

ABSTRACT
Sezary Syndrome is an aggressive T-cell Lymphoma involving blood, skin and lymphonodes Involvement of the CXCR4-SDF1 has been previously shown. We here present evidence also of the involvement of B-arrestin a downstream regulator of CXCR4, that is depleted and downregulated as well as a potential functional role for this depletion.
KEYWORDS: Leukemia/lymphoma, CTCL, B-arrestin, Copy Number Variation, CXCR4-SDF-1
Sezary Syndrome (SS) is a rare and aggressive cutaneous T cell-lymphoma (CTCL) characterized by the triad of erythroderma, lymphadenopathy and circulating malignant lymphocytes, named Sezary cells [1]. SS cells derive from central memory T-lymphocytes, long-lived cells with a high proliferative potential that actively recirculate between blood, lymph-node and skin [2]. SS is characterized by many genetic events such as mutations, copy number variations, unisomal parental disomy, noncoding RNA alterations, etc., who regulate many patterns such as DNA repair, cell cycle, apoptotic and TCR-signaling mechanisms [3]. Despite the many high-throughput data generated so far, a major genetic defect responsible for the SS disease initiation and/or progression is not yet identified.
An important role in SS pathogenesis and skin homing of malignant T-cells is sustained by the chemokine receptor CXCR4 who is up-regulated by SS cells and functionally able to drive chemotaxis toward its ligand SDF-1, abundantly observed in the skin of these patients [4]. CXCR4 is internalized from plasma membrane following SDF-1 binding by β-arrestin-2 (ARRB2). Upon this process, ARRB2 plays a crucial role for the CXCR4 degradation and recycling and, after these steps, it acts as signaling scaffold for the MAPK/ERK pathways involved in cell growth, differentiation and migration [5]. Interestingly, ARRB2 gene maps to the chromosomal region 17p13.2 that represents the most frequent chromosomal deletion observed in about 60% of SS, in the form of Iso(17q) [6] . This Iso(17q) is an abnormal chromosome 17 with two identical long (q) arms due to duplication and loss of the short (p) arm. We thus analyzed the copy number variation (CNV) of ARRB2 gene using Affymetrix array and droplet digital PCR conducted in the same cohort of 63 SS samples and three CTCL cell lines (Hut78, H9 and HH cells) investigated before [7]. We found the mono-allelic loss of ARRB2 in 38/63 samples (60%), in agreement with previous cytogenetic data. Hut78 cells showed a WT status while H9 cells displayed a CN gain of this gene. mRNA and protein expression of ARRB2 were evaluated by Real Time PCR and Western Blotting, and the majority of SS samples showed a consistent reduction of ARRB2 at mRNA (60%) and/or protein level (70%) respect healthy controls (HC). We also observed an enhanced ARRB2 protein level in H9 respect to Hut78 cells.
To determine if ARRB2 regulates the CXCR4 in SS, we next compared the CXCR4 expression through FACS analysis between H9 cells depleted of ARRB2 by small interfering RNA (siRNA) (51% of ARRB2 protein reduction) and scrambled control. Following SDF-1 stimulation, ARRB2 siRNA cells showed a higher percentage of CXCR4+ cells respect to control thus indicating that ARRB2 loss causes a defect in the CXCR4 internalization process in this system (Figure 1(a,b)). Finally, using a chemotaxis assay we observed that H9 cells with a CN gain of ARRB2 showed an enhanced migration response to SDF-1 respect to Hut78 with a WT status of ARRB2 (Figure1(c)). An enhanced response to SDF1 was also observed in scrambled H9 cells respect to depleted arrestin beta 2 siRNA H9 cells. In agreement with this finding, we also noted in depleted arrestin siRNA cells, a reduction of ERK and p38 phosphorylation, two key constituents of the MAPK cascade.
Figure 1.
