplemented with 50 μg/mL ampicillin and incubated overnight at 37 °C.
M. hyopneumoniae strain 232 was transformed with plasmid DNA by electroporation as described previously [29]. Briefly, 25 mL of modified Friis medium inoculated with 0.5 mL of M. hyopneumoniae stock culture was grown static for approximately 41 h at 37 °C. The M. hyopneumoniae were washed and incubated on ice for 30 min with 10 μg of plasmid DNA. M. hyopneumoniae were transformed by electroporation at 2.5 kV, 100 Ω and 25 μF. Afterwards, 900 μL ice-cold Friis medium was added and the transformations were incubated on ice for 15 min. Transformed M. hyopneumoniae were allowed to recover for 3 h in static incubation, at 37 °C, and were then cultured on Friis agar supplemented with 0.2 μg/mL tetracycline at 37 °C in 5% CO2 for 7–14 days. Transformed M. hyopneumoniae colonies were transferred to 1 mL Friis medium supplemented with 0.5 μg/mL tetracycline, and incubated at 37 °C until the culture became peach/orange in colour. Liquid cultures at this stage were stored at −70 °C.
Gentamicin survival assays
One hour gentamicin survival assay
Primary porcine alveolar macrophages (1 × 106 in 1 mL) that had been frozen (batch A) were seeded into 24-well cell-culture treated plates (Nunc Nunclon Delta, Thermo Fisher Scientific) and allowed to adhere in a humidified atmosphere of 5% CO2 at 37 °C, for at least 1 h.
A M. hyopneumoniae culture (20 mL) grown for approximately 32 h was centrifuged, and then briefly washed in sterile PBS. The M. hyopneumoniae cell pellet was resuspended in 10 mL tissue culture medium. E. coli DH5α was grown to 1 × 108 colony forming units (CFU)/mL, and 1 mL was centrifuged and washed in sterile PBS. The E. coli cell pellet was resuspended in 1 mL sterile PBS and then diluted 1 in 100 in tissue culture medium (10 mL).
Medium was removed from PAMs and replaced with either 1 mL of M. hyopneumoniae or E. coli inoculum, or tissue culture medium. The experiment was performed in duplicate and the intended multiplicity of infection (MOI) of bacteria/macrophage was 1:1. The inoculated PAMs were incubated in a humidified atmosphere of 5% CO2 at 37 °C for 1 h, and then the medium was removed and wells were washed very gently three times with sterile PBS. Tissue culture medium supplemented with or without 400 μg/mL gentamicin [31], was added to specified wells and the plate was incubated for a further 3 h. After incubation, culture supernatants of gentamicin-treated wells were collected and centrifuged at 16 000 × g for 2 min. Pellets were washed once in PBS, and then resuspended in 40 μL PBS and the entire volume was spread onto solid microbiological medium. The culture supernatants of wells without gentamicin were serially diluted 1 in 10 in PBS (1 mL) and 10 μL spots were inoculated onto solid microbiological medium.
After the culture supernatants had been removed, the wells were gently washed three times with warm sterile PBS. To wells inoculated with M. hyopneumoniae 400 μL of Friis medium was added, and 400 μL of LB broth to wells inoculated with E. coli. The well surfaces were scraped with sterile mini cell scrapers (Biotium, California, USA) and the contents were transferred to 2 mL microcentrifuge tubes containing a single tungsten carbide 3 mm bead (Qiagen, Manchester, UK). The tube contents were homogenised for 2 min at 30 cycles per second in a Retsch MM300 homogeniser (Retsch, Haan, Germany). It was previously confirmed that this method of homogenisation did not reduce the viability of M. hyopneumoniae. The homogenised samples that had not been treated with gentamicin, were serially diluted 1 in 10 in PBS (1 mL) and 10 μL spots were spotted onto solid microbiological medium. The entire volume (400 μL) of gentamicin-treated samples was spread onto 9 cm plates. Samples from uninoculated controls were plated to check for contaminating microorganisms.
Overnight gentamicin survival assay
Primary porcine alveolar macrophages (1 × 106 in 1 mL) that had been frozen (batch A), were seeded into 24-well cell-culture treated plates and were incubated for at least 1 h.
M. hyopneumoniae and E. coli were prepared similarly to the 1 h gentamicin survival assay, except the inoculum was reduced such that the starting MOI of bacteria/macrophage was 1:100.
The medium was removed from macrophages and replaced with either 1 mL of M. hyopneumoniae or E. coli inoculum. The macrophages and bacteria were incubated static in a humidified atmosphere of 5% CO2 at 37 °C for approximately 16 h, and then tissue culture medium supplemented with or without 400 µg/mL gentamicin was applied for 6 h. After the first 3 h of incubation the tissue culture medium ± gentamicin was refreshed. The experiment was performed in triplicate.
Flow cytometry
Flow cytometric analysis of GFP-transformed bacteria
Untransformed bacteria diluted 1 in 10 in PBS, were analysed on BD Accuri C6 flow cytometer (BD Bioscience, California, USA) to gate bacterial cells. GFP-transformed bacteria were also diluted 1 in 10 in PBS and analysed. Green fluorescent bacteria were detected by the FL1 detector. To distinguish fluorescent and non-fluorescent bacterial cells, a marker was drawn on the FL1 histogram plot of untransformed bacteria. This marker was then applied to FL1 histogram plots of GFP-transformed bacteria to establish the percentage of fluorescent bacteria.
Flow cytometric analysis of phagocytic uptake of GFP-labelled bacteria
Alveolar macrophages (5 × 105 in 0.5 mL per reaction) that had been frozen (batch A) were supplemented with or without 20 μg/mL cytochalasin D (CCD) (Sigma Aldrich, catalogue C2618). The macrophages (0.5 mL) were seeded into wells of a 24-well plate and incubated for 15 min at 37 °C prior to the addition of bacteria.
A 200 mL M. hyopneumoniae (pMHGFP-P97) culture was prepared by inoculating 0.5 mL stock culture into 200 mL Friis medium. This culture was grown for 122 h until the medium was orange in colour. The M. hyopneumoniae (pMHGFP-P97) culture was centrifuged at 9000 × g for 10 min at room temperature. The mycoplasma pellet was briefly washed in sterile PBS, and then resuspended in 4 mL tissue culture medium such that 200 μL was equal to 25 mL original culture. E. coli (pKENmut2) was grown until optical density600nm (OD600nm) 0.6, centrifuged at 2500 × g for 5 min, washed briefly in PBS and then diluted to 1.3 × 107 CFU/200 μL. The intended MOI of bacteria/PAM was 25:1. The macrophages, with and without CCD, were inoculated with 0.5 mL M. hyopneumoniae (pMHGFP-P97) or E. coli (pKENmut2); this halved the CCD concentration to 10 μg/mL. Some of the wells containing macrophages, with and without CDD, were mock-inoculated with tissue culture medium as the negative controls. The experiment was performed in triplicate. The macrophages were incubated with the bacteria for 1 h, static, in a humidified atmosphere of 5% CO2 at 37 °C. After incubation, the macrophages were gently scrapped from the well surface with mini cell scrappers, and then centrifuged at 400 × g for 5 min at room temperature in microcentrifuge tubes. The supernatants were removed and the macrophage cells pellets were resuspended in 500 μL FACSFlow. The macrophages were analysed immediately on a FACSCalibur flow cytometer (BD) using the FL1 detector. Uninoculated macrophages were used to gate non-fluorescent cells. The flow cytometric data were analysed in FlowJo version 10 software (FlowJo LLC, Oregon, USA).
