

ABSTRACT
Polyploid cells contain more than two homologous sets of chromosomes. The original observations of liver polyploidy date back to the 1940s, but functional roles for polyploid cells are still unclear. Liver polyploidy may influence regeneration, stress response, and cancer, although little evidence has established direct causal links between polyploidy and these biological phenotypes. In this review, we will introduce broad concepts about polyploidy including its distribution in nature and how polyploids form in normal and pathological situations. Then we will examine recent discoveries that have begun to clarify functionality and disease relevance of liver polyploidy. Finally, we will discuss implications and future directions of research about polyploidy in the liver.
KEYWORDS: Polyploidy, liver, hepatocyte, cell cycle, regeneration, cancer
Where is polyploidy found in the natural world?
Polyploid tissues and organisms contain cells with balanced duplications of all chromosomes. This duplication inevitably results in increased DNA content, potentially followed by an increased capacity for RNA and protein production, increased substrates for DNA repair and evolution, and increased centrosome number. These inevitable consequences could provide both substrates for evolution and adaptability while also introducing challenges for cell division. In fact, polyploidy in early prokaryotes may have protected against double strand breaks and deleterious recessive mutations in a highly oxygenated environment [1]. Polyploids are also common in the plant kingdom – all angiosperms underwent whole genome duplication events in the past [2,3] and 30–80% of all plants are polyploid [4,5]. Polyploidization in plants has been recognized as a centrally important evolutionary process that affected diversification and specification [6]. In plants, the success of angiosperms is at least in part due to gene or whole-genome duplication (WGD) events, which resulted in seed and flower gene diversification [7]. Species richness positively correlates with the frequency of polyploids [8], suggestive of how polyploidization contributed to plant speciation. It is likely that some lineages that underwent WGD events acquired survival advantages and evolved into novel polyploid species [9].
Polyploidy is less common in animals, possibly due to differences in development, genomic architecture, and sex determination [10]. Animals have more complex gene regulatory networks and tissue-specific expression patterns, which may have made genome reorganization more challenging after polyploidization events [10]. However, polyploid organisms still exist in all major taxonomic animal groups, particularly fish [11] and amphibians [12]. Of the 24,000 existing fish species in 57 orders, the majority (63%) fall into nine orders known to include polyploids [13]. As for amphibians, natural polyploids and spontaneous polyploid individuals (viable individuals that are polyploid, while the majority of the species is diploid) were found in 15 anuran families and 4 urodelan families [14]. Polyploidization may have also played a role in animal evolution since most vertebrate genomes evolved from WGD events [15]. While whole-body polyploids in mammals and birds are rare, a high frequency of polyploid cells can be found in mammalian organs such as the bone marrow [16], heart [17], skeletal muscle [18], brain [19], and liver [20].
Within individual animal tissues, dynamic polyploidization is also seen as a result of developmental processes, environmental stresses, or pathological conditions. As an example, early pregnancy induces the development of decidual polyploid cells [21]. At the site of blastocyst implantation, stromal cells undergo a process called decidualization, during which they extensively proliferate, differentiate, and polyploidize to remodel the endometrium in preparation for implantation [21]. This process involving polyploidization is crucial for successful embryo implantation and growth [22]. Polyploidy and aneuploidy are also observed in various stages of carcinogenesis. Polyploidization in cancer cells has been associated with tumor progression [23–25], potentially due to an association with chromosomal instability (CIN) [26].
How do cells become polyploid?
Described below are mechanisms that can lead to polyploidization during development or disease, including cell fusion and alternative cell cycling [27–28] (Figure 1).
Figure 1.
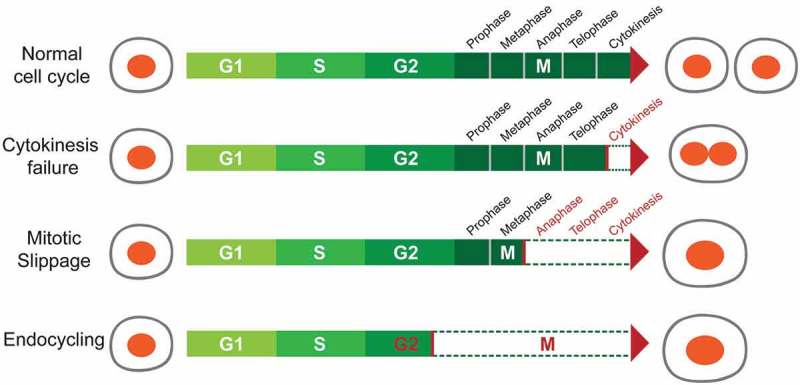
Mechanisms of polyploidization.
Polyploidization can be achieved via different mechanisms, including cytokinesis failure, mitotic slippage, and endocycling. Each mechanism involves deviations in how cells normally cycle.
(1). Cell fusion is the only mechanism that generates polyploid cells without requiring cell cycle entry. Fusion of two or more cells of the same type results in syncytial polyploid cells of the mammalian muscle, bone, and placenta. During embryonic development, myoblasts fuse together to form multi-nucleated fibers called myotubes, representing a terminally differentiated state [29]. Although its relevance is debated, cell fusion between hepatocytes has been observed in chimeric mouse transplantation models, albeit at low rates [30]. One study identified hepatocytes expressing markers from two separate donors, and reasoned that these were generated via fusion events. However, due to potential artifacts from extracellular vesicles and a lack of data regarding the ploidy of “fused” cells, the occurrence of hepatocyte fusion in the liver remains controversial.
(2). Endoreplication, also known as endoreduplication or endocycling, occurs when a cell replicates its DNA without entering mitosis. The resulting cellular progenies of endoreplication are usually mononucleated polyploid cells. In Drosophila, most larval tissues polyploidize via endoreplication. An extensively studied example is the giant salivary gland cell, which undergoes about 10 cycles of endoreplication, resulting in a final ploidy of > 1000 genome copies [31]. Another example in Drosophila are nurse cells, which become polyploid during oogenesis and provide maternal materials to oocytes [32]. In mammals, endoreplication occurs when trophoblast stem cells (TSC) differentiate into trophoblast giant cells (TGC), which are required for embryo implantation [33]. The abbreviated cell cycle where S and G phases alternate has been proposed as a way to accelerate TGC growth [34]. Outside of development, endoreplication also occurs in response to DNA damage. For example, in p53 null Mouse Embryonic Fibroblasts (MEFs), DNA damage signals from persistent telomere dysfunction were shown to inhibit mitotic entry, resulting in the formation of tetraploid cells through endoreplication [35].
(3). Mitotic slippage. During perturbed mitoses, i.e. when cells are treated with spindle inhibitors, cells are arrested at metaphase by the spindle assembly checkpoint (SAC), which is activated to ensure that each chromosome is properly attached. Prolonged SAC activation causes mitotic arrest and cell death, a phenomenon termed “mitotic catastrophe” [36]. Alternatively, a process called “mitotic slippage” occurs when arrested cells “slip” mitosis and enter the next interphase without undergoing chromosome segregation or cytokinesis. Since the satisfaction of SAC is not required for exiting mitosis [37], the arrested cells can “slip” mitosis when the exiting signal is activated, which requires the proteolysis of Cyclin B [38]. Thereby, cells undergoing “mitotic slippage”, also referred to as “postmitotic” cells, become polyploid. Tubulin binding agents such as taxanes, epothilones, and vinca alkaloids can activate the SAC to induce mitotic slippage [39,40]. However, how cells choose between “mitotic catastrophe” or “mitotic slippage” is still unclear [41,42].
