

ABSTRACT
Mutations in genes encoding components of the DNA damage response (DDR) are among the most frequent aberrations in human tumors. Moreover, a large array of human syndromes is caused by mutations in genes involved in DDR pathways. Among others, homologous recombination repair (HR) of DNA double-strand breaks (DSB) is frequently affected by disabling mutations. While impaired HR is clearly promoting tumorigenesis, it is also associated with an actionable sensitivity against PARP inhibitors. PARP inhibitors have recently received FDA approval for the treatment of breast- and ovarian cancer. However, as with all molecularly targeted agents, acquired resistance limits its use. Both pharmaco-genomic approaches and the study of human genome instability syndromes have led to a profound understanding of PARP inhibitor resistance. These experiments have revealed new insights into the molecular mechanisms that drive mammalian DSB repair. Here, we review recent discoveries in the field and provide a clinical perspective.
KEYWORDS: Double-strand break (DSB), genome instability, homologous recombination repair, non-homologous end joining, DSB repair pathway choice, PARP inhibitor
Introduction
Cancer remains a major medical challenge and the incidence rates will likely continue to increase in the Western world, as demographic changes will lead to aging societies. The arguably most important task of modern-day cancer research is the identification and molecular characterization of tumor-specific (epi)genetic aberrations, which provide entry routes for targeted therapeutic interventions. A number of oncogenic driver lesions, including mutant EGFR or EML4-ALK- and BCR-ABL rearrangements, constitute prime examples for such directly druggable cancer-specific genetic alterations [1–3]. Targeted inhibition of these oncogenically-rewired signaling pathways in oncogene-addicted malignancies has produced impressive clinical responses, particularly in sometimes extensively pre-treated patients. Unfortunately, a large number of oncogenic driver lesions exist for which no direct therapeutic targeting is currently available. Important examples of these drivers include transcription factors, such as MYC family members, non-kinase oncogenes, such as RAS family members, as well as mutationally or epigenetically silenced tumor suppressor genes, such as TP53 and RB1 [4–6]. Hence, novel therapeutic concepts have been developed to indirectly target these oncogenic driver lesions.
Mammalian cells have evolved distinct genome maintenance mechanisms
Following DNA damage, cells activate a complex signaling network, commonly referred to as the DNA damage response (DDR) [7]. DDR signaling induces cell cycle checkpoints and thus allows time for DNA repair, or, if the lesions are beyond repair capacity, leads to the activation of cell death pathways [7]. Perhaps not surprisingly, genes encoding for different DDR signaling components and particularly of DNA repair pathways, are among the most frequently mutated genes in human malignancies [4,8–10]. It has been proposed that the resulting molecular defects in genome maintenance pathways drive a so-called “mutator phenotype”, ultimately leading to the acquisition of additional cancer-promoting or -maintaining genomic aberrations [11–15]. While these mutationally-encoded genome maintenance defects likely contribute to the process of malignant transformation, they may also represent entry routes for cancer-specific therapeutic interventions. However, in order to selectively target cancerous cells, a fundamental understanding of the biological effects of these genome maintenance defects is of critical importance.
Mammalian cells evolved a series of distinct DNA repair mechanisms in order to remove structurally distinct DNA aberrations throughout the different phases of the cell cycle [9]. These pathways, which are extensively reviewed elsewhere [9,16–18], include mismatch repair (MMR), trans-lesion synthesis (TLS), the Fanconi anemia (FA) pathway and nucleotide excision repair (NER). NER in particular, has evolved to cope with helix-distorting lesions, such as those inflicted by cigarette smoke and UV light, as well as crosslinks between guanine bases induced by platinum-containing chemotherapies [19]. The ERCC1/XPF endonuclease complex mediates NER repair and in addition, excision repair cross-complementation group 1 (ERCC1) is involved in recombination mediated DNA repair, as well as repair of inter-strand crosslinks, such as those inflicted by cisplatin [20]. In line with this, reduced expression of ERCC1 is associated with cisplatin sensitivity in tumors [20]. Mutations in genes encoding for MMR pathway components are associated with high tumor mutational burden. The FDA has recently approved the immune checkpoint inhibitor pembrolizumab for the treatment of solid tumors harboring either microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) genetic aberrations [21].
Here, we will primarily focus on DNA double-strand break (DSB) repair. Human cells are equipped with two distinct dominant DSB repair pathways, namely the error-prone classical non-homologous end joining (cNHEJ) pathway and the error-free homologous recombination repair (HR) pathway [16]. HR and cNHEJ act as complementary partners for efficient DSB repair (Figure 1).
Figure 1.

Balancing DNA double-strand break (DSB) repair.
Homologous recombination repair (HR) and classical non-homologous end joining (cNHEJ) are orchestrated in a balanced DSB repair response. cNHEJ initiates with recognition of the DNA ends by the Ku70/Ku80 heterodimer, which then recruits the catalytic subunit of the DNA dependent protein kinase (DNA-PKcs). If necessary, ends can be trimmed by nucleases (e.g. Artemis) to create compatible ends. The ligation complex consisting of DNA ligase IV and XRCC4 ligates the ends. In contrast, HR is initiated by the nuclease MRE11, which generates together with CtIP and EXO1 single stranded DNA (ssDNA). The ssDNA is coated by RPA, which is subsequently replaced by RAD51 in a BRCA2-dependent fashion. RAD51 nucleoprotein filaments mediate strand invasion on the homologous sister chromatid. MRN (MRE11, RAD50, NBS1).
