

ABSTRACT
Basal-like breast cancer (BLBC) and triple-negative breast cancer (TNBC) are aggressive forms of human breast cancer with poor prognosis and limited treatment response. Molecular understanding of BLBC and TNBC biology is instrumental to improve detection and management of these deadly diseases. Tumor suppressors WW domain-containing oxidoreductase (WWOX) and TP53 are altered in BLBC and in TNBC. Nevertheless, the functional interplay between WWOX and p53 is poorly understood. In a recent study by Abdeen and colleagues, it has been demonstrated that WWOX loss drives BLBC formation via deregulating p53 functions. In this review, we highlight important signaling pathways regulated by WWOX and p53 that are related to estrogen receptor signaling, epithelial-to-mesenchymal transition, and genomic instability and how they impact BLBC and TNBC development.
KEYWORDS: BLBC, TNBC, breast cancer, genomic instability, fragile site, ER
Introduction
Human breast cancers are a group of heterogeneous diseases, harboring different genetic alterations and differentially responding to therapy. The classical molecular classification of breast cancer is mainly based on the expression of estrogen receptor (ER), progesterone receptor (PR) and the receptor tyrosine-protein kinase ErbB-2 (HER2). Breast cancer is hence classified into five subtypes: Luminal A (ER+ PR+, HER2- and Ki67-), Luminal B (ER+ PR±, mostly HER2- and Ki67+), HER2/ErbB2 subtype (ER- PR-, HER2+) where HER2 is usually amplified or overexpressed, normal-like subtype expressing adipose and other non-epithelial genes, basal-like subtype (BLBC) (ER- PR-, HER2- and high expression of basal genes such as cytokeratins CK-5 and CK-14 and EGFR/HER1) [1–3]. Most BLBCs are triple negative (TNBC); however, some ER-positive tumors and HER2-positive tumors display a basal-like gene expression profile. Other tumors are believed to acquire basal-like features through a trans-differentiation process known as EMT (epithelial-to-mesenchymal transition). BLBC is usually associated with increased aggressiveness, invasiveness and metastatic potential hence resulting in poor prognosis [4]. In 2007, Herschkowitz et al. introduced a new breast cancer subtype, called Caludin-low, which is characterized by the low expression of genes involved in tight junctions and cell-cell adhesion, including Claudins 3, 4, 7, Occludin, and E-cadherin [5].
Some of the most known genetic alterations in tumor suppressor genes in BLBC and TNBC are mutations in TP53, as well as the loss of RB1 and CDKN2A [6,7]. In fact, it has been reported previously that up to 80% of BLBC harbor TP53 mutations, which commonly include nonsense and frameshift mutations [8]. Recently and based on RNA-based method, it was reported that the RNA of 99.4% of BLBC cases harbor TP53 mutant-like status [9].
In a recent report, our group has demonstrated that tumor suppressor WWOX (WW domain-containing oxidoreductase) is frequently alerted in breast cancer and its targeted deletion in murine mammary epithelial glands drives the development of BLBC-like tumors via inactivation of Trp53 [10]. Several studies by a number of groups have further demonstrated that the WWOX locus is targeted in BLBC and TNBC tumors. For example, it has been shown that WWOX protein levels are reduced or absent in 96.6% of BLBC tumors [11]. More recently, Chang and colleagues have reported that WWOX protein expression is commonly absent in TNBC and this loss drives tumor metastasis [12]. Lately, our group has also determined that WWOX reduced expression and copy number variation are correlated with advanced stages of TNBC, further highlighting the significance of WWOX function in TNBC development [13]. Beside genomic rearrangements and loss of protein expression, hypermethylation of the regulatory region of WWOX has been documented in neoplastic but not in paired adjacent non-neoplastic tissues [14–16].
WWOX tumor suppressor functions have been proposed to be mediated via its protein adaptor capabilities, through which WWOX’s first WW domain binds with proline-rich motifs of partner proteins hence regulating their localization, stability, and transactivation [17–20]. The consequence of these interactions resulted in regulating cellular pathways including apoptosis [21], DNA repair [22,23], cellular metabolism [24,25] and others [26–30]. Of particular interest, WWOX physical and functional interaction with p53 has been proposed to enhance apoptosis in vitro (reviewed in [31,32]). It was also reported that WWOX enhances the cytotoxic function of tumor necrosis factor (TNF) by down-regulating apoptosis inhibitors Bcl-2 and Bcl-xL and up-regulating apoptotic p53 [33]. Our recent findings indicate that the WWOX-p53 functional interaction is important in BLBC and TNBC development [10] as shown previously in other malignancies including, glioblastoma [34] and osteosarcoma [35]. In this review article, the crosstalk of WWOX-p53 in aggressive breast cancer is discussed in an attempt to better understand the complexity of this fatal disease hoping this would help in improving its detection and management.
Wwox and Trp53 conditional knockout (cKO) in mice
Genetically engineered mouse (GEM) models of cancer have been developed to model tumors with genetic alterations that resemble human cancer. Similar to human BLBC, inactivation of WWOX or p53 in the mammary gland epithelium is associated with BLBC-like in murine models. Previous studies revealed that conditional ablation of Trp53 using MMTV-Cre and WAP-Cre transgenic mice in C57BL/6 strain displays high percentage of tumor incidence, though at a later stage of life; in the MMTV case high percentage of mice (47%-100%) developed mammary tumors with latency of 14.5 months (Average of two different MMTV-Cre models) [36]. These tumors were classified as BLBC [36].
The fact that WWOX DNA sequence is highly conserved suggests its essential role in physiology and explains the pathophysiologies associated with its loss. For this reason, we and others have modeled WWOX loss of expression in different animal models (reviewed in [37]). Our lab has pioneered in studying conventional and conditional deletion of murine Wwox GEM models and contributed several research studies supporting WWOX tumor suppressor function [10,38–44], WWOX’s role in cellular metabolism [25,43] and other functions [45,46].