Confocal microscopy
Visualising GFP-transformed M. hyopneumoniae
Liquid cultures of GFP-transformed M. hyopneumoniae were grown until orange (72 h) in colour. The cultures were centrifuged at 9000 × g for 10 min at room temperature, and washed once in 1 mL PBS. M. hyopneumoniae cells were mounted onto glass coverslips as previously described [14]. Briefly, M. hyopneumoniae cells were resuspended in 100 μL PBS for every 1 mL of original culture. Glass coverslips (22 × 22 mm, Chance Propper, Warley, UK) were coated with 0.01% poly-l-lysine (w/v) (Sigma Aldrich) and air-dried at room temperature. M. hyopneumoniae suspensions (100 μL) were spotted onto the poly-l-lysine coated coverslips and allowed to adhere for 30 min at room temperature. The coverslips were washed twice in PBS, then M. hyopneumoniae were fixed in 2% formaldehyde in PBS overnight at 4–8 °C. Formaldehyde was removed by three brief washes in PBS. M. hyopneumoniae cells were stained with 1 μg/mL 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Roche Diagnostics, Mannheim, Germany) in PBS for 30 min at room temperature in the dark, followed by two brief washes with PBS. The coverslips were air-dried, and then 5 μL of Vectashield (Vector Laboratories, California, USA) was spotted on the coverslip and a glass slide was placed on top. The edges of the coverslip were sealed with transparent nail varnish (Maybelline, New York, USA).
GFP-transformed M. hyopneumoniae mounted onto glass coverslips were viewed on a confocal scanning laser Leica SP5 microscope (Leica Microsystems, Wetzlar, Germany). The samples were viewed by 63× objective with 4× digital zoom. Blue light (405 nm) was used to excite DAPI to visualise nucleic acid, and blue light (488 nm) to excite and visualise GFP. Untransformed M. hyopneumoniae were used as the negative control.
Confocal microscopy analysis of porcine alveolar macrophages infected with GFP-labelled M. hyopneumoniae
Time-dependent experiments
Primary porcine alveolar macrophages that had been frozen (batch B) were seeded (2.5 × 105 cells per well) into an 8-chamber glass Lab-Tek Chamber Slide System (Nunc). The macrophages were incubated for at least 1 h prior to use.
A M. hyopneumoniae (pMHGFP-P97) culture that was orange in colour was centrifuged at 9000 × g for 10 min at room temperature. The mycoplasma pellet was washed briefly in PBS and then resuspended in tissue culture medium (supplemented with 0.5 μg/mL tetracycline hydrochloride) such that 200 μL was equal to 12.5 mL of original culture. When M. hyopneumoniae wild type strain 232 were used, the mycoplasma pellet was suspended in tissue culture medium such that 200 μL was equal to 5 mL of original culture. E. coli (pKENmut2) were grown until OD600nm 0.6. The E. coli were centrifuged at 2500 × g for 5 min at room temperature, and briefly washed in PBS. The E. coli cell pellet was resuspended and diluted in tissue culture medium (supplemented with 50 μg/mL ampicillin) such that 200 μL contained 2.5 × 106 CFU.
The tissue culture medium was removed from macrophages, and replaced with either 200 μL of M. hyopneumoniae (pMHGFP-P97), or M. hyopneumoniae wild type, or E. coli (pKENmut2). The macrophages and bacteria were incubated for the specified time in a humidified atmosphere of 5% CO2 at 37 °C. After incubation, the culture medium was removed from wells and each well was gently washed with warm PBS. The samples were fixed with 2% paraformaldehyde in PBS (Sigma Aldrich) overnight at 4–8 °C in the dark.
Following fixation, the wells were washed with PBS twice, and then 100 μL 0.5% Triton-X-100 in PBS was added, and the samples were incubated at room temperature for 15 min. The wells were then washed with PBS three times, and 5% bovine serum albumin in PBS was added and the samples were incubated at 4–8 °C for 30 min. The wells were then washed with PBS twice, and 200 μL of 1 μg/mL DAPI and 5 U/mL phalloidin-CF568 (Biotium) in PBS was added, and the samples were incubated at room temperature for 30 min in the dark. The wells were washed with PBS twice and then 200 μL of 1 μg/mL mouse IgG1 anti-GFP (Sigma Aldrich, catalogue SAB4600051) was applied to each well and incubated at 4–8 °C for 1 h in the dark. After this incubation, the wells were washed with PBS three times, and the chamber and gasket were removed from the slide. The slide was air-dried and 30 μL of Vectashield was spotted onto the slide. A glass coverslip was placed on top and the edges were sealed with transparent nail varnish.
The samples were viewed on a confocal scanning laser Leica SP5 microscope. The samples were viewed by 63× objective with 4× digital zoom. Blue light (405 nm) was used to excite DAPI to visualise nucleic acid, blue light (488 nm) to excite and visualise GFP, and yellow light (594 nm) to excite phalloidin-CF568 to visualise macrophage actin. Micrographs were captured at increments of 0.13 μm along the z-axis to produce z-stack images of macrophages. To determine if bacteria were inside macrophages, the z-stack micrographs were viewed in x, y and z-planes simultaneously using the computer software Volocity version 6.3 (PerkinElmer, Massachusetts, USA).
Opsonisation experiments
Opsonisation of M. hyopneumoniae (pMHGFP-P97) and E. coli (pKENmut2) was performed using preimmune porcine serum and convalescent porcine sera A, B and C. The preimmune and convalescent sera A were derived from the same pig at different times; the preimmune serum was collected before M. hyopneumoniae infection and serum A was collected 5 weeks after infection with M. hyopneumoniae strain 232 (supplied by Jessica Beddow). Serum B was derived from a pig infected with M. hyopneumoniae strain 325 (known as 9954 serum), and serum C was derived from a pig infected with M. hyopneumoniae strain 89 (known as 9965 serum) (supplied by Andrew Rycroft). The presence of M. hyopneumoniae-specific antibody in the convalescent sera was confirmed by Western Blot (data not shown). Heat-treated FBS was the negative control serum.
M. hyopneumoniae (pMHGFP-P97) cell pellets, each equivalent to 12.5 mL original culture, were resuspended in 100 μL of tissue culture medium supplemented with 1 μg/mL tetracycline hydrochloride. To separate M. hyopneumoniae (pMHGFP-P97) suspensions (100 μL), 100 μL of the different sera were added and incubated at 37 °C for 15 min. E. coli (pKENmut2) pellets, each equivalent to 2.5 × 106 CFU, were resuspended in 100 μL of tissue culture medium supplemented with 100 μg/mL ampicillin sodium salt. These E. coli (pKENmut2) suspensions were treated the same as M. hyopneumoniae (pMHGFP-P97). Macrophages (batch B) were inoculated with these 200 μL suspensions of bacteria and incubated for 2 h. All other experiment details were identical to the time-dependent experiments.
Results
Few viable M. hyopneumoniae were recovered from inside porcine macrophages
To evaluate the ability of porcine lung macrophages to engulf M. hyopneumoniae, cells were prepared by lavage of the lungs from a freshly killed pig known to be free of M. hyopneumoniae and other respiratory pathogens [26]. Primary porcine alveolar macrophages were incubated with either M. hyopneumoniae 232 or E. coli DH5α for 1 h before addition of gentamicin for 3 h to kill extracellular bacteria. Enumeration of bacteria in the cell supernatants showed complete destruction of the extracellular E. coli. However, a small proportion of M. hyopneumoniae (0.0045%) survived the treatment with gentamicin (Figure 1A). Internalised bacteria were then recovered by rupture of lung cells by mechanical homogenisation. Culture of the cell homogenate revealed 0.125% of the M. hyopneumoniae were viable compared to the control without antibiotic (Figure 1A). The effectiveness of gentamicin treatment and efficiency of macrophage phagocytic function was confirmed as no E. coli DH5α were recovered from supernatant but E. coli were recovered from the cell homogenate (Figure 1B).