HR usage is largely restricted to the S- and G2-phases of the cell cycle, as the HR mechanism requires the availability of an intact DNA template [22]. The initial step of the HR process involves resection of the DSB, which is mediated by a series of endo- and exonucleases, including MRE11, EXO1 and DNA2 [23] (Figure 1). In an initial step, MRE11 performs an endonucleolytic incision, which is followed by its exonuclease activity digesting 3’-5’ toward the DNA end coupled with EXO1/BLM activity carrying out 5’-3’ resection away from the end [24]. Importantly, MRE11 plays a key role in DNA end-resection by promoting the initiation of the resection process, rather than driving resection processivity [25,26]. Following DNA end resection, the resulting 3′- single-stranded DNA (ssDNA) overhang is coated by the ssDNA-binding protein RPA [27,28] which is subsequently replaced by RAD51 in an ATM/CHK2/BRCA1/BRCA2/PALB2-dependent fashion [29–32]. The resulting RAD51-decorated ssDNA overhang ultimately invades the intact sister chromatid [30–32]. RAD51 is critically involved in homology search, strand exchange, and Holliday junction formation [22].
Throughout the HR process, it is important to note that patients harboring heterozygous germline mutations in different HR-genes, such as BRCA1, BRCA2 and RAD51C, carry a substantially increased risk for the development of cancer [29,33–37]. Moreover, somatic protein-damaging mutations in BRCA1, BRCA2, ATM, CHEK2, RAD50, RAD51C and others have been recurrently identified in numerous cancer entities [38–43]. Altogether, these clinical observations underscore the enormous importance of the HR pathway for cancer prevention.
In contrast to the HR mechanism, cNHEJ-mediated DSB repair does not require the presence of an intact template for effective DNA repair [44,45]. Thus, the cNHEJ mechanism is preferentially employed during the G1-phase of the cell cycle, where no intact template is available for DSB repair [44]. During cNHEJ-mediated DSB repair, the non-catalytic subunits Ku70 and Ku80 initially form a heterodimer, which detects and engages the free DNA ends [45]. This Ku70-Ku80 complex subsequently recruits the proximal DDR kinase DNA-PKcs to the site of the break. DNA-PKcs activity is critical to promote XRCC4- and LIG4-mediated DSB sealing [45] (Figure 1). While cNHEJ is a highly effective DSB repair mechanism, it is also intrinsically error-prone. cNHEJ-mediated DSB repair relies on the presence of a free 5’-phosphate and a 3’-hydroxyl group at each end of the broken DNA. Consequently, the original DNA sequence can only be restored if the DNA ends can be re-ligated without prior resection. However, if the DSB is not resulting from a disrupted phosphodiester bond, but rather involves the pentose sugar backbone of the DNA structure, re-ligation cannot proceed prior to a limited end-processing. The necessary modifications of the free DNA ends, involving nucleolytic cleavage and gap filling of the ends, are inaccurate, rendering cNHEJ-mediated DSB repair highly mutagenic [45]. Beyond cNHEJ, where DSB repair occurs at regions of microhomology without extensive DSB end resection, alternative NHEJ mechanisms, such as microhomology-mediated end joining (MMEJ) exist [9,46]. The MMEJ pathway involves strand resection and annealing of short areas of homology [9,46]. Analogously, the single-strand annealing (SSA) mechanism also relies on strand resection, prior to annealing of larger areas of homology and subsequent flap-processing and ligation (for excellent reviews see [46–48]). SSA thus represents a RAD51-independent homology-directed DSB repair mechanism, which is typically employed to repair DSBs localized between repetitive DNA elements, where two homologous sequence stretches are situated on either side of the DSB on the same chromatid [9,49]. SSA-mediated repair is initiated by recruiting RPA and RAD52 to the 3′-ssDNA overhangs [9,49]. Complementary sequences up- and downstream of the DSB are then annealed by the RPA/RAD52/ssDNA ternary complex [9,49]. The annealing products are typically flanked by displaced non-homologous 3’ flap DNA fragments, which are removed by ERCC1/XPF and MSH2/MSH3 complexes, which are canonical components of the NER and MMR pathways, respectively [9,49]. SSA-mediated DSB repair, thus, ultimately leads to the deletion of the DNA stretch between the homologous DNA repeat sequences used for annealing [9,49]. Hence, the SSA pathway represents an error-prone homology-mediated DSB repair mechanism.
Cancer-associated defects in homologous recombination are associated with PARP1 inhibitor sensitivity
Defective genome maintenance mechanisms clearly contribute to malignant transformation. However, these same molecular aberrations can also be associated with actionable vulnerabilities. This is perhaps best exemplified by the selective toxicity of PARP inhibitors in BRCA1- or BRCA2-mutant cells [50,51]. BRCA1 and BRCA2 are critical components of the HR pathway [9]. This initial observation was quickly further extended and it was demonstrated that defects in additional HR components, such as RAD51, RPA1, ATM, CHK2, TOPBP1 and others, were also associated with PARP inhibitor sensitivity [52–55]. The clinical relevance of these observations is underscored by the recent approval of the PARP inhibitor olaparib in two clinical settings: for the treatment of adult patients with deleterious germline BRCA1- or BRCA2-mutant (gBRCAm), HER2-negative metastatic breast cancer that have previously received chemo- or endocrine therapy and for the fourth line treatment of adult patients with gBRCAm advanced ovarian cancer [56,57]. While PARP inhibitors have proven their clinical efficacy in HR-defective cancer entities, the exact mechanism of action is still debated. PARP1 itself constitutes a component of the base excision repair (BER) pathway, which mediates DNA single-strand break repair [58]. Given its role in BER, PARP inhibition was initially proposed to cause an accumulation of persistent SSBs, which are occasionally converted to DSBs during DNA replication [50]. More recent data, however, suggest that some PARP inhibitors induce trapping of PARP1 on the DNA, preventing auto-PARylation and PARP1 release from the site of the DNA lesion [59–62]. It was further hypothesized that this trapped PARP1 enzyme is the relevant toxic equivalent, reminiscent of topoisomerase II inhibitors, such as etoposide, which also “trap” their target enzyme on the DNA. In line with this hypothesis, PARP1-deficient cells display PARP inhibitor resistance [59,62–64]. Further, unligated Okazaki fragments that are trapped by PARP inhibitor require HR for their removal, either directly as single-strand gaps or following their conversion into DSBs by nucleases or DNA replication fork collapse [65]. Moreover, through the use of RNA interference screens, mouse models and the analysis of specimens derived from breast and ovarian cancer patients, it was recently shown that loss of poly(ADP-ribose) glycohydrolase (PARG), an enzyme which removes PARP-mediated PARylations, drives resistance against PARP inhibitors in BRCA2-deficient settings [66]. Mechanistically, PARG depletion restored PAR formation and, at least partially, rescued PARP1 signaling [66]. Intriguingly, PARG repression did not only promote PARP inhibitor resistance, but was also associated with enhanced sensitivity against ionizing radiation [66].