(4). Cytokinesis failure. Cytokinesis is the final step of the cell cycle, during which the mother cell divides into daughter cells. It requires anaphase spindle reorganization, formation of a cleavage furrow, and abscission of the midbody structure. Cytokinesis failure can lead to binucleated polyploid cells, or if the nuclei re-fuse after failed cytokinesis, it can also result in a mononucleated daughter cell [43]. As an example, platelet producing megakaryocytes go through anaphase and telophase, but then do not complete cytokinesis due to cleavage furrow regression, resulting in ploidies of up to 128n [44]. Polyploidization is essential for megakaryocyte function because enforcing cytokinesis in megakaryocytes by Cdc20 ablation leads to decreased ploidy and a reduction in platelet production [45]. In the context of Cdc20 loss, rescuing polyploidization by deleting Cdk1 or Cdk1/Cdk2 also restores platelet production [45]. This suggests that polyploidy itself is required for megakaryocyte function, rather than just the specific genes responsible for polyploidization. Another example of cytokinesis failure is in zebrafish epicardial regeneration. Cao et al. showed that the regenerating epicardium consists of two cell populations: polyploid “leader” cells at the front of regenerating tissues and diploid “follower” cells [46,47]. The polyploid cells are formed primarily via cytokinesis failure induced by increased mechanical tension. Although both polyploid leader cells and diploid follower cells were independently sufficient for epicardial regeneration, polyploid cells were more efficient at regeneration [46].
Characterization of polyploidy in the liver
The liver is an organ that carries out essential functions such as nutrient synthesis and distribution, storage of amino acids, lipids and carbohydrates, and detoxification of xenobiotics [48]. These functions are conducted primarily by hepatocytes, which account for 70–80% of cells within the liver. Hepatocytes are relatively quiescent with a turnover rate of 200–300 days, but are able to rapidly proliferate upon tissue loss or injury [49]. At birth, all hepatocytes have a single nucleus with 2n DNA content and are thus diploid. During postnatal development, hepatocytes become polyploid predominantly through cytokinesis failure, as demonstrated by live cell imaging and immunofluorescence staining on tissue sections [50,51]. Polyploid hepatocytes can take many forms: they can be tetraploid (binucleated with two 2n nuclei or mononucleated with one 4n nuclei) or octaploid (binucleated with two 4n nuclei or mononucleated with one 8n nuclei). The prevalence of polyploid hepatocytes increases with age, ultimately resulting in approximately 90% polyploid hepatocytes in adult mice and upwards of 50% in adult humans, as measured by flow cytometry and fluorescence in situ hybridization (FISH) [52,53]. In rodents, hepatocyte polyploidization starts around weaning, and is tightly associated with dietary changes and fluctuations in insulin/Akt signaling (Figure 2) [54,55]. Celton-Morizur et al. found that blocking insulin/PI3K/Akt signaling reduced the formation of binucleated hepatocytes in rats. Kinetically, polyploidy increases rapidly between weaning and maturation, followed by a more gradual elevation throughout adulthood, in part because proliferation slows down after 4 weeks of age.
Figure 2.
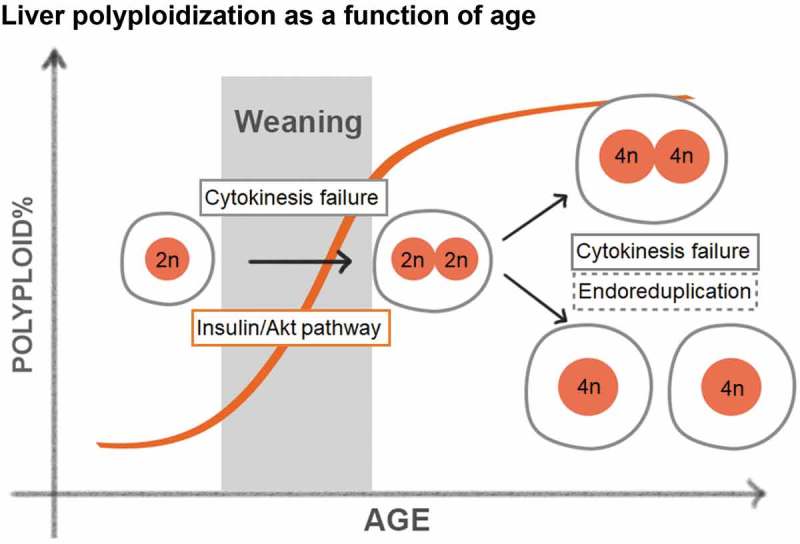
Liver polyploidization as a function of age.
Hepatocyte ploidy increases with age, and involves a rapid increase around the time of weaning. Polyploidization slows after maturation because proliferation rates decrease. Insulin/Akt is one pathway that controls polyploidization by promoting cytokinesis failure. Hepatocyte polyploidization can also occur through endoreduplication during later stages.
Liver ploidy also changes after injury and stress. 70% partial hepatectomy induces compensatory hepatocyte hypertrophy and hyperplasia associated with a reduction in diploid cells and an increase in mononucleated polyploid cells [56,57]. Miyaoka et al. examined regeneration after 70% hepatectomy, and suggested that the mechanisms of polyploidization during regeneration are distinct from the mechanisms during development. First, they found that hepatocytes entered S-phase (BrdU incorporation) but rarely entered mitosis (phospho-H3 staining), indicating that the cells might undergo endoreplication. Second, they proposed that the binuclear hepatocytes tend to undergo cell division to generate mononuclear hepatocytes [56]. Although this study was solely based on imaging tissue sections and lacked direct evidence for aberrant cell cycles, it demonstrated the remarkable plasticity of hepatocyte cell cycles. In addition to surgical resection, chemical, genetic, and metabolic injuries can also elicit ploidy changes. We observed increased liver polypoidy in mice after repeated administration of carbon tetrachloride (CCl4), a dry-cleaning toxin that causes centrilobular hepatocyte necrosis (unpublished). In a rat model of Wilson’s Disease, excessive hepatic copper accumulation is associated with delayed mitosis and increased ploidy [58]. Recently, Gentric et al. reported that hepatocyte ploidy increased in murine models of nonalcoholic fatty liver disease (NAFLD) vs. controls. They also found increased nuclear size in hepatocytes from patients with steatohepatitis (NASH) compared to healthy controls, suggesting polyploidization [59]. Furthermore, they determined that oxidative stress promoted polyploidization in NAFLD models, and antioxidants could revert the ploidy back to normal levels. Our group examined a functional role for ploidy in this setting by first altering mouse liver ploidy, then inducing fatty liver disease using high fat diet. We found that increased polyploidy did not alter the severity of steatosis, but did suppress tumorigenesis.
Polyploidy is one of many types of cellular heterogeneity among hepatocytes
An important question is if polyploid hepatocytes are more proliferative or differentiated than diploid hepatocytes. Results from recent liver stem cell studies shed some light on this topic. The hepatic lobule is the fundamental histologic unit of the liver and is classified to three concentric zones covering the portal to central vein axis. These zones are the periportal (zone 1), mid-zone (zone 2), and perivenous (zone 3). Gene expression changes across zones due to differences in blood flow, an oxygen gradient, Wnt signals from endothelial cells of the central vein, and other factors [60–62]. Intriguingly, hepatocyte ploidy also differs between zones. By using single molecule-based tissue imaging, Tanami et al. reconstructed the spatial zonation profile of liver ploidy: periportal zone 1 has the most diploid hepatocytes, mid-zone 2 has the most polyploid hepatocytes, and the perivenous zone 3 is intermediate [63].