Figure 1.

1 h phagocytosis assay of M. hyopneumoniae. Porcine alveolar macrophages were incubated with M. hyopneumoniae or E. coli for 1 h and then 400 µg/mL gentamicin was applied for 3 h. Samples were plated on to solid medium to enumerate bacteria. A M. hyopneumoniae recovered from culture supernatant or disrupted macrophages, with and without gentamicin treatment. B E. coli recovered from culture supernatant or disrupted macrophages, with and without gentamicin treatment.
To determine whether phagocytic activity was time-dependent, M. hyopneumoniae were incubated with lung macrophages for 16 h. Gentamicin (400 μg/mL) was then applied for 6 h. Extended exposure to gentamicin was intended to eliminate all extracellular M. hyopneumoniae. No M. hyopneumoniae were recovered from the culture supernatants following extended exposure to gentamicin. This indicated that all extracellular M. hyopneumoniae had been eliminated (Figure 2A). No M. hyopneumoniae were recovered from gentamicin-treated disrupted macrophages while numerous viable M. hyopneumoniae were recovered from untreated macrophages (Figure 2A). This result suggested that very few M. hyopneumoniae had been phagocytosed by macrophages.
Figure 2.

Overnight phagocytosis assay of M. hyopneumoniae. Porcine alveolar macrophages were incubated with M. hyopneumoniae or E. coli overnight (16 h) and then 400 µg/mL gentamicin was applied for 6 h. Samples were plated on to solid medium to enumerate bacteria. A M. hyopneumoniae recovered from culture supernatant or disrupted macrophages, with and without gentamicin treatment. B E. coli recovered from culture supernatant or disrupted macrophages, with and without gentamicin treatment. Error bars show standard error of the mean from one experiment performed in triplicate.
Expression and detection of green fluorescent protein in M. hyopneumoniae
In order to express GFP in M. hyopneumoniae, a plasmid carrying the GFP gene gfpmut2 was constructed from a plasmid known to be stably inherited in M. hyopneumoniae. The plasmid construct was based on pMHO-1 which contained the predicted origin of replication of M. hyopneumoniae strain 232, the tetM tetracycline resistance gene expressed under control of its natural promoter from Enterococcus faecalis, and the bla ampicillin resistance gene [29]. Plasmid pMHGFP-P97 was produced by cloning the gfpmut2 gene under control of the P97 gene promoter sequence, from M. hyopneumoniae strain 232, into plasmid pMHO-1 (Figure 3).
Figure 3.
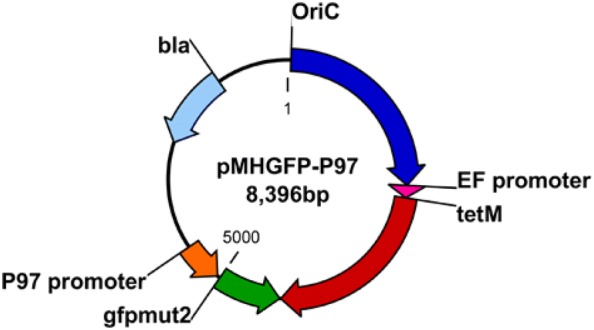
pMHGFP-P97 plasmid diagram. Oric = M. hyopneumoniae strain 232 predicited origin of replication, EF promoter = tetM gene promoter from Enterococcus faecalis, tetM = tetracycline resistance gene, gfpmut2 = green fluorescent protein gene, P97 promoter = predicted P97 gene promoter from M. hyopneumoniae strain 232, bla = ampicillin resistance gene.
M. hyopneumoniae strain 232 was electroporated with plasmid pMHGFP-P97 and transformants were recovered on selective medium. Transformed M. hyopneumoniae were viewed using a confocal laser scanning microscope for green fluorescence.
M. hyopneumoniae (pMHGFP-P97) cells fluoresced green, enabling visualization of individual mycoplasma cells (Figure 4).
Figure 4.
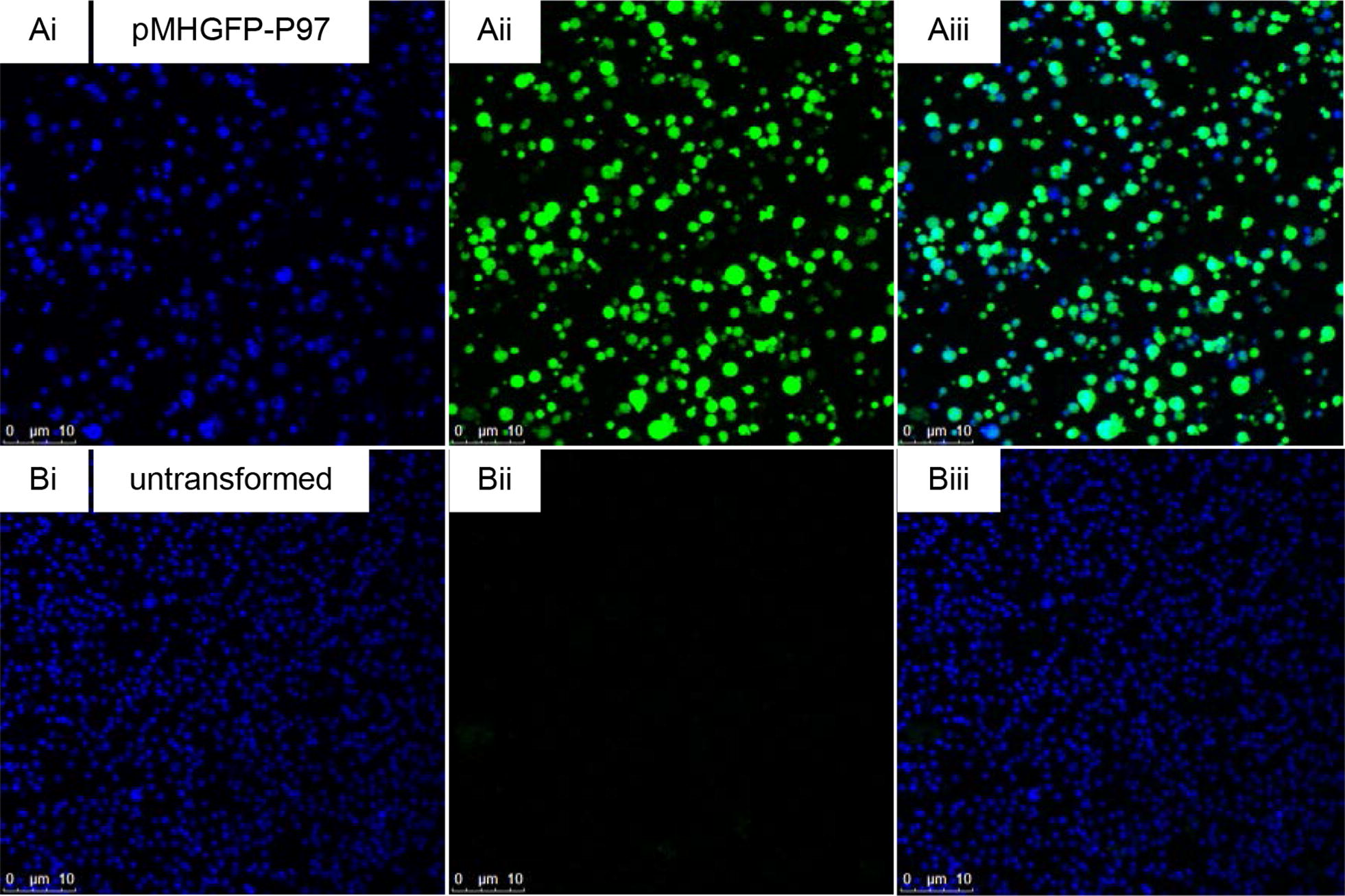
Confocal micrographs of M. hyopneumoniae (pMHGFP-P97). M. hyopneumoniae cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) and were excited by 405 nm and 488 nm lasers to visualise nucleic acid and green fluorescence, respectively. A (i) M. hyopneumoniae (pMHGFP-P97) cells stained by DAPI, (ii) M. hyopneumoniae (pMHGFP-P97) cells viewed on green fluorescence channel, (iii) overlay of i and ii. B (i) untransformed M. hyopneumoniae cells stained by DAPI, (ii) untransformed M. hyopneumoniae cells viewed on green fluorescence channel, (iii) overlay of i and ii.