Pharmaco-genetic approaches uncover novel regulators of DSB repair
As may have been expected, the clinical efficacy of PARP inhibitors for the treatment of patients harboring HR-defective tumors is limited by the occurrence of clinical resistance. The molecular characterization of PARP inhibitor resistance mechanisms over recent years, did not only provide therapeutic strategies to circumvent resistance, but also led to new mechanistic insights into the process of DSB repair. As the prerequisite for PARP inhibitor toxicity appears to be a defective HR mechanism, restoration of HR capacity and/or repression of NHEJ activity have emerged as PARP inhibitor resistance mechanisms in model systems and patients. The first evidence for such resistance mechanisms was provided by the observation that continued treatment of BRCA2-mutant cancer cells with olaparib or cisplatin led to the occurrence of resistant clones, which harbored secondary intra-genic BRCA2 mutations [67,68]. These intra-genic secondary aberrations led to the re-expression of HR-competent, PARP inhibitor resistance-mediating BRCA2 isoforms [67,68]. Similar secondary mutations were also reported in BRCA1- or BRCA2-mutant ovarian cancer patients, upon clinical manifestation of carboplatin resistance [67–69]. It is important to note that these secondary BRCA aberrations potentially mediate cross-resistance against platinum salts and PARP inhibitors [69]. This observation is particularly relevant for the sequential therapeutic management of patients with tumors harboring either somatic or germline BRCA mutations.
In addition to on-target resistance mechanisms, which directly affect one of the synthetic lethal partners (i.e. the BRCA gene or PARP1 itself), a series of off-target aberrations leading to PARP inhibitor resistance have recently emerged. For instance, the demethylase JMJD1C was recently shown to regulate the balance between HR and NHEJ [70]. Specifically, JMJD1C restricted the formation of RAD51 repair foci, which are a central feature of HR-mediated DSB repair [70]. Moreover, and in line with the notion that HR defects are associated with PARP inhibitor sensitivity, JMJD1C depletion promoted PARP inhibitor resistance [70]. Moreover, upregulation of RAD51 in BRCA1-defective cells is associated with resistance to PARP inhibitors [71]. RAD51 overexpression has been observed in a wide range of human cancers, notably in triple-negative breast cancer (TNBC) [72]. Using a genetic screen, Marzio and colleagues identified EMI1 as a modulator of PARP inhibitor sensitivity in TNBC particularly by targeting RAD51 for proteasome-mediated degradation [73]. Following genotoxic stress, the CHK1-mediated phosphorylation of RAD51 on Thr-309 increased its affinity for BRCA2 and decreased the affinity for the RAD51-EMI1 interaction, thereby allowing its accumulation and efficient HR [73]. Next to JMJD1C and EMI1, somatic loss of Tp53bp1 in Brca1- murine mammary tumors was shown to be associated with restoration of HR activity and pre-clinical resistance against olaparib [74]. In Brca1-deficient cancer cells, 53BP1 represses HR activity, which drives DSB repair towards the use of NHEJ, which in turn is cytotoxic, due to its inherent mutagenicity [75–77]. 53BP1 suppresses HR by preventing the nucleolytic resection of DSBs [75–77]. This function is dependent on interactions with PTIP and the downstream effector molecules RIF1 and REV7 [75–83]. Once 53BP1 function is abolished in Brca1-deficient settings, this HR suppression is relieved and PARP inhibitor-induced lesions are repaired efficiently with limited cytotoxicity [75]. The observation that loss of Tp53bp1 restores HR capacity in Brca1-deficient settings argues that BRCA1-independent HR activity is curtailed by 53BP1 [74]. Further support for this concept came from a report showing that loss of REV7 in murine and human BRCA1-deficient cells restores CTIP-dependent DSB end resection of DSBs, leading to HR re-activation and the subsequent manifestation of PARP inhibitor resistance [84]. It was further shown that REV7 is recruited to sites of DSBs in an γH2AX-MDC1-RNF8-RNF168-53BP1-RIF1-dependent fashion [84]. Thus, REV7 appears to operate downstream of 53BP1 in repressing BRCA1-independent HR activity. Overall, depletion of either 53BP1 or REV7 appears to provide synthetic viability to HR-defective cells.