Polyploidy is associated with terminal differentiation and functional maturity in megakaryocytes, cardiomyocytes [45], and TGCs [64]. Thus, polyploid cells are often considered less “stem-like”. However, putative liver stem or progenitor compartments have diverse ploidy levels. Since oval shaped cells near bile ducts were observed to expand in chronic liver diseases, many attempted to identify a non-hepatocyte “liver progenitor cell” population that gives rise to hepatocytes when hepatocyte proliferation is inhibited. Injury-induced cells expressing bile duct markers (Opn, Ck19, Lgr5, EpCAM, etc.), were thought to replenish the hepatocyte pool upon injury [65,66]. However, multiple rigorously performed lineage-tracing studies suggested that hepatocytes themselves were the major regenerative cell source during injury [67,68]. More recently, specific hepatocyte subpopulations were lineage-traced to identify cells that can self-renew under physiological conditions or upon injury. Wang et al. labeled Axin2+ hepatocytes surrounding the central vein and found that these cells could both self-renew and give rise to new hepatocytes in the absence of injury [60]. Consistent with their location in zone 3, Axin2+ cells were more likely to be diploid compared to their progeny, although 30–40% of this population was actually tetraploid. Font-Burgada et al. labeled hybrid periportal hepatocytes (HybHP) using a Sox9-CreER tracer, and found these cells did not expand under normal homeostasis, but became highly proliferative under multiple injury conditions [69]. Their results were supported by another group that used Mfsd2a to trace periportal hepatocytes [70]. These studies did not focus on ploidy profile, but these cellular populations are more likely to be diploid based on zonation. Lin et al. labeled a group of hepatocytes expressing higher levels of telomerase reverse transcriptase (Tert) that were found to regenerate during homeostasis and under injury conditions [71]. The Terthigh hepatocytes were equally distributed through all liver zones and comprised of a similar proportion of diploid and polyploid cells as Tertlow hepatocytes. These important studies used different lineage tracing reagents and insults but remain difficult to reconcile. As had been previously reported for another Sox9-CreERT2 knock-in mouse, transgenic reporter systems can introduce alterations in cellular proliferation and fitness [67,72,73]. Alternatively, it is possible that different injury models favor regeneration from distinct cell populations. Since many liver injuries cause damage in the pericentral area, where Cytochrome P450 enzymes are most highly expressed, it makes sense that there are reserve cell populations at the opposite zonal end to mediate regeneration under diverse conditions. Importantly, these studies do not consistently support the concept that polyploid hepatocytes are more terminally differentiated, less proliferative, or senescent than diploid hepatocytes.
Genetic regulators of liver ploidy
What dictates the timing of scheduled polyploidization? Because rodent liver polyploidization increases dramatically during the nursing to weaning transition, it has been hypothesized that food intake and nutrient signaling are effectors of when and how much polyploidzation occurs. Indeed, early weaning of rat and mouse pups leads to premature polyploidzation [54,74]. This effect can be recapitulated by manipulating insulin/PI3K/Akt signaling [54]. Inhibiting Akt activity decreases failed cytokinesis events during weaning, supporting the hypothesis that insulin signaling promotes polyploidization [54]. It is likely that other growth pathways that have effects on cell cycle machinery would also lead to detectable ploidy changes. Since polyploidization involves programmed cell cycle anomalies (cytokinesis failure and endoreplication), it is not surprising that perturbation of cell cycle regulators can alter ploidy. The E2F family comprises such a group of transcription factors involved in cell cycle transitions. The family contains activators such as E2f1, E2f2, E2f3, and repressors such as E2f7, E2f8. Indeed, the expression levels of E2f7 and E2f8 are elevated during hepatic polyploidization, suggesting their regulatory roles of polyploidization in normal physiological state. Simultaneous E2f8 and E2f7 deletion leads to the upregulation of cytokinesis genes and a completely diploid liver with few other physiologic phenotypes, discussed below [75]. On the other hand, E2f1 deficiency results in increased polyploidy via the downregulation of G2/M transcriptional programs [76,77]. Perturbations in other cell cycle regulators, though they are not necessarily contributing to natural hepatocyte polyploidization, also lead to ploidy changes. For example, Ccne1 and Ccne2 play antagonizing roles in liver ploidy regulation. Ccne1 deletion in mice results in endoreplication defects and decreased polyploidy, while Ccne2 deletion results in accelerated DNA synthesis and increased polyploidy [78]. Other regulators with roles in ploidy determination include p53 [79], p21 [80], Rb [81], Cdk1 [82], Skp2 [83], Ssu72 [84], and Survivin [85], and deficiencies of these genes all cause increased polyploidy. MicroRNAs also regulate ploidy. The direct targets of miR-122 include a set of cytokinesis genes, and miR-122 deletion results in a profound reduction of polyploid hepatocytes [86]. Though it might be tempting to study the functionality of polyploidy with these models, it would be difficult to dissociate contributions of ploidy from those of gene deletion. Multiple orthogonal methods of altering liver ploidy will likely be required to reach convincing conclusions about the functionality of ploidy states.
What is the function of polyploidy in the liver?
Polyploidy in the liver is both developmentally regulated and dynamically controlled in the context of cellular stress or disease. These observations made us wonder what functionality polyploidy plays in hepatocyte biology. One hypothesis is that polyploidization increases the metabolic capacity of hepatocytes such that they can meet the requirements of rapid growth [87]. In plants, endoreplication occurs in tissues that grow quickly and that have a high metabolic capacity [88]. Livers from Cdk1-/- mice, which contain dramatically polyploidized hepatocytes, were previously used to investigate the impact of cell size on metabolism and gene expression [89]. Cytoskeletal genes were upregulated, while mitochondrial and lipogenic genes were downregulated in Cdk1-/- hepatocytes. This could suggest that metabolic capacity is a function of ploidy, but it is also possible that Cdk1 deletion itself induced metabolic changes through cell cycle alterations independent of ploidy [90,91]. Microarray studies comparing diploid and polyploid hepatocytes from wild-type mice did not identify substantial gene expression changes [92]. Although polyploid plants and placental trophoblast cells exhibit altered gene expression in comparison to their corresponding diploid counterparts [93–95], transcriptional alterations are not a prominent feature of polyploid liver cells at least on the population level. Recently, Shalev Itzkovitz’s group examined transcriptional fidelity in the mammalian liver [96]. They developed a method based on single molecule FISH to quantitatively characterize the promoter states of hepatocytes in situ, and found that the promoters stochastically switched between closed and open states, leading to cell-to-cell variation in gene expression, or so-called “transcriptional noise”. Interestingly, polyploid hepatocytes exhibited less noise, suggesting a more buffered and tightly controlled gene expression. Finally, polyploid cells in other settings also exhibit under-replication, epigenetic changes, and chromatin remodeling alterations, topics that have been reviewed elsewhere [95]. Whether or not these changes occur or are functionally relevant in polyploid hepatocytes are still unknown.