M. hyopneumoniae (pMHGFP-P97) was analysed further analysis by flow cytometry. The effect of culture time on fluorescence intensity was determined. At each time point the fluorescent population was depicted by a broad peak suggesting that cells differed in fluorescence intensity. At 24 h post-inoculation, 17% of the M. hyopneumoniae (pMHGFP-P97) population were fluorescing green, this increased in intensity with incubation to 96 h (Figure 5). The intensity of fluorescence of M. hyopneumoniae (pMHGFP-P97) was considered sufficient as a label in further investigations.
Figure 5.

Flow cytometric analysis of M. hyopneumoniae (pMHGFP-P97) fluorescence over time. A M. hyopneumoniae (pMHGFP-P97) culture was analysed for green fluorescence over time by flow cytometry. The histogram FL1-A axis depicts green fluorescence intensity. On histograms plots, the vertical line indicates the boundary between fluorescent and non-fluorescent cells, to right of the line are fluorescent cells. A 24 h. B 48 h. C 72 h. D 96 h. E 168 h. F Untransformed M. hyopneumoniae. G Viable counts of M. hyopneumoniae (pMHGFP-P97) were performed from 24 to 96 h. Error bars show standard error of the mean.
Flow cytometric analysis showed M. hyopneumoniae was not phagocytosed by macrophages
A flow cytometric phagocytosis assay using GFP-labelled M. hyopneumoniae 232 (pMHGFP-P97) was used to investigate phagocytic uptake by porcine alveolar macrophages [32, 33]. The assay determines phagocytic uptake of fluorescent bacteria by comparing the fluorescence intensity of phagocytes that have been treated with and without cytochalasin D (CCD), an inhibitor of actin-dependent phagocytosis. Uptake of fluorescent bacteria is evident when CCD-treated phagocytes have lower fluorescence intensity than the untreated phagocytes, because CCD-treated phagocytes only have externally-associated bacteria. Alveolar macrophages treated with CCD, or left untreated, were incubated with M. hyopneumoniae (pMHGFP-P97) for 1 h. Green fluorescent E. coli (pKENmut2) was used as the control organism.
Untreated macrophages incubated with the positive control E. coli had significantly higher fluorescence intensity than CCD-treated macrophages (Figure 6). This result confirmed untreated macrophages had taken up E. coli (pKENmut2), and that phagocytosis was inhibited by CCD. In contrast, the mean fluorescence intensity of macrophages incubated with M. hyopneumoniae (pMHGFP-P97), with and without CCD, were very similar (Figure 6). The fluorescence intensity of CCD-treated macrophages was marginally higher than untreated macrophages, but this difference was not significant. It was suggested from these results that more M. hyopneumoniae (pMHGFP-P97) were bound to CCD-treated macrophages than untreated macrophages. It was therefore concluded that M. hyopneumoniae (pMHGFP-P97) had not been engulfed by primary macrophages.
Figure 6.

Geometric mean fluorescence intensities of macrophages, without and without CCD, after incubation with GFP-labelled M. hyopneumoniae and E. coli. The green fluorescence intensity of PAMs following incubation with GFP-labelled M. hyopneumoniae (M. hyopneumoniae (pMHGFP-P97)) or GFP-labelled E. coli (E. coli (pKENmut2)) was measured and normalised against uninoculated PAMs. Error bars show standard error of the mean from two separate experiments, 6 replicates in total. Significant mean differences were determined by independent T-test (PAMs ± CCD and E. coli) and Mann–Whitney U test (PAMs ± CCD and M. hyopneumoniae), ***p < 0.001. Porcine alveolar macrophages (PAMs), cytochalasin D (CCD).
Internalised M. hyopneumoniae detection by confocal microscopy
M. hyopneumoniae (pMHGFP-P97) was utilised in confocal microscopy experiments to look for evidence of uptake by porcine alveolar macrophages. Cells were incubated with M. hyopneumoniae (pMHGFP-P97) for 2, 4 and 6 h, and were examined for green-labelled mycoplasma localised in the cytoplasm. E. coli (pKENmut2) was again used as the positive control organism.
E. coli was observed inside macrophages at each time point, demonstrating the macrophages were phagocytic (Figure 7). Macrophages exposed to M. hyopneumoniae (pMHGFP-P97) exhibited numerous externally-bound mycoplasma cells, many of which appeared flattened against the macrophage cell-surface (Figure 7). A few M. hyopneumoniae (pMHGFP-P97) were also observed inside the macrophages. An average of 2 M. hyopneumoniae (pMHGFP-P97) cells were found inside macrophages after 2 and 4 h of incubation (Table 2). This increased to 4 intracellular M. hyopneumoniae (pMHGFP-P97) after 6 h of incubation. Untransformed M. hyopneumoniae, visualised by nucleic acid staining, behaved similarly to GFP-labelled M. hyopneumoniae (data not shown).
Figure 7.

External and internalised M. hyopneumoniae (pMHGFP-P97) following incubation with porcine alveolar macrophages. Confocal micrographs of macrophages incubated with M. hyopneumoniae (pMHGFP-P97) or the control, E. coli (pKENmut2), for 2, 4 and 6 h. Fluorescent stains show: macrophage actin cytoskeleton in red, nucleic acid in blue, and GFP-labelled bacteria in green. Yellow arrows indicate internal bacteria in the XYZ planes, white arrows indicate internal bacteria, and white triangles indicate external bacteria.
Table 2.
Number of M. hyopneumoniae (pMHGFP-P97) inside porcine alveolar macrophages following 2, 4 and 6 h of co-incubation
| Time (hours) | Avergae number of intracellular M. hyopneumoniae (pMHGFP-P97)/PAM |
|---|---|
| 2 | 2 ± 0.4 (35 PAMs) |
| 4 | 2 ± 0.6 (8 PAMs) |
| 6 | 4 ± 0.5 (35 PAMs) |
Standard error of the mean ± is shown, the number of porcine alveolar macrophages (PAMs) analysed is shown in parentheses.