Using these GEM models, it has been demonstrated that aged germline Wwox-heterozygous mice in mammary susceptible C3H genetic background develop mammary tumors with ~50% penetrance [40]. These mammary tumors were mostly ER-negative and PR-negative, expressing cytokeratin (CK)-14, hence reminiscent of the common WWOX inactivation in BLBC. Conditional knockout of Wwox in the mammary glands (using MMTV-Cre) resulted in mammary tumors (latency of 270 days) [10]. These tumors were also characterized as BLBC-like tumors: ER-negative, PR-negative and show high mRNA expression of the basal markers including Ck-14, Ck-17, Cav1, Cav2 and low expression of Foxa1 and Gata3 [10]. In the same study, conditional knockout of Trp53 in mammary glands (using MMTV-Cre transgenic line) resulted in mammary tumors (latency of 262.5 days) with basal-like features. Tumors of both conditional Wwox and Trp53 knockout mice were indistinguishable and displayed hallmarks of BLBC both at immunohistochemical staining and RNA sequencing [10]. Furthermore, when RNA of Wwox-knockout tumors wascompared to that of new and previous Trp53-knockout models [47], all the analyzed tumors clustered together. These findings indicate the existence of an intimate relationship between WWOX and p53 in BLBC and TNBC development.
WWOX, p53 and estrogen receptor (ER)
ER is considered as a powerful prognostic marker and an efficient target for the treatment of hormone-dependent breast cancer with antiestrogens [48,49]. ERs are ligand-activated transcription factors [50] and are known to have a central role in cell cycle regulation [51,52]. Two forms of ER exist: alpha and beta. Currently, only the ERα subtype is clinically considered for clinical decision-making and treatment in breast cancer [50]. ERα is expressed in the majority of breast tumors (with immunohistochemical staining in approximately two-thirds of breast tumors) [50]. Therefore, it is generally believed that breast tumors depend, at least initially, on the stimulatory effects of estrogens; however, many breast tumors eventually progress to an estrogen-independent growth phenotype [53]. Several factors have been proposed to contribute to this later phenomenon among which are increased expression of estrogen-regulated genes [54], activation of mitogen-activated protein kinase (MAPK) [55], overexpression of the vascular endothelial growth factor (VEGF) [56].
Several lines of evidence showed that there is a close link between WWOX and ER in both normal and cancer contexts. In cancer, WWOX is reduced/absent in BLBC and TNBC tumors (mostly negative for ER and PR) [40]. Absent WWOX expression significantly associates with poor distant disease-free survival when compared with patients that display normal WWOX expression. This association was maintained in the subgroup of ER-negative patients but not in ER-positive patients [57]. An independent study has also reported that there is a strong correlation between WWOX expression and ER; ~80% of ER-negative cases demonstrate loss or reduced expression of WWOX [58]. Moreover, patients with strong expression for WWOX are more sensitive to tamoxifen treatment, while patients with reduced expression of WWOX are tamoxifen-resistant [59]. Remarkably, WWOX depletion in human ER-positive MCF7 breast cancer cell line, using shRNA constructs or by CRISPR/Cas9, results in reduced ER expression and function [10,40]. Along this line, it was shown that in normal mouse mammary gland development WWOX protein levels are induced at the age of 3–4 weeks [42], parallel to ER that is known to be induced at the same time [60]. Additionally, ER conditional knockout mice (using MMTV-Cre) display severe-impaired ductal elongation and side branching [61], akin to Wwox cKO mice [42,62]. How WWOX modulates ER levels and activity is largely unknown, one possibility could be through regulation of ER co-activator WBP2 (WW domain-binding protein 2) that enhances ER function via YAP and/or Wnt signaling and reported to physically interact with WWOX [63–66]. Altogether, these clinical findings suggest that there is a positive correlation between WWOX and ER in normal tissues and imply WWOX is upstream to ER signaling. In ER+ breast cancer, ER signaling somehow progresses to be WWOX-independent, while in ER-cancer it seems that tumor cells initially lose WWOX and then lose ER expression.
On the other hand, it is known that ERα inhibits p53-mediated cell cycle arrest and apoptosis through binding p53 and repressing its transcriptional function (MCF7 cells, as well as in a mouse xenograft model) [67–69]. Moreover, Konduri et al. reported that ER plays an important role in the repression of p53-mediated transcriptional activity [70]. Instead, knockdown of ERα (in MCF7) resulted in decreased expression of p53 and its downstream targets, MDM2 and CDKN1A [71]. It was also demonstrated that ERα activates p53 transcription via binding to estrogen response element within the p53 promoter [71].
In our mammary-specific Wwox-cKO model, we presented evidence that the reduction in p53 levels is due to genomic focal deletion that was validated by PCR of gDNA [10]. Interestingly, when WWOX was knocked out in vitro (in MCF7 cells), p53 dysregulation was not associated with genomic deletion in the TP53 locus but likely was due to transcriptional repression [10]. This observation could imply that WWOX controls p53 transcription through regulation of ER. Further studies would be required to delineate the WWOX-ER-p53 axis to better uncover its dynamics in breast cancer development.
WWOX, p53, and EMT
Epithelial-to-mesenchymal transition (EMT) is defined by enhanced levels of the mesenchymal markers (vimentin, smooth-muscle actin, N-cadherin and cadherin-11), reduced expression of the epithelial marker (E-cadherin) [72], loss of cellular adhesion and changes in polarization of the cell and its cytoskeleton [73]. EMT was described to be associated with malignancy, invasion, and metastasis [73]. As BLBC and TNBC are the most aggressive breast cancer subtypes, they display high metastatic ability and mesenchymal features [73]. Sarrio et al. indeed reported an up-regulation of the EMT markers and reduction of the epithelial markers among basal-like tumors, suggesting that this mesenchymal transition may be related to the high aggressiveness and metastatic spread of the basal-like tumors [72]. Choi et al. also showed that BLBCs have high levels of the EMT markers [74].