Further insight into the molecular mechanisms by which the 53BP1-RIF1-REV7 pathway governs NHEJ-mediated DSB repair came from the recent discovery of the shieldin complex, consisting of REV7, SHLD1 (C20ORF196), SHLD2 (FAM35A) and SHLD3 (CTC-534A2.2) [85–90]. The different components of the shieldin complex were identified through mass spectrometry interaction screens with 53BP1 and REV7, but also through CRISPR/Cas9 synthetic viability screens in BRCA1-deficient cells exposed to PARP inhibitors [85–88]. The shieldin complex is recruited to DSBs in a 53BP1- and RIF1-dependent manner, and its SHLD2 subunit binds to single-stranded DNA via OB-fold domains, which are analogous to those found in the ssDNA-binding protein RPA [85–90]. ssDNA binding of SHLD2 was impaired when the two conserved Trp residues W489 and W640 were mutated to Ala [87]. Loss of individual shieldin subunits causes PARP inhibitor resistance in BRCA1-deficient cells and tumors, due to functional HR restoration [85–90]. Moreover, binding of ssDNA by SHLD2 is critical for shieldin function, consistent with a model in which shieldin protects DNA ends from excessive resection and to mediate 53BP1-dependent DNA repair [85–90] (Figure 2). Within the shieldin complex, SHLD3 directly interacts with REV7, through amino acids 28–83 in SHLD3 [85]. SHLD3 also binds to RIF1, thus physically linking the shieldin complex with the 53BP1-RIF1-REV7 axis [86]. Furthermore, the N-terminal region of SHLD2 mediates binding to SHLD3-REV7 [85]. The N-terminal amino acids 1–60 of SHLD2 were sufficient to interact with SHLD3-REV7 [85]. Particularly, amino acids 6–11 are critical for this interaction, as deletion of this amino acid stretch in SHLD2 abolished the interaction with SHLD3-REV7 [85]. Further interaction studies showed that the C-terminal SHLD2 domain (amino acids 650–835) is critical for interaction with SHLD1 [85]. Altogether, these biochemical data revealed that SHLD1/2/3 and REV7 interact within the shieldin complex. Functional experiments revealed that depletion of SHLD1, 2 or 3 facilitates excessive DSB resection, evidenced by increased RPA chromatin loading [85]. Moreover, loss of SHLD3 promoted CTIP-dependent increased RAD51 chromatin recruitment [85]. Intriguingly, it was also shown that loss of SHLD1 or 2 enhanced the cisplatin sensitivity of BRCA1-deficient cells [87]. Moreover, this increased cisplatin sensitivity in SHLD1- or 2-depleted cells was associated with increased formation of nuclear FANCD2 foci, a protein involved in the detection and repair of DNA crosslinks [9,87]. These observations indicate that, PARP inhibitor resistance due to loss of SHLD1 or 2 may be associated with cisplatin sensitivity. This is particularly important, as secondary BRCA mutations, which also constitute a PARP inhibitor resistance mechanism (see above), appear to mediate cross-resistance against platinum salts and PARP inhibitors [69].
Figure 2.
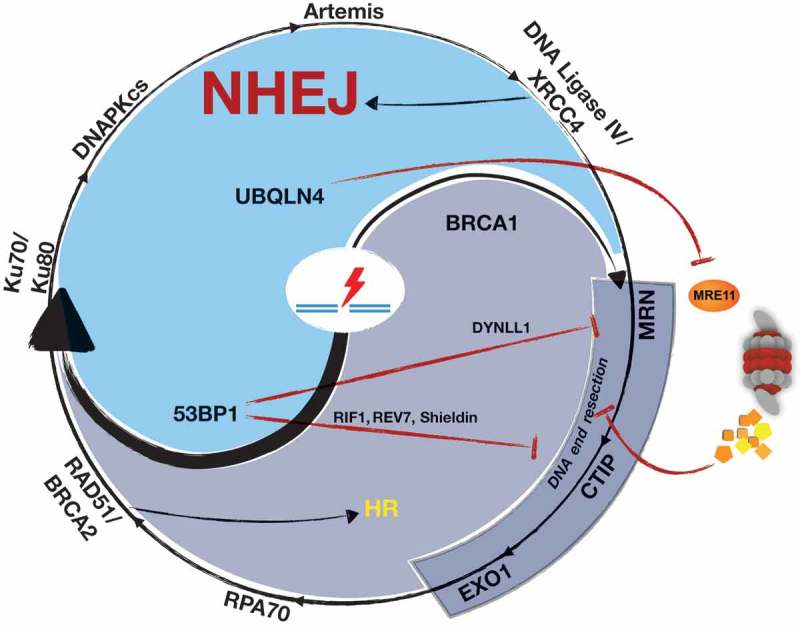
Inhibition of DNA end resection promotes cNHEJ.
DYNLL1, shieldin and UBQLN4 inhibit DNA end resection thereby promoting DSB repair by cNHEJ. The shieldin complex is recruited to DSBs in a 53BP1- and RIF1-dependent manner and protects DNA ends from excessive end resection. Loss of individual shieldin subunits, such as loss of the 53BP1-interaction partner DYNLL1, causes PARP inhibitor resistance in BRCA1-deficient cells due to re-enabling of DSB end resection and restoration of HR activity. UBQLN4 promotes the proteasomal degradation of MRE11 thereby creating a PARP inhibitor vulnerability in UBQLN4-overexpressing cancer cells.