Opsonisation did not enhance phagocytic uptake
To assess whether opsonisation improved engulfment, M. hyopneumoniae (pMHGFP-P97) were exposed to three different convalescent porcine sera (sera A, B and C) or preimmune serum prior to incubation with alveolar macrophages for 2 h. The convalescent sera were sources of complement and specific antibody; preimmune serum provided complement only. Heat-inactivated foetal bovine serum was used as the negative control.
Analysis of confocal micrographs revealed that on average, the number of intracellular M. hyopneumoniae (pMHGFP-P97) did not differ between samples containing the different porcine sera or heat-inactivated foetal bovine serum (Table 3). It was concluded that neither complement, nor M. hyopneumoniae-specific serum antibody, appeared to enhance phagocytic uptake of the mycoplasma by porcine alveolar macrophages.
Table 3.
Effect of opsonisation on internalisation of M. hyopneumoniae (pMHGFP-P97) by porcine alveolar macrophages
| Serum | Average number of intracellular M. hyopneumoniae (pMHGFP-P97) |
|---|---|
| Preimmune | 3 ± 0.5 (36 PAMs) |
| A | 3 ± 0.8 (24 PAMs) |
| B | 3 ± 0.5 (24 PAMs) |
| C | 3 ± 0.3 (38 PAMs) |
| Heat-inactivated foetal bovine | 2 ± 0.4 (35 PAMs) |
Standard error of the mean ± is shown, the number of porcine alveolar macrophages (PAMs) analysed is shown in parentheses. Sera A, B and C are convalescent M. hyopneumoniae serum, heat inactivated bovine serum served as the control.
Discussion
The aim of this study was to learn more about the interactions between M. hyopneumoniae and porcine macrophages. It was considered likely that M. hyopneumoniae may evade alveolar macrophage clearance mechanisms but few functional experiments investigating this have been reported. Investigations which have utilised macrophages focused primarily on cytokine production and changes in macrophage viability following exposure to M. hyopneumoniae or M. hyopneumoniae-cell fractions. For instance, Bai et al. [34] found that lipid-associated membrane proteins (LAMPs) derived from M. hyopneumoniae strain 232 exerted a cytopathic effect on the immortalised porcine alveolar macrophage cell line PAM 3D4/21 [34]. Although M. hyopneumoniae LAMPs induced apoptosis in more than 80% of PAM 3D4/21 cells, a much lesser effect was observed for whole live M. hyopneumoniae cells. Liu et al. [35] recently identified mhp390 (P68) as a surface membrane protein of M. hyopneumoniae which was able to induce caspase-3 activation, and thus apoptosis in primary porcine alveolar macrophages following 36 h of exposure.
In the present study, gentamicin survival experiments were conducted to ascertain whether viable M. hyopneumoniae could be recovered from inside porcine alveolar macrophages; which would suggest phagocytic uptake of this mycoplasma. The experimental results indicated very few viable M. hyopneumoniae were inside porcine alveolar macrophages. This could be interpreted that few M. hyopneumoniae had been engulfed, or that once inside macrophages M. hyopneumoniae were killed or few mycoplasmas remained viable. Since the macrophages appeared to engulf but not kill the control organism, E. coli DH5α, it was thought unlikely that M. hyopneumoniae had been killed by the macrophages. Therefore, it was concluded that very few M. hyopneumoniae had been engulfed by the macrophages. Other investigators have conducted similar experiments with different Mycoplasma species. Davis et al. [18] for example found that the number of inoculated M. pulmonis were not reduced following incubation with mouse alveolar macrophages. The investigators only observed a reduction in M. pulmonis co-cultured with macrophages when hyperimmune rabbit antiserum was applied. More recent studies have indicated M. pulmonis resists binding to the mouse alveolar macrophages cell line, MH-S cells, which prevents phagocytic uptake and subsequent killing [19, 36]. Results from the present gentamicin survival experiments indicated M. hyopneumoniae, like M. pulmonis, evaded uptake by host alveolar macrophages; at least in the absence of opsonins. A recent publication by Raymond et al. [37] also used an in vitro gentamicin survival method to show M. hyopneumoniae (up to 8%) can be found intracellularly in the porcine kidney epithelial cell line, PK-15.
Since the gentamicin survival experiments relied on the viability of M. hyopneumoniae to inform on phagocytic uptake, an additional approach was taken that did not measure mycoplasma viability. Flow cytometric and confocal microscopy experiments were set up to provide more information about the physical interactions between M. hyopneumoniae and macrophages. To perform these experiments M. hyopneumoniae was transformed with green fluorescent protein using the extrachromosomal plasmid pMHGFP-P97, comprising gfpmut2 controlled by the P97 gene promoter. Two additional plasmids were investigated to express GFP in M. hyopneumoniae; plasmid pMHGFP2, comprising gfpmut2 controlled by the spiralin gene promoter; and pMHGFP-tuf which contained gfpmut2 controlled by the tuf gene promoter of M. hyopneumoniae strain 232. Plasmid pMHGFP-tuf failed to transform M. hyopneumoniae and M. hyopneumoniae (pMHGFP2) did not fluoresce as brightly as M. hyopneumoniae (pMHGFP-P97) (data not shown).
Expression of GFP in M. hyopneumoniae was first reported by Ishag et al. [38] who used extrachromosomal plasmid pMD18- TOgfp to express GFP. The plasmid pMD18-TOgfp possessed a gfp gene under control of the P97 gene promoter sequence, and a tetM gene under control of an additional copy of the P97 gene promoter. In contrast, pMHGFP-P97 produced in this study only possessed one copy of the P97 promoter sequence to control expression of gfpmut2. Hence, pMHGFP-P97 may offer more stability than pMD18-TOgfp. The fluorescence intensity of M. hyopneumoniae (pMHGF-P97) cells was found to be dependent on time, at least 72 h of culture was required for maximal numbers of M. hyopneumoniae (pMHGFP-P97) cells to fluoresce. In addition, flow cytometric analysis of M. hyopneumoniae (pMHGFP-P97) revealed the fluorescence intensity of mycoplasma cells was heterogeneous, as indicated by the broad histogram peak. This may be related to different levels of intracellular GFP in differently aged M. hyopneumoniae (pMHGFP-P97) cells. Neither Ishag et al. [38] nor investigators expressing fluorescent protein in other mycoplasmas have noted a dependence of fluorescence intensity on culture time or fluorescence heterogeneity [39–41]. Despite the heterogeneous nature of M. hyopneumoniae (pMHGFP-P97) fluorescence, it was still suitable to use in downstream flow cytometry and confocal microscopy experiments.
Results from the flow cytometry experiments showed M. hyopneumoniae (pMHGFP-P97) became associated with macrophages within 1 h of co-incubation. Since the fluorescence intensities of M. hyopneumoniae (pMHGFP-P97)-infected macrophages treated with and without CCD were similar, it was inferred that all M. hyopneumoniae (pMHGFP-P97) were attached to the external surface of macrophages. An unexpected finding was that more M. hyopneumoniae (pMHGFP-P97) appeared bound to CCD-treated macrophages than untreated macrophages. This might be explained by an unanticipated side effect where CCD caused the macrophages to detach from the surface of the culture dish. Hence, while in suspension the CCD-treated macrophages may have encountered M. hyopneumoniae (pMHGFP-P97) more frequently than the adhered untreated macrophages. The confocal microscopy experiments also confirmed M. hyopneumoniae (pMHGFP-P97) adhered to the surface of macrophages, some mycoplasmas even appeared flattened to the macrophage cell surface. In contrast to results from the flow cytometry experiments, analysis of confocal micrographs revealed a small number of M. hyopneumoniae (pMHGFP-P97) were in-fact located in the cytoplasm of porcine alveolar macrophages. The slightly differing results of the confocal microscopy and flow cytometer experiments may have been due to lower sensitivity of the flow cytometer to detect internalisation of such a low number of M. hyopneumoniae (pMHGFP-P97). It is also noted that the gentamicin survival and flow cytometry experiments utilised macrophages originating from the same pig, whereas macrophages from another animal were used in the confocal microscopy experiments (both animals were derived from the same source at the same time). Consequently, variability in the activity of macrophages obtained from different animals should be considered. However, the phagocytic capability of both macrophage batches appeared comparable throughout this investigation, as demonstrated by engulfment of the control organism, E. coli, via three different methods.