Several lines of evidence demonstrate that inactivation of WWOX affects EMT, which might be related to tumor progression [10,13,75]. Gourley et al. reported that loss of WWOX during ovarian cancer development results in increased levels of membranous integrins and increased adhesion and migration of tumor cells on extracellular matrix and hence speculated that this would enhance locoregional peritoneal ovarian tumor spread and metastasis [76]. Consistent with this scenario, loss of WWOX protein expression has previously been correlated with advanced stage disease and poorer survival in ovarian cancer patients [58]. More recently, Khawaled et al. demonstrated that manipulation of WWOX expression in TNBC and BLBC cell lines modulates invasion, metastatic seeding, and colonization [13]. It was demonstrated that WWOX, through modulation of microRNAs, regulates the levels of both epithelial and mesenchymal markers hence antagonizing EMT and invasion of TNBC cells [13]. This has been suggested to be achieved, at least in part, through negative regulation of c-MYC expression and activity, resulting in miR-146a accumulation hence targeting the mesenchymal gene Fibronectin and supports the epithelial phenotype [13]. These findings and others suggest that WWOX tumor suppressor function impacts a plethora of pathways to antagonize invasion and metastasis [12].
p53 has been also described to regulate EMT, often through microRNAs [77–82]. For example, Ohtsuka et al. reported that loss of p53 induces EMT and cellular motility in gastric epithelial cells prior to the development of gastric cancer [83]. Additionally, it was shown that p53 alters ZEB1 and ZEB2 expression, transcription factors known to promote EMT, by upregulating microRNAs, including miR-200 and miR-192 family members [80]. Di Gennaro et al. demonstrated that p53 controls EMT and tumor cell invasion via miR-30a, which probably acts through upregulation of miR-200c [84]. p53 transactivates miR-200c through direct binding to its promoter [77]; when p53 is lost in mammary epithelial cells this results in decreased expression of miR-200c and in EMT activation [77,85]. Altogether, these findings suggest that both WWOX and p53 may play an important role in inhibiting the EMT pathway and subsequently inhibiting the breast cancer progression. Whether WWOX loss mediates p53 regulation of microRNAs in TNBC and BLBC is still unknown and shall be determined in future studies.
WWOX, p53, and genomic instability
Genome instability is considered as an enabling characteristic of almost all cancer types [86]. Genomic instability can be referred to an increased tendency of alterations in the genome during the life cycle of cells. These genomic alterations confer a selective advantage on subclones of cells, enabling their outgrowth and expansion, resulting in cancer [86]. Moreover, genomic instability is thought to play an important role in cancer resistance to therapy [87,88]. Disruption of the DNA damage response (DDR) machinery in human cells leads to genomic instability and an increased risk of cancer progression [86,88].
p53 has been shown to be involved in the DDR. Induction of DSBs activates ATM [89], which in turn phosphorylates p53 directly or indirectly, resulting in p53 accumulation and activation [90–93]. ATM could also phosphorylate MDM2, inhibiting the ability of MDM2 to ubiquitinate p53, thus leading to p53 stabilization [94]. The consequence of p53 accumulation and stabilization results in transcribing its target genes which include those important for cell cycle arrest (to allow repair of the DNA damage) or apoptosis (to eliminate cells with unrepaired DNA). The cell cycle arrest mediated by p53 activation is mainly regulated by p21/WAF [95] while apoptosis is mediated by different pro-apoptotic genes including Puma, Noxa, Bax, and others [95]. In case of p53 deficiency, unrepaired damage and genomic instability are observed [96]. In fact, p53-deficient mammary tumors display increased genomic instability with aneuploidy, amplifications, and deletions [97] and are associated with radioresistance [98].
WWOX has been also shown to play a direct role in the DDR. Upon DNA damage, WWOX levels are induced and accumulate in the cell nucleus, where it interacts with ATM and enhances its activation [22]. WWOX depletion results in reduced ATM signaling and reduced DNA repair, contributing to increased DSBs [10,99]. WWOX loss was also associated with increased chromosomal instability and increased DSBs [23]. Interestingly, the induction of DSBs in WWOX deficient cells is associated with impaired p53 accumulation and signaling [10,22]. More recently, it was shown that WWOX binds BRCA1 and regulates DNA repair [100]. Altogether, we believe that alteration of WWOX expression, which is associated with impaired checkpoint protein signaling, could impair p53 accumulation and activation resulting in improper DDR and genomic instability further contributing to BLBC and TNBC formation and progression.
The interplay between WWOX and p53 in breast carcinogenesis: conclusions and future perspective
Positive and/or negative regulators, mainly at the protein level, do control the p53 protein. Upon stress, kinases including ATM/ATR and Chk1/Chk2 phosphorylate p53, thus stabilize its protein product and promote its DNA binding ability [101]. Another positive regulation mechanism is recognized by increasing the p53 stability as shown by Freeman et al. demonstrating that PTEN stabilizes p53 by increasing its half-life [102]. On the other hand, Mdm2 negatively regulates p53 through the ubiquitin-proteasome system [103–105]. Mdm2 is transcriptionally induced by p53 highlighting a negative feedback loop mechanism in which p53 controls its own degradation. Other reports showed that microRNAs could also regulate p53 levels [106–108]. For example, miR-125b binds to 3′-UTR of the TP53 mRNA resulting in decreased mRNA level [107]. Knockdown of miR-125b induced apoptosis through increasing the levels of p53 in human lung fibroblasts [107]. MicroRNAs can also positively regulate p53 by targeting known negative regulators of p53, such as silent information regulator 1 (SIRT1) gene [109,110].