Next to SHLD1 and 2, the CRISPR/Cas9 synthetic viability screen in BRCA1-deficient cells exposed to PARP inhibitors revealed additional new suppressor candidate gene products, including TEN1, a component of the CST complex, consisting of CTC1, STN1 and TEN1 [87]. Consistent with this observation, it was recently shown that the CST complex, which is known to recruit Polα-primase to telomeres, operates downstream of the 53BP1-RIF1-REV7-shieldin axis, in order to repress DSB end resection [91,92]. Multiple interactions between the CST and shieldin complexes were reported, including interactions between CTC1 and SHLD1, STN1 and SHLD3, CTC1 and REV7, TEN1 and SHLD3, as well as STN1 with SHLD1, SHLD2 and REV7 [91]. Moreover, STN1 co-localizes with 53BP1 at IR-induced DSBs in a shieldin-dependent fashion [91]. Reminiscent of 53BP1, RIF1 or shieldin, CST depletion was shown to promote increased DSB resection [91]. Moreover, CST represses RAD51 loading and enhances the cytotoxicity of PARP inhibitors in BRCA1-deficient cells [91]. Further experiments revealed that Polα-primase inhibition abolishes the effect of PARP inhibitors in these cells. Collectively, these data may indicate that CST-Polα-driven nucleotide fill-in contributes to drive DSB repair towards NHEJ usage downstream of 53BP1, RIF1 and shieldin.
The nuclease Artemis emerged as a further effector molecule of 53BP1 in repressing HR [93]. Artemis is retained at sites of genotoxic damage through an interaction with PTIP. This interaction is largely dependent on DNA damage-induced and ATM-mediated Artemis phosphorylation on Thr-656 and involves the BRCT-2 domain of PTIP [93]. Artemis depletion, as well as reconstitution of Artemis-depleted cells with the nuclease-dead H35A/D37N Artemis mutant led to marked PARP inhibitor resistance in BRCA1-defective cells [93].
An additional suppressor candidate gene revealed by CRISPR/Cas9 synthetic viability screening in BRCA1-deficient cells exposed to PARP inhibitors was the known 53BP1-interaction partner DYNLL1 [87,94] (Figure 2). Loss of DYNLL1 was shown to enable DSB end resection and to restore HR activity in BRCA1-mutant cells, thereby promoting resistance to PARP inhibitors and cisplatin [94]. Moreover, elegant in vitro experiments in a cell-free DNA end resection assay system revealed that DYNLL1 repressed the end resection activity of the MRE11/RAD50/NBS1 (MRN) complex in the presence of BLM, DNA2 and RPA, likely mediated by a direct interaction with MRE11 [94]. Further, DYNLL1-deficient cells displayed an increased number of nuclear MRE11 foci, following olaparib treatment [94]. In addition to its interaction with MRE11, DYNLL1 was also found to drive the formation of multimeric 53BP1 complexes [95]. DYNLL1 binding was shown to stimulate 53BP1 oligomerization, and to facilitate its recruitment to DSB sites [95]. Lastly, not only deletion of Dynll1, but also deletion of its transcriptional regulator Asciz led to PARP inhibitor resistance, in Brca1-deficient murine breast cancer cells [95]. While the above-mentioned data on the 53BP1-RIF1-REV7-shieldin-CST axis and DYNLL1 provided important molecular insights into the regulation of resection initiation, less is known about mechanisms that regulate resection processivity. It was recently shown that DNA helicase B (HELB) lies at the heart of a feedback inhibition mechanism that curtails DSB resection [96]. HELB was demonstrated to be recruited to ssDNA through an interaction with RPA [96]. Moreover, HELB represses EXO1- and BLM-DNA2-dependent DNA end resection. Fully in line with a role in repressing DNA end resection, Helb depletion results in PARP inhibitor resistance in Brca1-deficient murine mammary tumor cells [96]. Thus, HELB emerges as a negative regulator of ongoing DNA end resection.
Human syndromes serve as a toolbox for the identification of novel regulators of DSB repair
Genetic screens and DNA sequencing analyses in the context of PARP inhibition revealed profound insight into the intricate biology of the DDR in general and into molecular mechanisms underlying PARP inhibitor resistance, in particular. Moreover, human syndromes also constitute a rich resource for understanding the biology of DNA repair. One of the most prominent examples is probably the discovery of bi-allelic ATM mutations as the underlying cause of Ataxia-telangiectasia [97]. Countless additional genome instability syndromes have been deciphered during the last decades (for a recent review, please refer to [98]). Here, we will particularly focus on the RIDDLE syndrome and the newly identified UBQLN4 deficiency syndrome, as the underlying biology offers important insight in the mechanisms of DSB repair [99,100]. RNF168, the gene mutated in RIDDLE syndrome, encodes an E3 ubiquitin ligase [100]. The role of RNF168 within the DDR was unraveled through a thorough investigation of RIDDLE syndrome patient-derived cells [100]. In response to genotoxic damage, RNF168 is recruited into repair foci nucleating around MDC1. These repair complexes form, once ATM is activated and mediates H2AX Ser139 phosphorylation. The phospho-peptide-binding BRCT domains of MDC1, which itself is phosphorylated by ATM, subsequently engage the pSer139 residue in γH2AX [7]. The ATM-dependent phospho-epitopes in MDC1 are required for the recruitment of the E3-ubiquitin ligase RING finger 8 (RNF8) into this multi-protein complex [7]. Particularly the FHA domain of RNF8 binds to MDC1 [101–103]. RNF8, together with the E2-conjugating enzyme UBC13, promotes ubiquitylation of H2AX, and possibly H2A [7]. RNF168, which also harbors robust ubiquitylation activity towards H2A-type histones, is also recruited into this super-complex in an RNF8-dependent manner [104]. RNF168 is recruited through motifs interacting with ubiquitin (MIUs) [100]. It is likely that RNF8 serves a priming function, providing initial ubiquitylation marks to drive subsequent RNF168 recruitment. Consistent with this model, RNF168 is recruited to sites of DNA damage after RNF8 [104]. Similar to RNF8, RNF168 employs UBC13 as an E2 partner to drive Lys63-linked poly-ubiquitylation of H2A and H2AX [100]. RNF168 was shown to enhance the ubiquitylation of H2A-type histones to facilitate the ubiquitylation-dependent recruitment of further proteins, such as 53BP1 and BRCA1 [104]. Overall, RNF168, which itself is recruited through binding to ubiquitylated H2A and H2AX, appears to further amplify H2A and H2AX ubiquitylation, leading to robust histone polyubiquitylation at DNA stretches surrounding DSB lesions. Ubiquitylated H2A is subsequently engaged by an additional protein complex consisting of RAP80 (receptor-associated protein 80), Abraxas and BRCA1, where RAP80 directly binds the ubiquitylated forms of H2A and H2AX around the DNA break through its ubiquitin-interaction motifs [103,105–107]. Thus, through a detailed molecular understanding of the RIDDLE syndrome, it became clear that initial phosphorylation of H2AX is translated into extensive ubiquitylation events around the damaged site, which in turn serve as docking sites for the recruitment of additional repair factors.