The relationship between polyploidy and cancer is context-specific. On the one hand, polyploidy may protect against transformation in a mutagenic environment. On the other hand, polyploidy is frequently observed in cancer cells and may represent a risk factor for chromosomal instability, which can lead to aneuploidy [97,98]. Since genome duplications in cancer cells are often accompanied by the deletion, amplification, or translocation of whole or parts of chromosomes [99,100], cancer cells are more often aneuploid rather than simply polyploid. In 1902, Theodore Boveri reasoned that carcinogenesis arose from aberrant mitosis and scrambled chromosomes [101]. David Pellman’s group contributed evidence to this idea by showing that tetraploidy is an intermediate state of cancer cells that leads to aneuploidy [26], a specific instance of Boveri’s hypothesis. Fujiwara directly tested this idea in p53 null mouse mammary epithelial cells (MMECs). In comparison to diploid MMECs, tetraploids generated by cytokinesis failure exhibited more frequent chromosomal missegregation and enhanced tumorigenicity [102]. Similarly, Davoli et al. showed that persistent telomere dysfunction and genome-wide DNA damage could induce cell tetraploidization in p53 deficient cells [35], and that tetraploid cells created via the telomere crisis exhibited enhanced transformation [103]. Boveri’s hypothesis has been further supported by many tumor ploidy and cell cycle studies [104–106], adding to a body of work that associates polyploidy and aneuploidy with cancer progression. Although these studies established a link between polyploidy and tumorigenesis, they were predominantly performed in p53 null cells or more generally, in immortalized cells that have the requisite genetic defects to propagate in a dish. Because tetraploidization can activate p53, which blocks entrance into S-phase [107,108], these studies likely reveal consequences of polyploidy that would otherwise not been observed in animals with intact p53. It is likely that polyploidy in the absence of p53 is a cancer driver, but this cannot be asserted for polyploidy in general.
Other studies have supported a tumor suppressive role for polyploidy, potentially due to reduced proliferative rates. Surprisingly, the fundamental question of how hepatic ploidy influences mitosis mechanics and proliferation rates has so far evaded definitive answers. Ganem et al. demonstrated that cytokinesis failure could trigger p53 and Hippo pathway activation, leading to growth inhibition within polyploid hepatocytes, but much of that evidence was examined in vitro [109]. Knouse et al. carefully quantified polyploid and diploid cells and their rates of division after partial hepatectomy and found that polyploids divide as much as diploids [110]. Moreover, E2f7/E2f8 knockout livers, which are predominantly diploid, did not exhibit a higher liver mass recovery rate after hepatectomy compared to wild-type livers [75]. The caveat here is that there are only 1–2 rounds of division after surgical resection so it is hard to discern small differences in proliferation rates. By using primary hepatocytes directly isolated from the mice livers, Wilkinson et al., recently showed that proliferation rates of E2f7/E2f8 knockout hepatocytes, which cannot become polyploid, are higher than that of wild-type polyploid hepatocytes [111]. Collectively, there is a robust body of work showing that polyploids do divide to generate hepatocytes of the same or higher ploidy, and that these divisions happen at a slightly lower rate than in diploids. There is still room for in vivo experiments to quantitatively measure cell division rates of wild-type diploids vs. polyploids. This is important because understanding the long-term consequences polyploidy is dependent on the understanding of how many daughter cells arise from various ploidy populations.
Our group showed that livers with more diploid cells are more likely to get cancer because these cells are more likely to lose all copies of important tumor suppressors [112]. To address the functionality of polyploidy in the liver, we built inducible tools to manipulate ploidy levels in vivo without permanent changes in genes [74]. By manipulating the weaning time and levels of Anln or E2f8 genes to change liver ploidy levels, we found that liver tumorigenesis was inversely correlated with initial polyploidy levels, indicating a tumor suppressive role for polyploidy (Figure 3). Moreover, the additional chromosomes in polyploid cells led to less frequent loss of heterozygosity (LOH), which helped to prevent tumor initiation (Figure 4). Normal liver polyploidy plays a protective role against mutagen induced tumorigenesis, perhaps because polyploids can accumulate more deleterious recessive mutations before completely losing heterozygosity.
Figure 3.
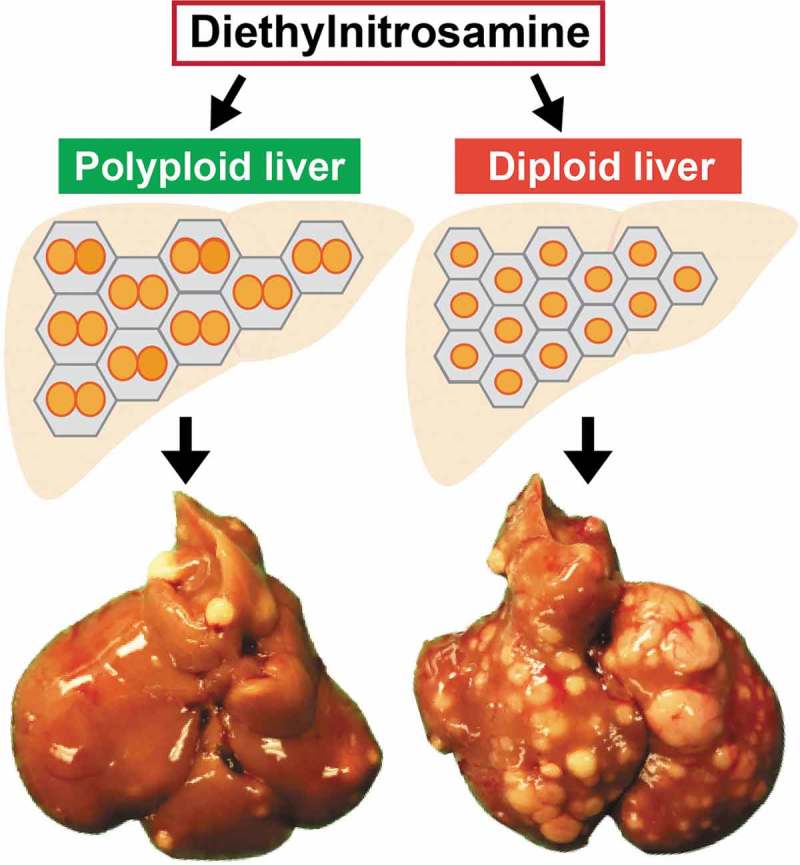
Polyploid hepatocytes protects the liver from mutagen-induced tumorigenesis.
Controlling liver polyploidization by manipulating weaning times or cytokinesis genes (i.e. Anln or E2f8) generated livers with different ploidy states. Induced polyploidization protected livers from mutagen-induced tumorigenesis while induced diploidization did the opposite. Figures adapted from [74].
Figure 4.
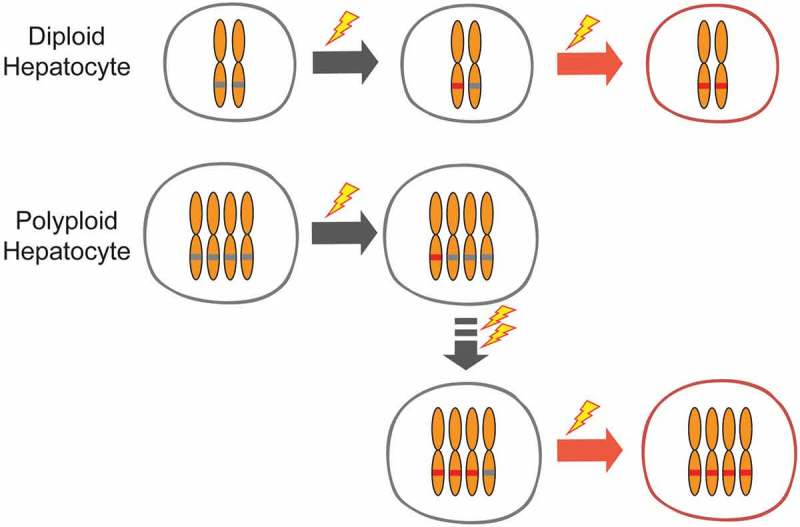
Tumor protection by polyploids is mediated through reduced loss of heterozygosity.
Polyploid cells prevent tumor initiation by impairing loss of heterozygosity. After initiating mutations occur, the remaining wild-type allele(s) in polyploid cells provide additional tumor suppressor gene copies. In diploid cells, a second mutation can lead to loss of heterozygosity.