Our recent findings demonstrate that WWOX, gene product of FRA16D, can also regulate p53 levels and activity. Expression of the WWOX gene is induced upon DNA damage to enhance DNA repair or apoptosis [22,23]. Furthermore, several microRNAs were shown to regulate WWOX expression. One example is overexpression of miR-29 in lung cancer cell lines which has been shown to restore normal patterns of DNA methylation and induce expression of WWOX hence suppressing tumorigenicity both in vitro and in vivo. Interestingly, miR-29 is induced in response to DNA damage and occurs in a p53-dependent manner [111] perhaps contributing to the suppression of breast carcinogenesis [112,113].
The similar behavior of WWOX and p53 suggests that both act in the same way, at least in breast cancer. Moreover, the current evidence suggests that WWOX is an upstream regulator of p53. We showed that WWOX deficiency results in p53 loss/reduction in vitro and in vivo [10,99]. Our recent study suggests new mechanisms of regulation of p53 that are mediated by WWOX action (Figure 1). The first mechanism we propose is p53 regulation through ER signaling; p53 is regulated by ER and WWOX depletion results in reduced ER levels hence it is possible to speculate that WWOX through regulating ER levels controls those of p53. It is possible that the reduction in ER levels in WWOX KO cells is mediated through WBP2 protein. In fact, it was reported that WBP2 is essential for proper activation of PR and ER [63]. Intriguingly, WBP2 contains a PPxY motif that associates with WW domain protein YAP [114], leading to ER and PR transactivation. As WWOX is known to interact with PPxY-containing proteins as well [26], it is possible that WWOX may associate with WBP2 and regulate ER/PR signaling. Consistent with this scenario, it was shown that the WBP2–WWOX interaction attenuates the transactivation functions of ER [115]. It is also possible that WWOX may associate with transcription factors that regulate ER gene transcription. Further studies should shed light on such plausible mechanisms.
Figure 1.
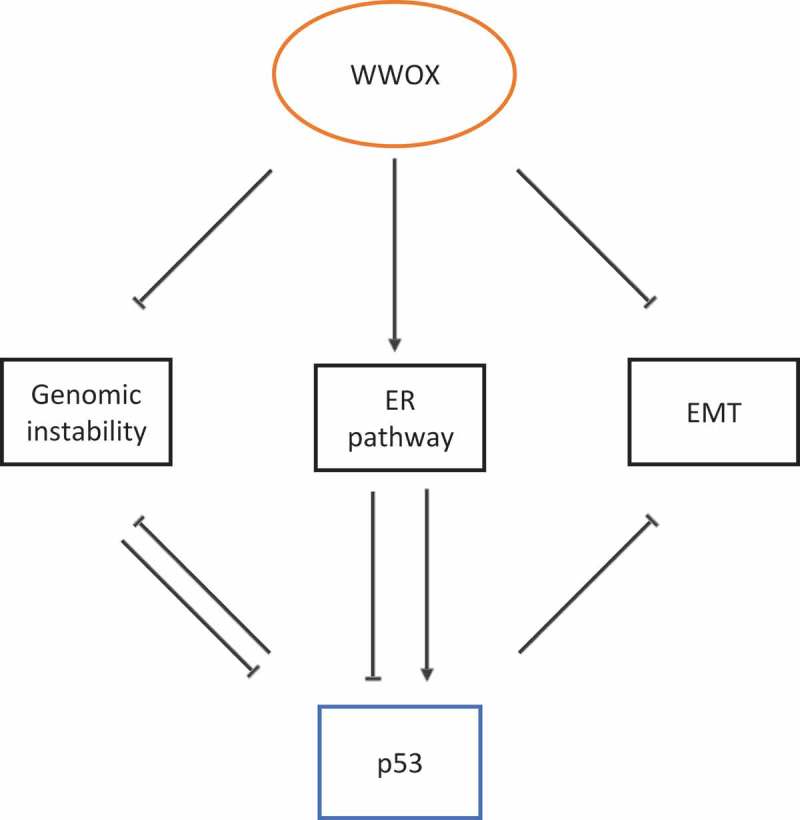
WWOX regulates p53 function, model of action and scenarios. I. WWOX modulates the DNA damage response pathway, antagonizes genomic instability and inhibits loss of p53. II. WWOX modulates ER expression and function, which in turn positively or negatively affects p53. III. WWOX inhibits the epithelial-to-mesenchymal transition (EMT). p53 is known to inhibit both genomic instability and EMT.
Mammary tumors developed in Wwox and Trp53 mouse models display enhanced expression of EMT markers and high EMT scores (compared to other mammary tumor mouse models) [10]. Up till now, the role of both WWOX and p53 in EMT is reported to be mediated by microRNAs. Whether WWOX has an upstream effect on p53 in the EMT program is still to be investigated. Recently, it has been reported that WWOX negatively regulates c-Myc, a master regulator of microRNA expression [13]. Some of these microRNAs may regulate p53 mRNA levels, though this is yet to be shown.