It appeared that uptake of M. hyopneumoniae by porcine alveolar macrophages was limited and that this mycoplasma may even resist significant uptake. Powell and Clyde [42] found that in the absence of antibody M. pneumoniae also readily associated with the membrane of guinea pig alveolar macrophages, but were rarely found inside macrophages. The addition of hyperimmune rabbit antiserum however led to uptake of M. pneumoniae [42]. In the present study, the gentamicin survival, flow cytometry and initial confocal microscopy experiments had been performed in the absence of host opsonins. To address this, M. hyopneumoniae (pMHGFP-P97) were opsonised with porcine serum containing complement, or convalescent serum containing complement and anti-M. hyopneumoniae antibody. M. hyopneumoniae (pMHGFP-P97) were not susceptible to the bactericidal activity of serum complement, unlike the control E. coli (pKENmut2) which was killed by exposure to serum (data not shown). Analysis of confocal micrographs suggested serum complement did not enhance phagocytic uptake of M. hyopneumoniae (pMHGFP-P97), and contrary to expectation neither did the convalescent sera. It was not entirely surprising that a pathogenic organism like M. hyopneumoniae exhibited resistance to the bactericidal activity of complement. Although M. hyopneumoniae appeared resistant to complement killing, deposition of complement onto the surface of M. hyopneumoniae cells was not determined in this study and could not be ruled out.
M. hyopneumoniae convalescent serum had been purposefully excluded from some experiments to mimic the in vivo environment of the naïve host, and provide more insight into the initial interactions between M. hyopneumoniae and alveolar macrophages. It was surprising to find that antibody from three different convalescent sera did not promote engulfment of M. hyopneumoniae. Other investigators have reported swift uptake of mycoplasmas such as M. dispar, M. pulmonis, M. ovipneumoniae, M. pneumoniae and M. bovis by macrophages following antibody opsonisation [17, 18, 21, 22, 42]. A study by Bonnefois et al. [40] investigated uptake of fluorescently labelled M. bovis by monocyte-derived macrophages using confocal microscopy. The fluorescent M. bovis were opsonised with pooled bovine convalescent serum (ten animals), and within 30 min of co-incubation green fluorescent clumps of M. bovis were observed inside the macrophages. In light of these findings for M. bovis it was expected that convalescent serum would promote uptake of M. hyopneumoniae (pMHGFP-P97), but this was not the case. Although M. hyopneumoniae (pMHGFP-P97) were present inside macrophages, the number of intracellular mycoplasmas did not differ in the presence of naïve or convalescent serum; and intracellular M. hyopneumoniae (pMHGFP-P97) were located discreetly in the macrophage cytoplasm. It remains to be determined the mechanism by which the mycoplasmas entered macrophages. It was assumed that intracellular M. hyopneumoniae resulted from phagocytic uptake, yet a recent publication has offered a potential alternative mechanism. Raymond et al. [37] have suggested that M. hyopneumoniae appears to enter PK-15 cells by exploiting clathrin- and caveolae-mediated endocytosis pathways; these pathways are available in porcine alveolar macrophages [43, 44]. The intracellular M. hyopneumoniae were apparently able to survive fusion of endocytic vesicles with lysosomes and escape into the cell cytoplasm. So far, the authors provide only limited evidence that M. hyopneumoniae might invade respiratory epithelial cells in vivo [37]. However, the data presented by Raymond et al. [37] do challenge the long-held belief that M. hyopneumoniae is a strict extracellular pathogen.
It is not clear why convalescent sera from experimentally-infected pigs failed to promote phagocytosis of M. hyopneumoniae (pMHGFP-P97) in the present investigation. Two of the convalescent sera, sera B and C, were raised against heterologous strains of M. hyopneumoniae compared to M. hyopneumoniae (pMHGFP-P97) used in the present study. Hence, it is possible that strain differences prevented efficient opsonisation. However, convalescent serum A was produced following infection with M. hyopneumoniae strain 232, so should have contained antibody recognising surface antigens of M. hyopneumoniae (pMHGFP-P97). It is conceivable that the porcine host did not produce effective opsonising antibody isotypes IgG1 and IgG3 in this instance. This raises questions about the efficacy of the porcine serum response to M. hyopneumoniae infection. Anecdotally, production of antibody following M. hyopneumoniae infection can take many weeks and does not correlate well with resolution of disease. It must be noted that in this investigation the opsonising powers of mucosal antibody isotypes IgA1 and IgA2 were not examined. Experimental evidence has suggested that humoral immunity alone is not enough to overcome M. hyopneumoniae infection, a cell-mediated immune response is also required [8, 45, 46]. An alternative explanation may be that M. hyopneumoniae is preventing antibody opsonophagocytosis in some way. An antibody capture and cleavage system, designated MIB-MIP, has been identified in M. mycoides subsp. capri which may function as a virulence mechanism [47]. The authors identified potential homologues of the MIB-MIP system in other Mycoplasma spp. which included M. hyopneumoniae. The expression and functionality of a putative M. hyopneumoniae MIB-MIP system requires further investigation to determine whether this potential virulence mechanism aides M. hyopneumoniae in the evasion of humoral effector functions such as opsonophagocytosis.
In conclusion, results indicate that M. hyopneumoniae is not readily engulfed by primary porcine alveolar macrophages, even in the presence of convalescent porcine serum. To strengthen further this observation of an antiphagocytic property, different strains of M. hyopneumoniae could be evaluated following exposure to a larger panel of convalescent serum, including serum from vaccinated animals; this would also aid analysis of the opsonising power of the humoral response against M. hyopneumoniae. Overall, this investigation has shed more light on the interactions between M. hyopneumoniae and porcine alveolar macrophages, in particular it has identified a possible mechanism of immune evasion by this important pig pathogen.
Acknowledgements
Dr Andrew Hibbert and Dr Benjamin R Raymond are acknowledged for their advice and assistance with the confocal microscopy aspect of this investigation. Dr Jessica Beddow and Laura Richards are thanked for their assistance in the collection of primary porcine alveolar macrophages.
Authors’ contributions
Experiments were conceived by ASD and ANR. GAM, EJ, LC and SC made substantial contributions to experimental design. Experiments were performed by ASD. LC and SC made substantial contributions to data acquisition. The manuscript was written by ASD, revised by ANR and approved by all authors. All authors read and approved the final manuscript.
Funding
This study was funded by the Royal Veterinary College, heritage-Merial (now Boehringer Ingelheim) and the Biotechnology and Biological Sciences Research Council (BB/G020744/1).
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alannah S. Deeney, Email: adeeney@rvc.ac.uk
Gareth A. Maglennon, Email: garethadam.maglennon@astrazeneca.com
Ludivine Chapat, Email: Ludivine.CHAPAT@boehringer-ingelheim.com.