If polyploid hepatocytes frequently reduce their DNA content through reductive divisions, then the genetic buffering mechanism would not be protective because these divisions could also lead to tumor suppressor loss. In fact, Lucchetta et al. observed that in the Drosophila intestine, polyploid enterocytes go through ploidy reduction, or amitosis, to generate diploid stem cells under conditions of drastic stem cell depletion [113]. They also showed that amitosis could lead to LOH. Therefore, ploidy reducing events other than meiosis can occur in somatic cells. In mammalian livers, the frequency and mechanisms of ploidy reduction are not well understood. Duncan et al., showed that a small fraction (~4%) of polyploid hepatocytes cultured in vitro could divide by forming multipolar spindles or dual spindles, resulting in daughter cells with reduced DNA content, a phenomenon they termed “reductive mitosis” [114]. They also showed that polyploid hepatocytes undergo reductive divisions after transplantation into Fah null mice [115]. It is worth pointing out that, both the stem cell depletion in Drosophila intestine and the hepatocyte transplantation into Fah null liver are intense selection processes, which might force more reductive divisions than would otherwise occur in intact, normal tissues. Another question is whether reductive divisions generate aneuploid daughter cells. As there is currently no known mechanism that ensures chromosome segregation fidelity during reductive divisions, this process is associated with the production of aneuploid cells. In vitro, reductive divisions occur via multipolar spindle formation and double mitosis, which often results in lagging chromosomes, missegregation, and aneuploidy. Occasionally, the two nuclei in a binucleated tetraploid hepatocyte undergo mitosis separately and generate 4 mononucleated daughter cells [114], a process that does not involve multipolar spindle formation. Thus, reductive division can happen through various mechanisms in vitro, but whether it occurs, how it occurs, and whether it relates to aneuploidy formation in vivo are still unclear.
Independent of how aneuploidy might arise, the actual frequency of aneuploid cells in the liver seems highly dependent on the assays and cellular states. By single nuclei sequencing, the percentage of aneuploid chromosomes was non-detectable in non-injured mouse liver, about 0.2% in post-regeneration livers, and about 1% in in vitro expanded hepatocytes (48h expansion) [110,116]. In contrast, with FISH, the aneuploidy frequency was less than 1% in freshly isolated hepatocytes, and about 20% in in vitro expanded hepatocytes (5 d expansion) [114]. One consistent observation from these studies is that the in vitro expansion leads to more aneuploid cells, which is in part due to the absence of proper tissue architecture in vitro [110,116]. In fact, proliferating polyploid hepatocytes were frequently observed in vivo and could also form multipolar spindles, but these multipolar spindles were almost always congressed into bipolar spindles during anaphase, leading to equatorial divisions [110]. Another interesting observation is that, liver regeneration following hepatectomy does result in more aneuploid cells (from non-detectable to ~0.2%) [110], suggesting a low rate of aneuploidy formation during hepatocyte proliferation. As the liver injuries in real life are mostly chronic injuries, which induce multiple rounds of cell proliferation, whether a significant population of aneuploid cells will emerge after chronic injury, and whether this will pose a higher cancer risk or better adaptation are important questions to be addressed [117]. Furthermore, clarifying the actual rate of aneuploid cells in healthy and diseased human livers will be an important finding for the field. Following that, determining if and how polyploids maintain mitotic fidelity will be critical. To resolve these complex issues, intravital imaging of hepatocyte proliferation and long-term lineage-tracing experiments will be needed to truly understand the frequency, mechanics, and long-term impact of ploidy divisions in physiologic settings.
Finally, polyploidy may promote adaptation to stressful environments. For example, tetraploid yeast underwent accelerated environmental adaptation compared to diploid and haploid strains. This was mediated by beneficial aneuploidy, concerted chromosome loss (random loss of multiple chromosomes so that cell ploidy returned to a nearly diploid level [118]), and point mutations [119]. Some beneficial mutations were selected for in tetraploid yeast. In addition, the polyploid Arabidopsis exhibited higher potassium concentration in leaves, thus enhancing their tolerance to salinity and survival after NaCl treatment [120]. In the liver, Duncan et al. argued that polyploidy and aneuploidy allowed for positive selection and adaptation to physiologic stressors [114,121]. Although the prevalence of aneuploidy in normal hepatocytes is debated [110,116], a small pool of aneuploid cells does appear to exist in the liver, especially after regeneration [110]. This is hypothesized to increase genetic diversity within a tissue to facilitate adaptation to stress, but additional evidence to support this hypothesis is needed [119]. Many functional roles of polyploidy in liver have been proposed, but these hypotheses are just beginning to be tested in models with relevance to human disease.
Concluding remarks
The rarity of polyploidy in animals has raised important questions about the purpose of maintaining polyploidy in particular tissues. Are there adaptive features of polyploid cells? Do polyploid cells acquire unique functions when compared to diploid cells? Is polyploidy deleterious in certain circumstances? Reasonable hypotheses are just beginning to be tested. For example, polyploid cells may play roles in normal and regenerative growth, but these roles may vary between cell types. This is exemplified by the enhanced surface coverage by polyploid epicardial tissues [46], but decreased regenerative capacity of polyploid cardiomyocytes [122]. Also, polyploid hepatocytes might be buffered from tumor suppressor LOH and cancer [74], but polyploid cancer cells might suffer from chromosomal or genomic instability [102,103]. The role of polyploidy, like aneuploidy, is likely to be cell-type and context-dependent with respect to cancer. Whether or not “professional polyploid cells” such as hepatocytes possess specialized mechanisms to deal with the cell cycle consequences of multiple centrosomes is an open question.
As one of the few mammalian organs that are highly polyploid, the liver exhibits ploidy changes under stressful conditions such as surgical resection [57], nonalcoholic steatohepatitis [59], and carbon tetrachloride injury, indicative of an association between ploidy change and stress response. Moreover, it is likely that liver polyploidy plays a role in buffering against deleterious mutations and carcinogenesis [74]. These observations suggest that dynamic changes in ploidy were retained in this organ to adapt to disease. For these reasons, understanding the behavior of ploidy populations is going to be more than academic. There are still many unanswered mysteries about hepatic ploidy. How do polyploid hepatocytes proliferate and are ploidy reducing cell divisions common? How do polyploid hepatocytes resolve the issue of multipolar spindles formed during mitosis [110,114]? What are the differences between diploid and polyploid hepatocytes on the levels of chromosome structure, chromatin condensation, transcription, and translation? What conditions are favorable for diploid hepatocytes vs polyploid hepatocytes? Although polyploidy is protective against liver cancer when there are acute mutagenic insults, are they still protective under real world chronic injury and proliferation scenarios (Figure 5)? These questions will help us better understand the physiological significance of liver polyploidy, as well as the evolutionary or functional advantages of polyploidy in other mammalian organs.
Figure 5.
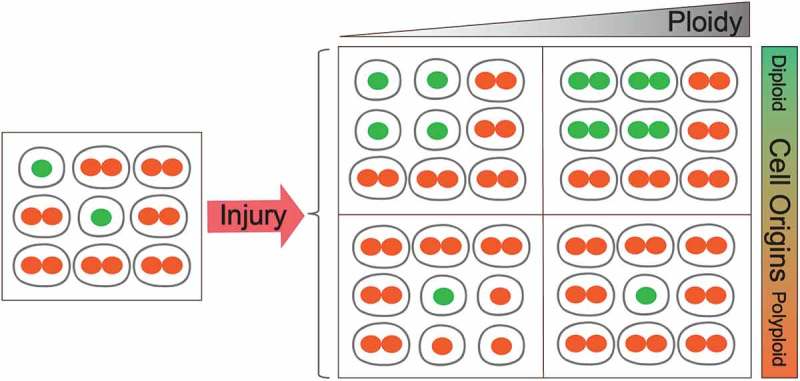
Possible fates of diploid and polyploid hepatocytes during chronic liver injury and regeneration.