The third possible mechanism of action for WWOX is through maintaining genome stability. The role of WWOX or p53 in the DDR is heavily investigated, but the connection between both of them is poorly studied. The Aqeilan’s lab showed that in response to induction of DSBs or DNA single-strand breaks (SSBs), WWOX accumulates and promotes DNA repair through the activation of ATM or ATR [22,23]. Indeed, WWOX loss is associated with reduced ATM/ATR activation and substrate phosphorylation, such as CHK2 and CHK1 phosphorylation, respectively [22,23]. Reduced ATM signaling could result in reduced p53 phosphorylation [91] and impaired DDR in WWOX-deficient cells. Indeed, we observed that ionizing radiation of WWOX–depleted MCF7 cells results in reduced ATM phosphorylation and reduced nuclear p53 accumulation [10], suggesting that WWOX may affect p53 levels through ATM. It was also reported by Ouchi et al. that BRCA1 coimmunoprecipitates with p53 and acts as a p53 coactivator, which enhances p53-dependent gene expression [116]. Moreover, BRCA1 and/or TP53 alterations are associated with higher homologous recombination deficiency score [117]. The Huebner lab has recently shown that WWOX, via its first WW domain, interacts with BRCA1 and proposed that the BRCA1–WWOX complex supports non-homologous end-joining pathway as the dominant DSB repair pathway in WWOX-sufficient cells [100]. Whether WWOX affects p53’s level/function through BRCA1, is still to be investigated.
Since cloning of the TP53 gene [118], thousands of articles were published describing its critical roles, functions, and partners in cancer and biology. Consequently, p53 is known today as the master regulator of many cellular pathways [119,120]. Here we introduce a new regulator of p53, WWOX. This regulation occurs at three levels: DNA, RNA, and protein.
Funding Statement
This work was supported by the FP7 Ideas: European Research Council [682118]; Israel Science Foundation [1574/15].
Acknowledgments
The Aqeilan lab is supported by European Research Council (ERC)-Consolidator Grant under the European Union’s Horizon 2020 research and innovation program (grant agreement No. 682118) and Israel Science Foundation (grant agreement No 1574/15).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. [DOI] [PubMed] [Google Scholar]
- [2].Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dai X, Li T, Bai Z, et al. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res. 2015;5(10):2929–2943. [PMC free article] [PubMed] [Google Scholar]
- [4].Gusterson B, Eaves CJ.. Basal-like breast cancers: from pathology to biology and back again. Stem Cell Reports. 2018;10(6):1676–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Herschkowitz JI, Simin K, Weigman VJ, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8(5):R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Knudsen ES, McClendon AK, Franco J, et al. RB loss contributes to aggressive tumor phenotypes in MYC-driven triple negative breast cancer. Cell Cycle. 2015;14(1):109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jiang Z, Jones R, Liu JC, et al. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle. 2011;10(10):1563–1570. [DOI] [PubMed] [Google Scholar]
- [8].Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Williams LA, Butler EN, Sun X, et al. TP53 protein levels, RNA-based pathway assessment, and race among invasive breast cancer cases. NPJ Breast Cancer. 2018;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Abdeen SK, Ben-David U, Shweiki A, et al. Somatic loss of WWOX is associated with TP53 perturbation in basal-like breast cancer. Cell Death Dis. 2018;9(8):832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Guler G, Huebner K, Himmetoglu C, et al. Fragile histidine triad protein, WW domain-containing oxidoreductase protein Wwox, and activator protein 2gamma expression levels correlate with basal phenotype in breast cancer. Cancer. 2009;115(4):899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chang R, Song L, Xu Y, et al. Loss of Wwox drives metastasis in triple-negative breast cancer by JAK2/STAT3 axis. Nat Commun. 2018;9(1):3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Khawaled S, Suh SS, Abdeen SK, et al. WWOX inhibits metastasis of triple-negative breast cancer cells via modulation of microRNAs. Cancer Res. 2019. [DOI] [PubMed] [Google Scholar]
- [14].Iliopoulos D, Guler G, Han SY, et al. Fragile genes as biomarkers: epigenetic control of WWOX and FHIT in lung, breast and bladder cancer. Oncogene. 2005;24(9):1625–1633. [DOI] [PubMed] [Google Scholar]
- [15].Iliopoulos D, Fabbri M, Druck T, et al. Inhibition of breast cancer cell growth in vitro and in vivo: effect of restoration of Wwox expression. Clin Cancer Res. 2007;13(1):268–274. [DOI] [PubMed] [Google Scholar]
- [16].Gardenswartz A, Aqeilan RI. WW domain-containing oxidoreductase’s role in myriad cancers: clinical significance and future implications. Exp Biol Med. 2014;239(3):253–263. [DOI] [PubMed] [Google Scholar]
- [17].Abu-Remaileh M, Joy-Dodson E, Schueler-Furman O, et al. Pleiotropic functions of tumor suppressor WWOX in normal and cancer cells. J Biol Chem. 2015;290(52):30728–30735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aqeilan RI. Role of common fragile sites and corresponding genes in cancer development. Cell Mol Life Sci. 2014;71(23):4487–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Salah Z, Alian A, Aqeilan RI. WW domain-containing proteins: retrospectives and the future. Front Biosci. 2012;17:331–348. [DOI] [PubMed] [Google Scholar]