The importance of ubiquitylation events within the DDR is further underscored by the recent discovery of the UBQLN4 deficiency syndrome [99]. The detailed genetic analysis of two consanguineous families revealed a deleterious UBQLN4 mutation to be the underlying cause of an autosomal recessive syndrome reminiscent of genome instability disorders [99]. Further analysis of cell lines derived from these patients revealed that UBQLN4 deficiency leads to delayed DSB repair [99]. UBQLN4 is a proteasomal shuttle factor, which was phosphorylated by ATM on Ser318 in response to genotoxic damage [99]. It was further shown that UBQLN4, upon Ser318 phosphorylation, binds to ubiquitylated MRE11 and that loss of UBQLN4 led to chromatin retention of MRE11 [99]. Moreover, UBQLN4 deficiency promoted non-physiological HR activity in vitro and in vivo, leading to excessive DSB end resection evidenced by a markedly increased number of RPA foci following DNA damage [99]. Conversely, 53BP1 foci formation was repressed in UBQLN4-defective cells [99]. Furthermore, UBQLN4 overexpression was shown to repress HR and to instead favor NHEJ [99] (Figure 2). ATM is generally thought of as being involved in promoting HR, thus the observation that UBQLN4 promotes MRE11 turnover at the damaged chromatin in an ATM-dependent fashion suggests a negative feedback loop, in which ATM also limits HR activity through UBQLN4-mediated MRE11 removal from the break site [99,108]. It was further reported that UBQLN4 overexpression is associated with poor overall survival in a number of cancer entities, including neuroblastoma, ovarian cancer, breast cancer, lung adenocarcinoma and melanoma [99]. Moreover, and fully in line with the reported HR defect in UBQLN4-overexpression settings, UBQLN4 overexpression was shown to be associated with marked PARP1 inhibitor sensitivity [99]. Thus, the molecular dissection of the UBQLN4 deficiency syndrome revealed new mechanistic insight into the regulation of DSB repair pathway choice and paves the way for an UBQLN4 expression-based stratification of patients for the treatment with PARP inhibitors.
Conclusion and clinical perspective
Over the last years, we have witnessed the development and approval of PARP inhibitors, as the first targeted agents that exploit defects in HR-mediated repair. The clinical efficacy most likely reaches beyond those patients that harbor BRCA1 or BRCA2 mutations, but likely spans a large spectrum of BRCAness malignancies. Moreover, we have also seen the emergence of numerous distinct resistance mechanisms. Given this complexity of lesions that predict response and resistance to PARP inhibitors, precise diagnostic tools are required that cover more aberrations than simple BRCA1/2 profiling. It remains a matter of debate, which technology is best suited to comprehensively detect patients that likely benefit from PARP inhibition [109]. One approach are large targeted sequencing panels, which cover a broad spectrum of exons from various genes, that are associated with PARP inhibitor sensitivity, when mutated [109]. In an extension of this approach, more recent developments aim at combined profiling of mutations and aberrant methylation patterns [110]. Moreover, several structural rearrangement signatures have been developed to derive scores that help to identify tumor samples that harbor an HR defect [109]. Lastly, functional assays have been developed. These approaches typically aim to score RAD51 foci formation in the tumor compared to a matched normal control. Reduced RAD51 foci formation can be interpreted as a failure to effectively execute the HR process [109]. Similarly, diagnostic tools have to be developed that accurately identify tumors that harbor PARP inhibitor resistance-mediating aberrations. As restoration of HR capacity appears to be a prominent underlying mechanism of PARP inhibitor resistance, not only panel sequencing approaches, but also functional assays to detect RAD51 may be viable option in this regard.
Another critically important issue evolves around tumor heterogeneity. While in BRCA1/2-mutant familial breast- and ovarian cancer all tumor cells are likely to harbor bi-allelic BRCA mutations, this may not be the case in sporadic cases, where PARP inhibitor sensitivity-mediating aberrations, such as ATM mutations may be subclonal. Based on these considerations, it might be useful to carefully develop combination strategies that eradicate BRCAness clones through a PARP inhibitor component, and also contain agents that target the remaining tumor bulk.
Funding Statement
This work was supported by the German-Israeli Foundation for Research and Development (I-65-412.20-2016 to HCR), the Deutsche Forschungsgemeinschaft (KFO-286-RP2/CP1 to HCR and JA2439/1-1 to RDJ), the Else Kröner-Fresenius Stiftung (2014-A06 to HCR, 2016_Kolleg.19 to RDJ), the Deutsche Krebshilfe (1117240 to HCR) and the German Ministry of Education and Research (BMBF e:Med 01ZX1303A to HCR).
Acknowledgments
We thank the members of the Reinhardt lab for helpful discussions. We apologize to our colleagues for the omission of many important contributions to the field, and their references, due to space limitations.