The cellular fates of hepatocytes with different ploidies is currently unknown. During liver regeneration after acute or chronic injury, it is known that newly formed hepatocytes can be derived from either diploid (green) or polyploid (orange) cells. It is less clear if the overall ploidy levels increase or decrease, since it is possible for ploidy states of individual cells to increase or decrease. This figure outlines the possible outcomes when cells with different ploidy states contribute to regeneration. Upper left: diploid cells divide more than polyploid cells and diploid daughter cells are more frequently generated, ultimately resulting in livers with decreased ploidy. Upper right: diploid cells polyploidize through cytokinesis failure or endoreduplication, resulting in livers with increased polyploidy. Lower left: polyploid cells divide more than diploid cells and diploid daughter cells are generated through reductive divisions, ultimately resulting in livers with decreased ploidy. Upper right: polyploid cells divide more than diploid cells, and polyploid daughter cells are generated, resulting in livers with increased ploidy.
Glossary box

Funding Statement
This work was supported by the National Institutes of Health [R01CA190525] and the Cancer Prevention and Research Institute of Texas [RP180268].
Disclosure statement
No potential conflict of interest was reported by the authors.
Grant support
H.Z. was supported by the Pollack Foundation, a Burroughs Wellcome Career Award for Medical Scientists, and a CPRIT Individual Investigator Award (RP180268).
References
- [1].Markov AV, Kaznacheev IS.. Evolutionary consequences of polyploidy in prokaryotes and the origin of mitosis and meiosis. Biol Direct. 2016;11:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Weiss-Schneeweiss H, Emadzade K, Jang T-S, et al. Evolutionary consequences, constraints and potential of polyploidy in plants. Cytogenet Genome Res. 2013;140(2–4):137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jiao Y, Wickett NJ, Ayyampalayam S, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473(7345):97–100. [DOI] [PubMed] [Google Scholar]
- [4].Rieseberg LH, Willis JH. Plant speciation. Science. 2007;317(5840):910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Meyers LA, Levin DA. On the abundance of polyploids in flowering plants. Evolution. 2006;60(6):1198–1206. [PubMed] [Google Scholar]
- [6].Adams KL, Wendel JF. Polyploidy and genome evolution in plants. Curr Opin Plant Biol. 2005;8(2):135–141. [DOI] [PubMed] [Google Scholar]
- [7].De Bodt S, Maere S, Van de Peer Y. Genome duplication and the origin of angiosperms. Trends Ecol Evol. 2005;20(11):591–597. [DOI] [PubMed] [Google Scholar]
- [8].Otto SP, Whitton J. Polyploid incidence and evolution. Annu Rev Genet. 2000;34:401–437. [DOI] [PubMed] [Google Scholar]
- [9].Van de Peer Y, Mizrachi E, Marchal K. The evolutionary significance of polyploidy. Nat Rev Genet. 2017;18(7):411–424. [DOI] [PubMed] [Google Scholar]
- [10].Wertheim B, Beukeboom LW, van de Zande L. Polyploidy in animals: effects of gene expression on sex determination, evolution and ecology. Cytogenet Genome Res. 2013;140(2–4):256–269. [DOI] [PubMed] [Google Scholar]
- [11].Leggatt RA, Iwama GK. Occurrence of polyploidy in the fishes. Rev Fish Biol Fish. 2003;13(3):9. [Google Scholar]
- [12].Tymowska J, Kobel HR. Karyotype analysis of Xenopus muelleri (Peters) and Xenopus laevis (Daudin), Pipidae. Cytogenetics. 1972;11(4):270–278. [PubMed] [Google Scholar]
- [13].Le Comber S, Smith C. Polyploidy in fishes: patterns and processes. Biological Journal of the Linnean Society. 2004;82(4):11. [Google Scholar]
- [14].Schmid M, Evans BJ, Bogart JP. Polyploidy in Amphibia. Cytogenet Genome Res. 2015;145(3–4):315–330. [DOI] [PubMed] [Google Scholar]
- [15].Dehal P, Boore JL. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005;3(10):e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Winkelmann M, Pfitzer P, Schneider W. Significance of polyploidy in megakaryocytes and other cells in health and tumor disease. Klin Wochenschr. 1987;65(23):1115–1131. [DOI] [PubMed] [Google Scholar]
- [17].Liu Z, Yue S, Chen X, et al. Regulation of cardiomyocyte polyploidy and multinucleation by CyclinG1. Circ Res. 2010;106(9):1498–1506. [DOI] [PubMed] [Google Scholar]
- [18].Ishikawa H, Bischoff R, Holtzer H. Mitosis and intermediate-sized filaments in developing skeletal muscle. J Cell Biol. 1968;38(3):538–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Frade JM. Somatic tetraploidy in vertebrate neurons: implications in physiology and pathology. Commun Integr Biol. 2010;3(2):201–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Celton-Morizur S, Desdouets C. Polyploidization of liver cells. Adv Exp Med Biol. 2010;676:123–135. [DOI] [PubMed] [Google Scholar]
- [21].Sroga JM, Ma X, Das SK. Developmental regulation of decidual cell polyploidy at the site of implantation. Front Biosci (Schol Ed). 2012;4:1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mori M, Kitazume M, Ose R, et al. Death effector domain-containing protein (DEDD) is required for uterine decidualization during early pregnancy in mice. J Clin Invest. 2011;121(1):318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fei F, Zhang D, Yang Z, et al. The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J Exp Clin Cancer Res. 2015;34:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mosieniak G, Sliwinska MA, Alster O, et al. Polyploidy formation in doxorubicin-treated cancer cells can favor escape from senescence. Neoplasia. 2015;17(12):882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nguyen HG, Ravid K. Polyploidy: mechanisms and cancer promotion in hematopoietic and other cells. Adv Exp Med Biol. 2010;676:105–122. [DOI] [PubMed] [Google Scholar]
- [26].Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol. 2004;5(1):45–54. [DOI] [PubMed] [Google Scholar]
- [27].Ovrebo JI, Edgar BA. Polyploidy in tissue homeostasis and regeneration. Development. 2018;145(14):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gentric G, Desdouets C. Polyploidization in liver tissue. Am J Pathol. 2014;184(2):322–331. [DOI] [PubMed] [Google Scholar]
- [29].Yaffe D, Feldman M. The formation of hybrid multinucleated muscle fibers from myoblasts of different genetic origin. Dev Biol. 1965;11:300–317. [DOI] [PubMed] [Google Scholar]
- [30].Faggioli F, Sacco MG, Susani L, et al. Cell fusion is a physiological process in mouse liver. Hepatology. 2008;48(5):1655–1664. [DOI] [PubMed] [Google Scholar]
- [31].Hammond MP, Laird CD. Control of DNA replication and spatial distribution of defined DNA sequences in salivary gland cells of Drosophila melanogaster. Chromosoma. 1985;91(3–4):279–286. [DOI] [PubMed] [Google Scholar]
- [32].Royzman I, Hayashi-Hagihara A, Dej KJ, et al. The E2F cell cycle regulator is required for Drosophila nurse cell DNA replication and apoptosis. Mech Dev. 2002;119(2):225–237. [DOI] [PubMed] [Google Scholar]
- [33].Ullah Z, Kohn MJ, Yagi R, et al. Differentiation of trophoblast stem cells into giant cells is triggered by p57/Kip2 inhibition of CDK1 activity. Genes Dev. 2008;22(21):3024–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gonçalves CR, Antonini S, Vianna-Morgante AM, et al. Developmental changes in the ploidy of mouse implanting trophoblast cells in vitro. Histochem Cell Biol. 2003;119(3):189–198. [DOI] [PubMed] [Google Scholar]
- [35].Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell. 2010;141(1):81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mansilla S, Priebe W, Portugal J. Mitotic catastrophe results in cell death by caspase-dependent and caspase-independent mechanisms. Cell Cycle. 2006;5(1):53–60. [DOI] [PubMed] [Google Scholar]