- [20].Salah Z, Aqeilan RI. WW domain interactions regulate the Hippo tumor suppressor pathway. Cell Death Dis. 2011;2:e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Aqeilan RI, Pekarsky Y, Herrero JJ, et al. Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc Natl Acad Sci U S A. 2004;101(13):4401–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Abu-Odeh M, Salah Z, Herbel C, et al. WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc Natl Acad Sci U S A. 2014;111(44):E4716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Abu-Odeh M, Hereema NA, Aqeilan RI. WWOX modulates the ATR-mediated DNA damage checkpoint response. Oncotarget. 2016;7(4):4344–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Abu-Remaileh M, Aqeilan RI. Tumor suppressor WWOX regulates glucose metabolism via HIF1alpha modulation. Cell Death Differ. 2014;21(11):1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Abu-Remaileh M, Abu-Remaileh M, Akkawi R, et al. WWOX somatic ablation in skeletal muscles alters glucose metabolism. Mol Metab. 2019;22:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Salah Z, Aqeilan R, Huebner K. WWOX gene and gene product: tumor suppression through specific protein interactions. Future Oncol. 2010;6(2):249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Aldaz CM, Ferguson BW, Abba MC. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim Biophys Acta. 2014;1846(1):188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Richards RI, Choo A, Lee CS, et al. WWOX, the chromosomal fragile site FRA16D spanning gene: its role in metabolism and contribution to cancer. Exp Biol Med. 2015;240(3):338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schrock MS, Huebner K. WWOX: a fragile tumor suppressor. Exp Biol Med. 2015;240(3):296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fu J, Qu Z, Yan P, et al. The tumor suppressor gene WWOX links the canonical and noncanonical NF-kappaB pathways in HTLV-I Tax-mediated tumorigenesis. Blood. 2011;117(5):1652–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chang JY, He RY, Lin HP, et al. Signaling from membrane receptors to tumor suppressor WW domain-containing oxidoreductase. Exp Biol Med (Maywood). 2010;235(7):796–804. [DOI] [PubMed] [Google Scholar]
- [32].Chang NS. A potential role of p53 and WOX1 in mitochondrial apoptosis (review). Int J Mol Med. 2002;9(1):19–24. [PubMed] [Google Scholar]
- [33].Chang NS, Pratt N, Heath J, et al. Hyaluronidase induction of a WW domain-containing oxidoreductase that enhances tumor necrosis factor cytotoxicity. J Biol Chem. 2001;276(5):3361–3370. [DOI] [PubMed] [Google Scholar]
- [34].Chiang MF, Chou PY, Wang WJ, et al. Tumor suppressor WWOX and p53 alterations and drug resistance in glioblastomas. Front Oncol. 2013;3:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Del Mare S, Husanie H, Iancu O, et al. WWOX and p53 dysregulation synergize to drive the development of osteosarcoma. Cancer Res. 2016;76(20):6107–6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lin SC, Lee KF, Nikitin AY, et al. Somatic mutation of p53 leads to estrogen receptor alpha-positive and -negative mouse mammary tumors with high frequency of metastasis. Cancer Res. 2004;64(10):3525–3532. [DOI] [PubMed] [Google Scholar]
- [37].Tanna M, Aqeilan RI. Modeling WWOX loss of function in vivo: what have we learned? Front Oncol. 2018;8:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Aqeilan RI, Trapasso F, Hussain S, et al. Targeted deletion of Wwox reveals a tumor suppressor function. Proc Natl Acad Sci U S A. 2007;104(10):3949–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Aqeilan RI, Hagan JP, Aqeilan HA, et al. Inactivation of the Wwox gene accelerates forestomach tumor progression in vivo. Cancer Res. 2007;67(12):5606–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Abdeen SK, Salah Z, Maly B, et al. Wwox inactivation enhances mammary tumorigenesis. Oncogene. 2011;30(36):3900–3906. [DOI] [PubMed] [Google Scholar]
- [41].Abdeen SK, Del Mare S, Hussain S, et al. Conditional inactivation of the mouse Wwox tumor suppressor gene recapitulates the null phenotype. J Cell Physiol. 2013;228(7):1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Abdeen SK, Salah Z, Khawaled S, et al. Characterization of WWOX inactivation in murine mammary gland development. J Cell Physiol. 2013;228(7):1391–1396. [DOI] [PubMed] [Google Scholar]
- [43].Abu-Remaileh M, Khalaileh A, Pikarsky E, et al. WWOX controls hepatic HIF1alpha to suppress hepatocyte proliferation and neoplasia. Cell Death Dis. 2018;9(5):511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kurek KC, Del Mare S, Salah Z, et al. Frequent attenuation of the WWOX tumor suppressor in osteosarcoma is associated with increased tumorigenicity and aberrant RUNX2 expression. Cancer Res. 2010;70(13):5577–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aqeilan RI, Hassan MQ, de Bruin A, et al. The WWOX tumor suppressor is essential for postnatal survival and normal bone metabolism. J Biol Chem. 2008;283(31):21629–21639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Aqeilan RI, Hagan JP, de Bruin A, et al. Targeted ablation of the WW domain-containing oxidoreductase tumor suppressor leads to impaired steroidogenesis. Endocrinology. 2009;150(3):1530–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhu M, Yi M, Kim CH, et al. Integrated miRNA and mRNA expression profiling of mouse mammary tumor models identifies miRNA signatures associated with mammary tumor lineage. Genome Biol. 2011;12(8):R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dunnwald LK, Rossing MA, Li CI. Hormone receptor status, tumor characteristics, and prognosis: a prospective cohort of breast cancer patients. Breast Cancer Res. 2007;9(1):R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Radmacher MD, Simon R. Estimation of tamoxifen’s efficacy for preventing the formation and growth of breast tumors. J Natl Cancer Inst. 2000;92(1):48–53. [DOI] [PubMed] [Google Scholar]
- [50].Dahlman-Wright K, Cavailles V, Fuqua SA, et al. International union of pharmacology. LXIV. Estrogen receptors. Pharmacol Rev. 2006;58(4):773–781. [DOI] [PubMed] [Google Scholar]
- [51].Lamb R, Lehn S, Rogerson L, et al. Cell cycle regulators cyclin D1 and CDK4/6 have estrogen receptor-dependent divergent functions in breast cancer migration and stem cell-like activity. Cell Cycle. 2013;12(15):2384–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].JavanMoghadam S, Weihua Z, Hunt KK, et al. Estrogen receptor alpha is cell cycle-regulated and regulates the cell cycle in a ligand-dependent fashion. Cell Cycle. 2016;15(12):1579–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jensen EV, Jordan VC. The estrogen receptor: a model for molecular medicine. Clin Cancer Res. 2003;9(6):1980–1989. [PubMed] [Google Scholar]