Steve Crussard, Email: Steve.CRUSSARD@boehringer-ingelheim.com.
Edmond Jolivet, Email: Edmond.JOLIVET@boehringer-ingelheim.com.
Andrew N. Rycroft, Email: arycroft@rvc.ac.uk
References
- 1.Kobisch M, Friis NF. Swine mycoplasmoses. Rev Sci Tech. 1996;15:1569–1605. doi: 10.20506/rst.15.4.983. [DOI] [PubMed] [Google Scholar]
- 2.Maes D, Deluyker H, Verdonck M, Castryck F, Miry C, Lein A, Vrijens B, de Kruif A. The effect of vaccination against Mycoplasma hyopneumoniae in pig herds with a continuous production system. Zentralbl Veterinarmed B. 1998;45:495–505. doi: 10.1111/j.1439-0450.1998.tb00820.x. [DOI] [PubMed] [Google Scholar]
- 3.Pointon AM, Byrt D, Heap P. Effect of enzootic pneumonia of pigs on growth performance. Aust Vet J. 1985;62:13–18. doi: 10.1111/j.1751-0813.1985.tb06032.x. [DOI] [PubMed] [Google Scholar]
- 4.Ciprian A, Pijoan C, Cruz T, Camacho J, Tortora J, Colmenares G, Lopez-Revilla R, de la Garza M. Mycoplasma hyopneumoniae increases the susceptibility of pigs to experimental Pasteurella multocida pneumonia. Can J Vet Res. 1988;52:434–438. [PMC free article] [PubMed] [Google Scholar]
- 5.Maes D, Verdonck M, Deluyker H, de Kruif A. Enzootic pneumonia in pigs. Vet Q. 1996;18:104–109. doi: 10.1080/01652176.1996.9694628. [DOI] [PubMed] [Google Scholar]
- 6.Kennedy G. Virus pneumonia of pigs. Iowa State Univ Vet. 1960;22:81–84. [Google Scholar]
- 7.Blanchard B, Vena MM, Cavalier A, Le Lannic J, Gouranton J, Kobisch M. Electron microscopic observation of the respiratory tract of SPF piglets inoculated with Mycoplasma hyopneumoniae. Vet Microbiol. 1992;30:329–341. doi: 10.1016/0378-1135(92)90020-T. [DOI] [PubMed] [Google Scholar]
- 8.Maes D, Segales J, Meyns T, Sibila M, Pieters M, Haesebrouck F. Control of Mycoplasma hyopneumoniae infections in pigs. Vet Microbiol. 2008;126:297–309. doi: 10.1016/j.vetmic.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng ZX, Shao GQ, Liu MJ, Wu XS, Zhou YQ, Gan Y. Immune responses to the attenuated Mycoplasma hyopneumoniae 168 strain vaccine by intrapulmonic immunization in piglets. Agric Sci China. 2010;9:423–431. doi: 10.1016/S1671-2927(09)60113-9. [DOI] [Google Scholar]
- 10.Maes D, Sibila M, Kuhnert P, Segales J, Haesebrouck F, Pieters M. Update on Mycoplasma hyopneumoniae infections in pigs: knowledge gaps for improved disease control. Transbound Emerg Dis. 2018;65(Suppl 1):110–124. doi: 10.1111/tbed.12677. [DOI] [PubMed] [Google Scholar]
- 11.Maes D. Vaccination against Mycoplasma hyopneumoniae infection in pigs: room for improvement. Vet J. 2014;200:214–215. doi: 10.1016/j.tvjl.2014.02.023. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Young TF, Ross RF. Identification and characterization of a Mycoplasma hyopneumoniae adhesin. Infect Immun. 1995;63:1013–1019. doi: 10.1128/iai.63.3.1013-1019.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu T, Minion FC. Molecular analysis of the P97 cilium adhesin operon of Mycoplasma hyopneumoniae. Gene. 1998;214:13–23. doi: 10.1016/S0378-1119(98)00247-9. [DOI] [PubMed] [Google Scholar]
- 14.Robinson MW, Buchtmann KA, Jenkins C, Tacchi JL, Raymond BB, To J, Roy Chowdhury P, Woolley LK, Labbate M, Turnbull L, Whitchurch CB, Padula MP, Djordjevic SP. MHJ_0125 is an M42 glutamyl aminopeptidase that moonlights as a multifunctional adhesin on the surface of Mycoplasma hyopneumoniae. Open Biol. 2013;3:130017. doi: 10.1098/rsob.130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarocki VM, Santos J, Tacchi JL, Raymond BB, Deutscher AT, Jenkins C, Padula MP, Djordjevic SP. MHJ_0461 is a multifunctional leucine aminopeptidase on the surface of Mycoplasma hyopneumoniae. Open Biol. 2015;5:140175. doi: 10.1098/rsob.140175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy K, Travers P, Walport M, Janeway C. Janeway’s immunobiology. 8. New York: Garland Science; 2012. Chapter 1: basic concepts in immunology; pp. 5–7. [Google Scholar]
- 17.Howard CJ, Taylor G, Collins J, Gourlay RN. Interaction of Mycoplasma dispar and Mycoplasma agalactiae subsp. bovis with bovine alveolar macrophages and bovine lacteal polymorphonuclear leukocytes. Infect Immun. 1976;14:11–17. doi: 10.1128/iai.14.1.11-17.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis JK, Delozier KM, Asa DK, Minion FC, Cassell GH. Interactions between murine alveolar macrophages and Mycoplasma pulmonis in vitro. Infect Immun. 1980;29:590–599. doi: 10.1128/iai.29.2.590-599.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw BM, Simmons WL, Dybvig K. The Vsa shield of Mycoplasma pulmonis is antiphagocytic. Infect Immun. 2012;80:704–709. doi: 10.1128/IAI.06009-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Busolo F, Tonellato L, Scremin L, Tonin E, Bertoloni G, Franceschi C. Phagocytosis of Mycoplasma pneumoniae and Acholeplasma laidlawii measured as inhibition of [3H]uridine uptake by macrophages. J Immunol Methods. 1986;90:235–240. doi: 10.1016/0022-1759(86)90080-3. [DOI] [PubMed] [Google Scholar]
- 21.Almeida RA, Wannemuehler MJ, Rosenbusch RF. Interaction of Mycoplasma dispar with bovine alveolar macrophages. Infect Immun. 1992;60:2914–2919. doi: 10.1128/iai.60.7.2914-2919.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al-Kaissi A, Alley MR. Electron microscopic studies of the interaction between ovine alveolar macrophages and Mycoplasma ovipneumoniae in vitro. Vet Microbiol. 1983;8:571–584. doi: 10.1016/0378-1135(83)90006-8. [DOI] [PubMed] [Google Scholar]
- 23.L’Ecuyer C, Switzer WP. Virus pneumonia of pigs; attempts at propagation of the causative agent in cell cultures and chicken embryos. Can J Comp Med Vet Sci. 1963;27:91–99. [PMC free article] [PubMed] [Google Scholar]
- 24.Maré CJ, Switzer WP. New species: Mycoplasma hyopneumoniae; a causative agent of virus pig pneumonia. Vet Med Small Anim Clin. 1965;60:841–846. [PubMed] [Google Scholar]
- 25.Cook BS, Beddow JG, Manso-Silvan L, Maglennon GA, Rycroft AN. Selective medium for culture of Mycoplasma hyopneumoniae. Vet Microbiol. 2016;195:158–164. doi: 10.1016/j.vetmic.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cullen JM, Rycroft AN. Phagocytosis by pig alveolar macrophages of Actinobacillus pleuropneumoniae serotype 2 mutant strains defective in haemolysin II (ApxII) and pleurotoxin (ApxIII) Microbiology. 1994;140:237–244. doi: 10.1099/13500872-140-2-237. [DOI] [PubMed] [Google Scholar]