- [37].Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7(5):637–651. [DOI] [PubMed] [Google Scholar]
- [38].Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16(12):1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Russell P, Hennessy BT, Li J, et al. Cyclin G1 regulates the outcome of taxane-induced mitotic checkpoint arrest. Oncogene. 2012;31(19):2450–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tsuda Y, Iimori M, Nakashima Y, et al. Mitotic slippage and the subsequent cell fates after inhibition of Aurora B during tubulin-binding agent-induced mitotic arrest. Sci Rep. 2017;7(1):16762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dalton WB, Yang VW. Role of prolonged mitotic checkpoint activation in the formation and treatment of cancer. Future Oncol. 2009;5(9):1363–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ohashi A. Different cell fates after mitotic slippage: from aneuploidy to polyploidy. Mol Cell Oncol. 2016;3(2):e1088503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rengstl B, Newrzela S, Heinrich T, et al. Incomplete cytokinesis and re-fusion of small mononucleated Hodgkin cells lead to giant multinucleated Reed-Sternberg cells. Proc Natl Acad Sci U S A. 2013;110(51):20729–20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lordier L, Jalil A, Aurade F, et al. Megakaryocyte endomitosis is a failure of late cytokinesis related to defects in the contractile ring and Rho/Rock signaling. Blood. 2008;112(8):3164–3174. [DOI] [PubMed] [Google Scholar]
- [45].Trakala M, Rodríguez-Acebes S, Maroto M, et al. Functional reprogramming of polyploidization in megakaryocytes. Dev Cell. 2015;32(2):155–167. [DOI] [PubMed] [Google Scholar]
- [46].Cao J, Wang J, Jackman CP, et al. Tension creates an endoreplication wavefront that leads regeneration of epicardial tissue. Dev Cell. 2017;42(6):600–615. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Spiro Z, Heisenberg CP. Regeneration tensed up: polyploidy takes the lead. Dev Cell. 2017;42(6):559–560. [DOI] [PubMed] [Google Scholar]
- [48].Lima JP. [Anatomy and physiology of the liver secretory apparatus]. Arq Gastroenterol. 1980;17(3):149–160. [PubMed] [Google Scholar]
- [49].Michalopoulos GK. Principles of liver regeneration and growth homeostasis. Compr Physiol. 2013;3(1):485–513. [DOI] [PubMed] [Google Scholar]
- [50].Margall-Ducos G, Celton-Morizur S, Couton D, et al. Liver tetraploidization is controlled by a new process of incomplete cytokinesis. J Cell Sci. 2007;120(Pt 20):3633–3639. [DOI] [PubMed] [Google Scholar]
- [51].Guidotti J-E, Brégerie O, Robert A, et al. Liver cell polyploidization: a pivotal role for binuclear hepatocytes. J Biol Chem. 2003;278(21):19095–19101. [DOI] [PubMed] [Google Scholar]
- [52].Duncan AW. Aneuploidy, polyploidy and ploidy reversal in the liver. Semin Cell Dev Biol. 2013;24(4):347–356. [DOI] [PubMed] [Google Scholar]
- [53].Duncan AW, Hanlon Newell AE, Smith L, et al. Frequent aneuploidy among normal human hepatocytes. Gastroenterology. 2012;142(1):25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Celton-Morizur S, Merlen G, Couton D, et al. The insulin/Akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. J Clin Invest. 2009;119(7):1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Celton-Morizur S, Merlen G, Couton D, et al. Polyploidy and liver proliferation: central role of insulin signaling. Cell Cycle. 2010;9(3):460–466. [DOI] [PubMed] [Google Scholar]
- [56].Miyaoka Y, Ebato K, Kato H, et al. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr Biol. 2012;22(13):1166–1175. [DOI] [PubMed] [Google Scholar]
- [57].Faktor VM, Uryvaeva IV. Progressive polyploidy in mouse liver following repeated hepatectomy. Tsitologiia. 1975;17(8):909–916. [PubMed] [Google Scholar]
- [58].Yamada T, Sogawa K, Kim JK, et al. Increased polyploidy, delayed mitosis and reduced protein phosphatase-1 activity associated with excess copper in the Long Evans Cinnamon rat. Res Commun Mol Pathol Pharmacol. 1998;99(3):283–304. [PubMed] [Google Scholar]
- [59].Gentric G, Maillet V, Paradis V, et al. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J Clin Invest. 2015;125(3):981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang B, Zhao L, Fish M, et al. Self-renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver. Nature. 2015;524(7564):180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kietzmann T. Metabolic zonation of the liver: the oxygen gradient revisited. Redox Biol. 2017;11:622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Benhamouche S, Decaens T, Godard C, et al. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell. 2006;10(6):759–770. [DOI] [PubMed] [Google Scholar]
- [63].Tanami S, Ben-Moshe S, Elkayam A, et al. Dynamic zonation of liver polyploidy. Cell Tissue Res. 2017;368(2):405–410. [DOI] [PubMed] [Google Scholar]
- [64].Cross JC. Genetic insights into trophoblast differentiation and placental morphogenesis. Semin Cell Dev Biol. 2000;11(2):105–113. [DOI] [PubMed] [Google Scholar]
- [65].Huch M, Dorrell C, Boj SF, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494(7436):247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Raven A, Lu W-Y, Man TY, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature. 2017;547(7663):350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tarlow BD, Finegold MJ, Grompe M. Clonal tracing of Sox9+ liver progenitors in mouse oval cell injury. Hepatology. 2014;60(1):278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Schaub JR, Malato Y, Gormond C, et al. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Rep. 2014;8(4):933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Font-Burgada J, Shalapour S, Ramaswamy S, et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell. 2015;162(4):766–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Pu W, Zhang H, Huang X, et al. Mfsd2a+ hepatocytes repopulate the liver during injury and regeneration. Nat Commun. 2016;7:13369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lin S., Nascimento EM, Gajera CR, et al. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature; 2018;556(7700):244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Furuyama K, Kawaguchi Y, Akiyama H, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet. 2011;43(1):34–41. [DOI] [PubMed] [Google Scholar]
- [73].Carpentier R, Suñer RE, van Hul N, et al. Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology. 2011;141(4):1432–8, 1438 e1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zhang S, Zhou K, Luo X, et al. The polyploid state plays a tumor-suppressive role in the liver. Dev Cell. 2018;44(4):447–459. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Pandit SK, Westendorp B, Nantasanti S, et al. E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol. 2012;14(11):1181–1191. [DOI] [PubMed] [Google Scholar]
- [76].Conner EA, Lemmer ER, Sánchez A, et al. E2F1 blocks and c-Myc accelerates hepatic ploidy in transgenic mouse models. Biochem Biophys Res Commun. 2003;302(1):114–120. [DOI] [PubMed] [Google Scholar]
- [77].Chen HZ, Ouseph MM, Li J, et al. Canonical and atypical E2Fs regulate the mammalian endocycle. Nat Cell Biol. 2012;14(11):1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Nevzorova YA, Tschaharganeh D, Gassler N, et al. Aberrant cell cycle progression and endoreplication in regenerating livers of mice that lack a single E-type cyclin. Gastroenterology. 2009;137(2):691–703, 703 e1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kurinna S, Stratton SA, Coban Z, et al. p53 regulates a mitotic transcription program and determines ploidy in normal mouse liver. Hepatology. 2013;57(5):2004–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Sheahan S, Bellamy COC, Treanor L, et al. Additive effect of p53, p21 and Rb deletion in triple knockout primary hepatocytes. Oncogene. 2004;23(8):1489–1497. [DOI] [PubMed] [Google Scholar]