- [54].Brunner N, Boulay V, Fojo A, et al. Acquisition of hormone-independent growth in MCF-7 cells is accompanied by increased expression of estrogen-regulated genes but without detectable DNA amplifications. Cancer Res. 1993;53(2):283–290. [PubMed] [Google Scholar]
- [55].Coutts AS, Murphy LC. Elevated mitogen-activated protein kinase activity in estrogen-nonresponsive human breast cancer cells. Cancer Res. 1998;58(18):4071–4074. [PubMed] [Google Scholar]
- [56].Guo P, Fang Q, Tao HQ, Schafer CA, Fenton BM, Ding I, et al Overexpression of vascular endothelial growth factor by MCF-7 breast cancer cells promotes estrogen-independent tumor growth in vivo. Cancer Res. 2003;63(15):4684–4691. [PubMed] [Google Scholar]
- [57].Aqeilan RI, Donati V, Gaudio E, et al. Association of Wwox with ErbB4 in breast cancer. Cancer Res. 2007;67(19):9330–9336. [DOI] [PubMed] [Google Scholar]
- [58].Nunez MI, Ludes-Meyers J, Abba MC, et al. Frequent loss of WWOX expression in breast cancer: correlation with estrogen receptor status. Breast Cancer Res Treat. 2005;89(2):99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Guler G, Iliopoulos D, Guler N, et al. Wwox and Ap2gamma expression levels predict tamoxifen response. Clin Cancer Res. 2007;13(20):6115–6121. [DOI] [PubMed] [Google Scholar]
- [60].Manavathi B, Samanthapudi VS, Gajulapalli VN. Estrogen receptor coregulators and pioneer factors: the orchestrators of mammary gland cell fate and development. Front Cell Dev Biol. 2014;2:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Feng Y, Manka D, Wagner KU, et al. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci U S A. 2007;104(37):14718–14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ferguson BW, Gao X, Kil H, et al. Conditional Wwox deletion in mouse mammary gland by means of two Cre recombinase approaches. PLoS One. 2012;7(5):e36618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Dhananjayan SC, Ramamoorthy S, Khan OY, et al. WW domain binding protein-2, an E6-associated protein interacting protein, acts as a coactivator of estrogen and progesterone receptors. Mol Endocrinol. 2006;20(10):2343–2354. [DOI] [PubMed] [Google Scholar]
- [64].Lim SK, Orhant-Prioux M, Toy W, et al. Tyrosine phosphorylation of transcriptional coactivator WW-domain binding protein 2 regulates estrogen receptor alpha function in breast cancer via the Wnt pathway. Faseb J. 2011;25(9):3004–3018. [DOI] [PubMed] [Google Scholar]
- [65].Chen S, Wang H, Huang YF, et al. WW domain-binding protein 2: an adaptor protein closely linked to the development of breast cancer. Mol Cancer. 2017;16(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].McDonald CB, Buffa L, Bar-Mag T, et al. Biophysical basis of the binding of WWOX tumor suppressor to WBP1 and WBP2 adaptors. J Mol Biol. 2012;422(1):58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Liu W, Ip MM, Podgorsak MB, et al. Disruption of estrogen receptor alpha-p53 interaction in breast tumors: a novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res Treat. 2009;115(1):43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Liu W, Konduri SD, Bansal S, et al. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J Biol Chem. 2006;281(15):9837–9840. [DOI] [PubMed] [Google Scholar]
- [69].Sayeed A, Konduri SD, Liu W, et al. Estrogen receptor alpha inhibits p53-mediated transcriptional repression: implications for the regulation of apoptosis. Cancer Res. 2007;67(16):7746–7755. [DOI] [PubMed] [Google Scholar]
- [70].Konduri SD, Medisetty R, Liu W, et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci U S A. 2010;107(34):15081–15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Berger CE, Qian Y, Liu G, et al. p53, a target of estrogen receptor (ER) alpha, modulates DNA damage-induced growth suppression in ER-positive breast cancer cells. J Biol Chem. 2012;287(36):30117–30127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sarrio D, Rodriguez-Pinilla SM, Hardisson D, et al. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68(4):989–997. [DOI] [PubMed] [Google Scholar]
- [73].Felipe Lima J, Nofech-Mozes S, Bayani J, et al. EMT in breast carcinoma-A review. J Clin Med. 2016;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Pomp V, Leo C, Mauracher A, et al. Differential expression of epithelial-mesenchymal transition and stem cell markers in intrinsic subtypes of breast cancer. Breast Cancer Res Treat. 2015;154(1):45–55. [DOI] [PubMed] [Google Scholar]
- [75].Pluciennik E, Nowakowska M, Pospiech K, et al. The role of WWOX tumor suppressor gene in the regulation of EMT process via regulation of CDH1-ZEB1-VIM expression in endometrial cancer. Int J Oncol. 2015;46(6):2639–2648. [DOI] [PubMed] [Google Scholar]
- [76].Gourley C, Paige AJ, Taylor KJ, et al. WWOX gene expression abolishes ovarian cancer tumorigenicity in vivo and decreases attachment to fibronectin via integrin alpha3. Cancer Res. 2009;69(11):4835–4842. [DOI] [PubMed] [Google Scholar]
- [77].Chang CJ, Chao CH, Xia W, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13(3):317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kim NH, Kim HS, Li XY, et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011;195(3):417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Siemens H, Jackstadt R, Hunten S, et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10(24):4256–4271. [DOI] [PubMed] [Google Scholar]
- [80].Kim T, Veronese A, Pichiorri F, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208(5):875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ma L, Weinberg RA. MicroRNAs in malignant progression. Cell Cycle. 2008;7(5):570–572. [DOI] [PubMed] [Google Scholar]
- [82].Shi XB, Tepper CG, deVere White RW. Cancerous miRNAs and their regulation. Cell Cycle. 2008;7(11):1529–1538. [DOI] [PubMed] [Google Scholar]