- 27.Piriou-Guzylack L, Salmon H. Membrane markers of the immune cells in swine: an update. Vet Res. 2008;39:54. doi: 10.1051/vetres:2008030. [DOI] [PubMed] [Google Scholar]
- 28.Maglennon GA, Cook BS, Deeney AS, Bossé JT, Peters SE, Langford PR, Maskell DJ, Tucker AW, Wren BW, Rycroft AN, Consortium BT Transposon mutagenesis in Mycoplasma hyopneumoniae using a novel mariner-based system for generating random mutations. Vet Res. 2013;44:124. doi: 10.1186/1297-9716-44-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maglennon GA, Cook BS, Matthews D, Deeney AS, Bossé JT, Langford PR, Maskell DJ, Tucker AW, Wren BW, Rycroft AN, Consortium BT Development of a self-replicating plasmid system for Mycoplasma hyopneumoniae. Vet Res. 2013;44:63. doi: 10.1186/1297-9716-44-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 31.Burki S, Gaschen V, Stoffel MH, Stojiljkovic A, Frey J, Kuehni-Boghenbor K, Pilo P. Invasion and persistence of Mycoplasma bovis in embryonic calf turbinate cells. Vet Res. 2015;46:53. doi: 10.1186/s13567-015-0194-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gille C, Spring B, Tewes L, Poets CF, Orlikowsky T. A new method to quantify phagocytosis and intracellular degradation using green fluorescent protein-labeled Escherichia coli: comparison of cord blood macrophages and peripheral blood macrophages of healthy adults. Cytom A. 2006;69:152–154. doi: 10.1002/cyto.a.20222. [DOI] [PubMed] [Google Scholar]
- 33.Gille C, Leiber A, Mundle I, Spring B, Abele H, Spellerberg B, Hartmann H, Poets CF, Orlikowsky TW. Phagocytosis and postphagocytic reaction of cord blood and adult blood monocyte after infection with green fluorescent protein-labeled Escherichia coli and group B Streptococci. Cytom B Clin Cytom. 2009;76:271–284. doi: 10.1002/cyto.b.20474. [DOI] [PubMed] [Google Scholar]
- 34.Bai F, Ni B, Liu M, Feng Z, Xiong Q, Xiao S, Shao G. Mycoplasma hyopneumoniae-derived lipid-associated membrane proteins induce apoptosis in porcine alveolar macrophage via increasing nitric oxide production, oxidative stress, and caspase-3 activation. Vet Immunol Immunopathol. 2013;155:155–161. doi: 10.1016/j.vetimm.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Zhou D, Yuan F, Liu Z, Duan Z, Yang K, Guo R, Li M, Li S, Fang L, Xiao S, Tian Y. Surface proteins mhp390 (P68) contributes to cilium adherence and mediates inflammation and apoptosis in Mycoplasma hyopneumoniae. Microb Pathog. 2018;126:92–100. doi: 10.1016/j.micpath.2018.10.035. [DOI] [PubMed] [Google Scholar]
- 36.Shaw BM, Daubenspeck JM, Simmons WL, Dybvig K. EPS-I polysaccharide protects Mycoplasma pulmonis from phagocytosis. FEMS Microbiol Lett. 2013;338:155–160. doi: 10.1111/1574-6968.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raymond BBA, Turnbull L, Jenkins C, Madhkoor R, Schleicher I, Uphoff CC, Whitchurch CB, Rohde M, Djordjevic SP. Mycoplasma hyopneumoniae resides intracellularly within porcine epithelial cells. Sci Rep. 2018;8:17697. doi: 10.1038/s41598-018-36054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishag HZ, Liu MJ, Yang RS, Xiong QY, Feng ZX, Shao GQ. A replicating plasmid-based vector for GFP expression in Mycoplasma hyopneumoniae. Genet Mol Res. 2016;15:gmr7832. doi: 10.4238/gmr.15027832. [DOI] [PubMed] [Google Scholar]
- 39.Tulum I, Yabe M, Uenoyama A, Miyata M. Localization of P42 and F(1)-ATPase α-subunit homolog of the gliding machinery in Mycoplasma mobile revealed by newly developed gene manipulation and fluorescent protein tagging. J Bacteriol. 2014;196:1815–1824. doi: 10.1128/JB.01418-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonnefois T, Vernerey MS, Rodrigues V, Totte P, Puech C, Ripoll C, Thiaucourt F, Manso-Silvan L. Development of fluorescence expression tools to study host–mycoplasma interactions and validation in two distant mycoplasma clades. J Biotechnol. 2016;236:35–44. doi: 10.1016/j.jbiotec.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 41.Balish MF, Santurri RT, Ricci AM, Lee KK, Krause DC. Localization of Mycoplasma pneumoniae cytadherence-associated protein HMW2 by fusion with green fluorescent protein: implications for attachment organelle structure. Mol Microbiol. 2003;47:49–60. doi: 10.1046/j.1365-2958.2003.03282.x. [DOI] [PubMed] [Google Scholar]
- 42.Powell DA, Clyde WA., Jr Opsonin-reversible resistance of Mycoplasma pneumoniae to in vitro phagocytosis by alveolar macrophages. Infect Immun. 1975;11:540–550. doi: 10.1128/iai.11.3.540-550.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galindo I, Cuesta-Geijo MA, Hlavova K, Munoz-Moreno R, Barrado-Gil L, Dominguez J, Alonso C. African swine fever virus infects macrophages, the natural host cells, via clathrin- and cholesterol-dependent endocytosis. Virus Res. 2015;200:45–55. doi: 10.1016/j.virusres.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 44.Ning P, Gao L, Zhou Y, Hu C, Lin Z, Gong C, Guo K, Zhang X. Caveolin-1-mediated endocytic pathway is involved in classical swine fever virus Shimen infection of porcine alveolar macrophages. Vet Microbiol. 2016;195:81–86. doi: 10.1016/j.vetmic.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 45.Thacker EL, Thacker BJ, Kuhn M, Hawkins PA, Waters WR. Evaluation of local and systemic immune responses induced by intramuscular injection of a Mycoplasma hyopneumoniae bacterin to pigs. Am J Vet Res. 2000;61:1384–1389. doi: 10.2460/ajvr.2000.61.1384. [DOI] [PubMed] [Google Scholar]
- 46.Xiong Q, Wei Y, Feng Z, Gan Y, Liu Z, Liu M, Bai F, Shao G. Protective efficacy of a live attenuated Mycoplasma hyopneumoniae vaccine with an ISCOM-matrix adjuvant in pigs. Vet J. 2014;199:268–274. doi: 10.1016/j.tvjl.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 47.Arfi Y, Minder L, Di Primo C, Le Roy A, Ebel C, Coquet L, Claverol S, Vashee S, Jores J, Blanchard A, Sirand-Pugnet P. MIB-MIP is a mycoplasma system that captures and cleaves immunoglobulin G. Proc Natl Acad Sci USA. 2016;113:5406–5411. doi: 10.1073/pnas.1600546113. [DOI] [PMC free article] [PubMed] [Google Scholar]