- [81].Mayhew CN, Bosco EE, Fox SR, et al. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res. 2005;65(11):4568–4577. [DOI] [PubMed] [Google Scholar]
- [82].Diril MK, Ratnacaram CK, Padmakumar VC, et al. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci U S A. 2012;109(10):3826–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Minamishima YA, Nakayama K, Nakayama K. Recovery of liver mass without proliferation of hepatocytes after partial hepatectomy in Skp2-deficient mice. Cancer Res. 2002;62(4):995–999. [PubMed] [Google Scholar]
- [84].Kim S-H, Jeon Y, Kim H-S, et al. Hepatocyte homeostasis for chromosome ploidization and liver function is regulated by Ssu72 protein phosphatase. Hepatology. 2016;63(1):247–259. [DOI] [PubMed] [Google Scholar]
- [85].Li D, Cen J, Chen X, et al. Hepatic loss of survivin impairs postnatal liver development and promotes expansion of hepatic progenitor cells in mice. Hepatology. 2013;58(6):2109–2121. [DOI] [PubMed] [Google Scholar]
- [86].Hsu SH, Delgado ER, Otero PA, et al. MicroRNA-122 regulates polyploidization in the murine liver. Hepatology. 2016;64(2):599–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol. 2011;27:585–610. [DOI] [PubMed] [Google Scholar]
- [88].Lee HO, Davidson JM, Duronio RJ. Endoreplication: polyploidy with purpose. Genes Dev. 2009;23(21):2461–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Miettinen TP, Pessa HKJ, Caldez MJ, et al. Identification of transcriptional and metabolic programs related to mammalian cell size. Curr Biol. 2014;24(6):598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Salazar-Roa M, Malumbres M. Fueling the cell division cycle. Trends Cell Biol. 2017;27(1):69–81. [DOI] [PubMed] [Google Scholar]
- [91].Ewald JC, Kuehne A, Zamboni N, et al. The yeast cyclin-dependent kinase routes carbon fluxes to fuel cell cycle progression. Mol Cell. 2016;62(4):532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lu P, Prost S, Caldwell H, et al. Microarray analysis of gene expression of mouse hepatocytes of different ploidy. Mamm Genome. 2007;18(9):617–626. [DOI] [PubMed] [Google Scholar]
- [93].Yoo MJ, Liu X, Pires JC, et al. Nonadditive gene expression in polyploids. Annu Rev Genet. 2014;48:485–517. [DOI] [PubMed] [Google Scholar]
- [94].Corbel C, Diabangouaya P, Gendrel A-V, et al. Unusual chromatin status and organization of the inactive X chromosome in murine trophoblast giant cells. Development. 2013;140(4):861–872. [DOI] [PubMed] [Google Scholar]
- [95].Schoenfelder KP, Fox DT. The expanding implications of polyploidy. J Cell Biol. 2015;209(4):485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Bahar Halpern K, Tanami S, Landen S, et al. Bursty gene expression in the intact mammalian liver. Mol Cell. 2015;58(1):147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Zack TI, Schumacher SE, Carter SL, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45(10):1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kaneko Y, Knudson AG. Mechanism and relevance of ploidy in neuroblastoma. Genes Chromosomes Cancer. 2000;29(2):89–95. [DOI] [PubMed] [Google Scholar]
- [99].Gusnanto A, Wood HM, Pawitan Y, et al. Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics. 2012;28(1):40–47. [DOI] [PubMed] [Google Scholar]
- [100].Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Shackney SE, Smith CA, Miller BW, et al. Model for the genetic evolution of human solid tumors. Cancer Res. 1989;49(12):3344–3354. [PubMed] [Google Scholar]
- [102].Fujiwara T, Bandi M, Nitta M, et al. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437(7061):1043–1047. [DOI] [PubMed] [Google Scholar]
- [103].Davoli T, de Lange T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell. 2012;21(6):765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Reid BJ, Barrett MT, Galipeau PC, et al. Barrett’s esophagus: ordering the events that lead to cancer. Eur J Cancer Prev. 1996;5(Suppl 2):57–65. [DOI] [PubMed] [Google Scholar]
- [105].Levine DS, Sanchez CA, Rabinovitch PS, et al. Formation of the tetraploid intermediate is associated with the development of cells with more than four centrioles in the elastase-simian virus 40 tumor antigen transgenic mouse model of pancreatic cancer. Proc Natl Acad Sci U S A. 1991;88(15):6427–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Margolis RL, Lohez OD, Andreassen PR. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J Cell Biochem. 2003;88(4):673–683. [DOI] [PubMed] [Google Scholar]
- [107].Andreassen PR, Lohez OD, Lacroix FB, et al. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell. 2001;12(5):1315–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Kuffer C, Kuznetsova AY, Storchova Z. Abnormal mitosis triggers p53-dependent cell cycle arrest in human tetraploid cells. Chromosoma. 2013;122(4):305–318. [DOI] [PubMed] [Google Scholar]
- [109].Ganem NJ, Cornils H, Chiu S-Y, et al. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell. 2014;158(4):833–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Knouse KA, Lopez KE, Bachofner M, et al. Chromosome segregation fidelity in epithelia requires tissue architecture. Cell. 2018;175(1):200–211. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Wilkinson P.D., Delgado ER, Alencastro F, et al. The polyploid state restricts hepatocyte proliferation and liver regeneration. Hepatology. 2019;69(3):1242–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Zhang S, Li L, Kendrick SL, et al. TALEN-mediated somatic mutagenesis in murine models of cancer. Cancer Res. 2014;74(18):5311–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Lucchetta EM, Ohlstein B. Amitosis of polyploid cells regenerates functional stem cells in the drosophila intestine. Cell Stem Cell. 2017;20(5):609–620. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Duncan AW, Taylor MH, Hickey RD, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467(7316):707–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wang MJ, Chen F, Li J-X, et al. Reversal of hepatocyte senescence after continuous in vivo cell proliferation. Hepatology. 2014;60(1):349–361. [DOI] [PubMed] [Google Scholar]
- [116].Knouse KA, Wu J, Whittaker CA, et al. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci U S A. 2014;111(37):13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Duncan AW, Soto-Gutierrez A. Liver repopulation and regeneration: new approaches to old questions. Curr Opin Organ Transplant. 2013;18(2):197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Bouchonville K, Forche A, Tang KES, et al. Aneuploid chromosomes are highly unstable during DNA transformation of Candida albicans. Eukaryot Cell. 2009;8(10):1554–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Selmecki AM, Maruvka YE, Richmond PA, et al. Polyploidy can drive rapid adaptation in yeast. Nature. 2015;519(7543):349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Chao D-Y, Dilkes B, Luo H, et al. Polyploids exhibit higher potassium uptake and salinity tolerance in Arabidopsis. Science. 2013;341(6146):658–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Duncan AW, Hanlon Newell AE, Bi W, et al. Aneuploidy as a mechanism for stress-induced liver adaptation. J Clin Invest. 2012;122(9):3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].González-Rosa JM, Sharpe M, Field D, et al. Myocardial polyploidization creates a barrier to heart regeneration in zebrafish. Dev Cell. 2018;44(4):433–446. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]