- [83].Ohtsuka J, Oshima H, Ezawa I, et al. Functional loss of p53 cooperates with the in vivo microenvironment to promote malignant progression of gastric cancers. Sci Rep. 2018;8(1):2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Di Gennaro A, Damiano V, Brisotto G, et al. A p53/miR-30a/ZEB2 axis controls triple negative breast cancer aggressiveness. Cell Death Differ. 2018;(12):2165–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Radojicic J, Zaravinos A, Vrekoussis T, et al. MicroRNA expression analysis in triple-negative (ER, PR and Her2/neu) breast cancer. Cell Cycle. 2011;10(3):507–517. [DOI] [PubMed] [Google Scholar]
- [86].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- [87].Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220–228. [DOI] [PubMed] [Google Scholar]
- [88].Hainaut P, Plymoth A. Targeting the hallmarks of cancer: towards a rational approach to next-generation cancer therapy. Curr Opin Oncol. 2013;25(1):50–51. [DOI] [PubMed] [Google Scholar]
- [89].Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14(4):197–210. [PubMed] [Google Scholar]
- [90].Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281(5383):1674–1677. [DOI] [PubMed] [Google Scholar]
- [91].Canman CE, Lim DS, Cimprich KA, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281(5383):1677–1679. [DOI] [PubMed] [Google Scholar]
- [92].Chehab NH, Malikzay A, Stavridi ES, et al. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci U S A. 1999;96(24):13777–13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Hirao A, Kong YY, Matsuoka S, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287(5459):1824–1827. [DOI] [PubMed] [Google Scholar]
- [94].Cheng Q, Chen J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle. 2010;9(3):472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6(3):a026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yeo CQX, Alexander I, Lin Z, et al. p53 maintains genomic stability by preventing interference between transcription and replication. Cell Rep. 2016;15(1):132–146. [DOI] [PubMed] [Google Scholar]
- [97].Donehower LA, Godley LA, Aldaz CM, et al. Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Genes Dev. 1995;9(7):882–895. [DOI] [PubMed] [Google Scholar]
- [98].Lee JM, Bernstein A. p53 mutations increase resistance to ionizing radiation. Proc Natl Acad Sci U S A. 1993;90(12):5742–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hazan I, Abu-Odeh M, Hofmann TG, et al. WWOX guards genome stability by activating ATM. Mol Cell Oncol. 2015;2(4):e1008288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Schrock MS, Batar B, Lee J, et al. Wwox-Brca1 interaction: role in DNA repair pathway choice. Oncogene. 2017;36(16):2215–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137(4):609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Freeman DJ, Li AG, Wei G, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3(2):117–130. [DOI] [PubMed] [Google Scholar]
- [103].Juven-Gershon T, Oren M. Mdm2: the ups and downs. Mol Med. 1999;5(2):71–83. [PMC free article] [PubMed] [Google Scholar]
- [104].Freedman DA, Wu L, Levine AJ. Functions of the MDM2 oncoprotein. Cell Mol Life Sci. 1999;55(1):96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Haupt Y, Maya R, Kazaz A, et al. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–299. [DOI] [PubMed] [Google Scholar]
- [106].Hu W, Chan CS, Wu R, et al. Negative regulation of tumor suppressor p53 by microRNA miR-504. Mol Cell. 2010;38(5):689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Le MT, Teh C, Shyh-Chang N, et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23(7):862–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Feng Z, Zhang C, Wu R, et al. Tumor suppressor p53 meets microRNAs. J Mol Cell Biol. 2011;3(1):44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105(36):13421–13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Park SY, Lee JH, Ha M, et al. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol. 2009;16(1):23–29. [DOI] [PubMed] [Google Scholar]
- [111].Ugalde AP, Ramsay AJ, de la Rosa J, et al. Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. Embo J. 2011;30(11):2219–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Rostas JW 3rd, Pruitt HC, Metge BJ, et al. microRNA-29 negatively regulates EMT regulator N-myc interactor in breast cancer. Mol Cancer. 2014;13:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Muluhngwi P, Alizadeh-Rad N, Vittitow SL, et al. The miR-29 transcriptome in endocrine-sensitive and resistant breast cancer cells. Sci Rep. 2017;7(1):5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Sudol M, Chen HI, Bougeret C, et al. Characterization of a novel protein-binding module–the WW domain. FEBS Lett. 1995;369(1):67–71. [DOI] [PubMed] [Google Scholar]
- [115].Buffa L, Saeed AM, Nawaz Z. Molecular mechanism of WW-domain binding protein-2 coactivation function in estrogen receptor signaling. IUBMB Life. 2013;65(1):76–84. [DOI] [PubMed] [Google Scholar]
- [116].Ouchi T, Monteiro AN, August A, et al. BRCA1 regulates p53-dependent gene expression. Proc Natl Acad Sci U S A. 1998;95(5):2302–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Knijnenburg TA, Wang L, Zimmermann MT, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018;23(1):239–54 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Oren M, Levine AJ. Molecular cloning of a cDNA specific for the murine p53 cellular tumor antigen. Proc Natl Acad Sci U S A. 1983;80(1):56–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Menendez D, Inga A, Resnick MA. The biological impact of the human master regulator p53 can be altered by mutations that change the spectrum and expression of its target genes. Mol Cell Biol. 2006;26(6):2297–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Farnebo M, Bykov VJ, Wiman KG. The p53 tumor suppressor: a master regulator of diverse cellular processes and therapeutic target in cancer. Biochem Biophys Res Commun. 2010;396(1):85–89. [DOI] [PubMed] [Google Scholar]