Disclosure statement
H.C.R received consulting fees from Abbvie, AstraZeneca, Vertex, and Merck and research funding from Gilead. The remaining authors have no potential conflict of interest.
References
- [1].Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. [DOI] [PubMed] [Google Scholar]
- [2].Sharma SV, Bell DW, Settleman J, et al. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. [DOI] [PubMed] [Google Scholar]
- [3].Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reinhardt HC, Jiang H, Hemann MT, et al. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle. 2009;8:3112–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dietlein F, Kalb B, Jokic M, et al. A synergistic interaction between Chk1- and MK2 inhibitors in KRAS-mutant cancer. Cell. 2015;162:146–159. [DOI] [PubMed] [Google Scholar]
- [6].George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Reinhardt HC, Yaffe MB.. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol. 2013;14:563–580. [DOI] [PubMed] [Google Scholar]
- [8].Ciriello G, Miller ML, Aksoy BA, et al. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dietlein F, Thelen L, Reinhardt HC. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014;30:326–339. [DOI] [PubMed] [Google Scholar]
- [10].Bailey MH, Tokheim C, Porta-Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371–385 e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. [DOI] [PubMed] [Google Scholar]
- [12].Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci U S A. 2003;100:776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Loeb LA, Bielas JH, Beckman RA. Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 2008;68:3551–3557. discussion 3557. [DOI] [PubMed] [Google Scholar]
- [15].Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–346. [DOI] [PubMed] [Google Scholar]
- [16].Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. [DOI] [PubMed] [Google Scholar]
- [17].Waters LS, Minesinger BK, Wiltrout ME, et al. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73:134–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–1485. [DOI] [PubMed] [Google Scholar]
- [20].Jokic M, Vlasic I, Rinneburger M, et al. Ercc1 deficiency promotes tumorigenesis and increases cisplatin sensitivity in a Tp53 context-specific manner. Mol Cancer Res. 2016;14:1110–1123. [DOI] [PubMed] [Google Scholar]
- [21].Marcus L, Lemery SJ, Keegan P, et al. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res. 2019 Feb 20. pii: clincanres.4070.2018. doi: 10.1158/1078-0432.CCR-18-4070. [Epub ahead of print. PMID:30787022. DOI:10.1158/1078-0432.CCR-18-4070. [DOI] [PubMed] [Google Scholar]
- [22].Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. [DOI] [PubMed] [Google Scholar]
- [23].Nimonkar AV, Genschel J, Kinoshita E, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shibata A, Moiani D, Arvai AS, et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell. 2014;53:7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rass E, Grabarz A, Plo I, et al. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–824. [DOI] [PubMed] [Google Scholar]
- [26].Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. [DOI] [PubMed] [Google Scholar]
- [27].Lyndaker AM, Alani E. A tale of tails: insights into the coordination of 3’ end processing during homologous recombination. Bioessays. 2009;31:315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7:739–750. [DOI] [PubMed] [Google Scholar]
- [31].Krejci L, Altmannova V, Spirek M, et al. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40:5795–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. [DOI] [PubMed] [Google Scholar]
- [33].Venkitaraman AR. Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat Rev Cancer. 2004;4:266–276. [DOI] [PubMed] [Google Scholar]
- [34].Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42:410–414. [DOI] [PubMed] [Google Scholar]
- [35].Al-Sukhni W, Rothenmund H, Borgida AE, et al. Germline BRCA1 mutations predispose to pancreatic adenocarcinoma. Hum Genet. 2008;124:271–278. [DOI] [PubMed] [Google Scholar]
- [36].Bartsch DK, Gress TM, Langer P. Familial pancreatic cancer–current knowledge. Nat Rev Gastroenterol Hepatol. 2012;9:445–453. [DOI] [PubMed] [Google Scholar]
- [37].Greer JB, Whitcomb DC. Role of BRCA1 and BRCA2 mutations in pancreatic cancer. Gut. 2007;56:601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].T. C. G. A. R. Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44:47–52. [DOI] [PubMed] [Google Scholar]
- [43].Knittel G, Rehkamper T, Korovkina D, et al. Two mouse models reveal an actionable PARP1 dependence in aggressive chronic lymphocytic leukemia. Nat Commun. 2017;8:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lees-Miller SP, Meek K. Repair of DNA double strand breaks by non-homologous end joining. Biochimie. 2003;85:1161–1173. [DOI] [PubMed] [Google Scholar]
- [46].McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet. 2013;47:433–455. [DOI] [PubMed] [Google Scholar]
- [48].Frit P, Barboule N, Yuan Y, et al. Alternative end-joining pathway(s): bricolage at DNA breaks. DNA Repair (Amst). 2014;17:81–97. [DOI] [PubMed] [Google Scholar]
- [49].Iyama T, Wilson DM 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst). 2013;12:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. [DOI] [PubMed] [Google Scholar]
- [51].Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. [DOI] [PubMed] [Google Scholar]
- [52].McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. [DOI] [PubMed] [Google Scholar]
- [53].Moudry P, Watanabe K, Wolanin KM, et al. TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J Cell Biol. 2016;212:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schmitt A, Knittel G, Welcker D, et al. ATM deficiency is associated with sensitivity to PARP1- and ATR inhibitors in lung adenocarcinoma. Cancer Res. 2017;77:3040–3056. [DOI] [PubMed] [Google Scholar]
- [55].Perkhofer L, Schmitt A, Romero Carrasco MC, et al. ATM deficiency generating genomic instability sensitizes pancreatic ductal adenocarcinoma cells to therapy-induced DNA damage. Cancer Res. 2017;77:5576–5590. [DOI] [PubMed] [Google Scholar]
- [56].Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523–533. [DOI] [PubMed] [Google Scholar]
- [57].Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Murai J, Huang SY, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8:362ps317. [DOI] [PubMed] [Google Scholar]