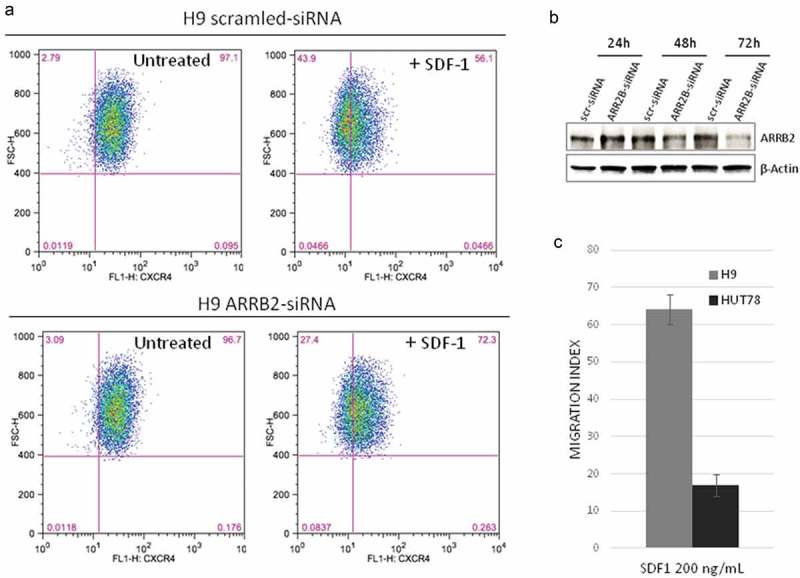
ARRB2 Depletion decreases CXCR4 internalization and migration index upon SDF-1 treatment.
(A) H9 cells depleted ofARRB2 by siRNA transfection (lower panel) show an increased number of CXCR4+cells upon SDF-1 treatment, when compared to scrambled control (upper panel). One representative experiment is shown in figure (n = 4, 66 ± 8,57 vs 54,93 ± 6,05; P = 0.039). (B) Western blot analysis confirms a reduction of ARRB2 protein in siRNA-transfected H9 cells respect to control (scr-siRNA) after 48 and 72 hours. (C) H9 cells (ARRB2 CN gain) show a higher migration response to SDF-1 than Hut78 cells (ARRB2W1) (n.4, FC = 3,85, P < 0.001).
In this scenario, the loss of ARRB2 sheds new light on the role of CXCR4-SDF-1 in SS, reinforcing the importance of this axis, already supported by multiple findings: the SS cells do not express SDF-1 mRNA, but rather they seem to uptake the SDF-1 released from epithelial, dendritic and endothelial cells of the skin. Thus, SS cells move toward skin through a chemotactic gradient [4]. This hypothesis is further sustained by the lack from SS cell surface of CD26 peptidase able to cleave and inactivate SDF-1, by the versican overexpression, a proteoglycan that enhances the SS locomotion toward SDF-1 [8] and by the comparable levels of plasmatic SDF-1 found between normal and SS individuals. In this context, the frequent loss of ARRB2 can further affect the trafficking ability of SS cells that, unlike their physiological counterpart, may persist longer in the skin causing the intense pruritus, lichenification, exfoliation and edema observed in this lymphoma. Beyond migration, we recently also demonstrated that SDF-1 activates the TORC1signaling and promotes proliferation in primary skin-derived SS cells [8]. In conclusion, ARRB2 loss can further immobilize the SS cells in the skin contributing to their cellular energy supply and proliferation stimuli provided by mTORC1 pathway activation.
Funding Statement
This study was supported by the Associazione Italiana per la Ricerca sul Cancro (IG17048 to M.G.N).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Olsen EA, Whittaker S, Kim YH, et al. Clinical end points and response criteria in mycosis fungoides and Sézary Syndrome: a consensus statement of the international society for cutaneous lymphomas, the United States cutaneous lymphoma consortium, and the cutaneous lymphoma task force of the european organisation for research and treatment of cancer. J Clin Oncol. 2011;29:2598–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Campbell JJ, Clark RA, Watanabe R, et al. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010. August 5;116(5):767–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Choi J, Goh G, Walradt T, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015. September;47(9):1011–1019. Epub 2015 Jul 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Narducci MG, Scala E, Bresin A, et al. Skin homing of Sézary cells involves SDF-1-CXCR4 signaling and down-regulation of CD26/dipeptidylpeptidase IV. Blood. 2006. February 1;107(3):1108–1115. Epub 2005 Oct 4. [DOI] [PubMed] [Google Scholar]
- [5].Song Q, Ji Q, Li Q The role and mechanism of B-arrestin in cancer invasion and metastasis (review). Int J Mol Med. 2018. Feb;41(2):631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Caprini E, Diks SH, Den Dunnen WFA, et al. Kinome profiling in pediatric brain tumors as a new approach for target discovery. Cancer Res. 2009. November 1;69(21):8438–8446. [DOI] [PubMed] [Google Scholar]
- [7].Cristofoletti C, Bresin A, Picozza M, et al Blood and skin-derived Sezary cells: differences in proliferation-index, activation of PI3K/AKT/mTORC1 pathway and its prognostic relevance. Leukemia. 2018. December 5 DOI: 10.1038/s41375-018-0305-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fujii K, Page BDG, Kraft IL, et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia. 2015. October;29(3):586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]