- [62].Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Pettitt SJ, Rehman FL, Bajrami I, et al. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS One. 2013;8:e61520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pettitt SJ, Krastev DB, Brandsma I, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hanzlikova H, Kalasova I, Demin AA, et al. The importance of poly(ADP-Ribose) polymerase as a sensor of unligated okazaki fragments during DNA replication. Mol Cell. 2018;71:319–331 e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gogola E, Duarte AA, de Ruiter JR, et al. Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell. 2018;33:1078–1093 e1012. [DOI] [PubMed] [Google Scholar]
- [67].Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. [DOI] [PubMed] [Google Scholar]
- [68].Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Watanabe S, Watanabe K, Akimov V, et al. JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1-mediated chromatin response to DNA breaks. Nat Struct Mol Biol. 2013;20:1425–1433. [DOI] [PubMed] [Google Scholar]
- [71].Liu Y, Burness ML, Martin-Trevino R, et al. RAD51 mediates resistance of cancer stem cells to PARP inhibition in triple-negative breast cancer. Clin Cancer Res. 2017;23:514–522. [DOI] [PubMed] [Google Scholar]
- [72].Wiegmans AP, Yap PY, Ward A, et al. Differences in expression of key DNA damage repair genes after epigenetic-induced BRCAness dictate synthetic lethality with PARP1 inhibition. Mol Cancer Ther. 2015;14:2321–2331. [DOI] [PubMed] [Google Scholar]
- [73].Marzio A, Puccini J, Kwon Y, et al. The F-box domain-dependent activity of EMI1 regulates PARPi sensitivity in triple-negative breast cancers. Mol Cell. 2019;73:224–237 e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Jaspers JE, Kersbergen A, Boon U, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3:68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–470. [DOI] [PubMed] [Google Scholar]
- [76].Boersma V, Moatti N, Segura-Bayona S, et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature. 2015;521:537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Bunting SF, Callen E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Munoz IM, Jowsey PA, Toth R, et al. Phospho-epitope binding by the BRCT domains of hPTIP controls multiple aspects of the cellular response to DNA damage. Nucleic Acids Res. 2007;35:5312–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Zimmermann M, Lottersberger F, Buonomo SB, et al. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science. 2013;339:700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Feng L, Fong KW, Wang J, et al. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem. 2013;288:11135–11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–883. [DOI] [PubMed] [Google Scholar]
- [82].Di Virgilio M, Callen E, Yamane A, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Chapman JR, Barral P, Vannier JB, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Xu G, Chapman JR, Brandsma I, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Gupta R, Somyajit K, Narita T, et al. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018;173:972–988 e923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Noordermeer SM, Adam S, Setiaputra D, et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018;560:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Dev H, Chiang TW, Lescale C, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol. 2018;20:954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Ghezraoui H, Oliveira C, Becker JR, et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature. 2018;560:122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Tomida J, Takata KI, Bhetawal S, et al. FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. Embo J. 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Findlay S, Heath J, Luo VM, et al. SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. Embo J. 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Mirman Z, Lottersberger F, Takai H, et al. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature. 2018;560:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Barazas M, Annunziato S, Pettitt SJ, et al. The CST complex mediates end protection at double-strand breaks and promotes PARP inhibitor sensitivity in BRCA1-deficient cells. Cell Rep. 2018;23:2107–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wang J, Aroumougame A, Lobrich M, et al. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014;28:2693–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].He YJ, Meghani K, Caron MC, et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature. 2018;563:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Becker JR, Cuella-Martin R, Barazas M, et al. The ASCIZ-DYNLL1 axis promotes 53BP1-dependent non-homologous end joining and PARP inhibitor sensitivity. Nat Commun. 2018;9:5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Tkac J, Xu G, Adhikary H, et al. HELB is a feedback inhibitor of DNA end resection. Mol Cell. 2016;61:405–418. [DOI] [PubMed] [Google Scholar]
- [97].Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. [DOI] [PubMed] [Google Scholar]
- [98].Terabayashi T, Hanada K. Genome instability syndromes caused by impaired DNA repair and aberrant DNA damage responses. Cell Biol Toxicol. 2018;34:337–350. [DOI] [PubMed] [Google Scholar]
- [99].Jachimowicz RD, Beleggia F, Isensee J, et al. UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Cell. 2019;176:505–519 e522. [DOI] [PubMed] [Google Scholar]
- [100].Stewart GS, Panier S, Townsend K, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–434. [DOI] [PubMed] [Google Scholar]
- [101].Huen MS, Grant R, Manke I, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Mailand N, Bekker-Jensen S, Faustrup H, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. [DOI] [PubMed] [Google Scholar]
- [103].Kolas NK, Chapman JR, Nakada S, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Doil C, Mailand N, Bekker-Jensen S, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. [DOI] [PubMed] [Google Scholar]
- [105].Wang B, Matsuoka S, Ballif BA, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wu J, Huen MS, Lu LY, et al. Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol Cell Biol. 2009;29:849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. [DOI] [PubMed] [Google Scholar]
- [108].Jachimowicz RD, Reinhardt HC. UBQLN4 promotes non-homologous end joining by repressing DNA end-resection. Mol Cell Oncol. 2019;6:1575692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–120. [DOI] [PubMed] [Google Scholar]
- [110].Grimm C, Fischer A, Farrelly AM, et al. Combined targeted re-sequencing of cytosine DNA methylation and mutations of DNA repair genes with potential use for PARP1 inhibitor sensitivity testing. J Mol Diagn. 2018;21:198–213. [DOI] [PubMed] [Google Scholar]