

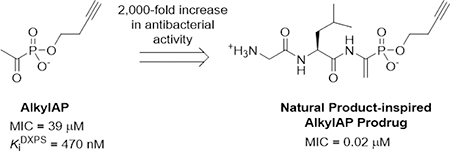
Abstract
To fight the growing threat of antibiotic resistance, new antibiotics are required that target essential bacterial processes other than protein, DNA/RNA, and cell wall synthesis which constitute the majority of currently used antibiotics. 1-Deoxy-d-xylulose-5-phosphate (DXP) synthase is a vital enzyme in bacterial central metabolism feeding into the de novo synthesis of thiamine diphosphate (ThDP), pyridoxal phosphate (PLP), and essential isoprenoid precursors, isopentenyl diphosphate and dimethylallyl diphosphate. While potent and selective inhibitors of DXP synthase in vitro activity have been discovered, their antibacterial activity is modest. To improve the antibacterial activity of selective alkyl acetylphosphonate (alkylAP) inhibitors of DXP synthase, we have synthesized peptidic enamide prodrugs of alkylAPs inspired by the natural product dehydrophos, a prodrug of methyl acetylphosphonate. This prodrug strategy achieves dramatic increases in activity against Gram-negative pathogens for two alkylAPs, butyl acetylphosphonate and homopropargyl acetylphosphonate, decreasing MICs against E. coli by 33- and nearly 2000-fold, respectively. Antimicrobial studies and LC-MS/MS analysis of alkylAP-treated E. coli establish that the increased potency of prodrugs is due to increased accumulation of alkylAP inhibitors of DXP synthase via transport of the prodrug through the OppA peptide permease and subsequent amide hydrolysis. This work demonstrates the promise of targeting DXP synthase for the development of novel antibacterial agents.
Keywords: bacterial central metabolism, phosphonate prodrug, 1-deoxy-d-xylulose 5-phosphate synthase, bacterial metabolic branch point, OppA peptide permease, Dehydrophos
Graphical Abstract
Introduction
1-Deoxy-d-xylulose-5-phosphate (DXP) synthase is an essential enzyme in bacterial central metabolism and a potential new target under investigation in our laboratory for the development of novel antibacterial strategies to combat antibiotic resistance. It catalyzes the thiamin diphosphate (ThDP)-dependent decarboxylative condensation of pyruvate and d-glyceraldehdye-3-phosphate (d-GAP) to form DXP, a branchpoint intermediate that feeds into the de novo biosynthesis of thiamin diphosphate (ThDP), pyridoxal phosphate (PLP), and the methyl erythritol phosphate (MEP) pathway to essential isoprenoids. DXP synthase is present in most pathogenic bacteria, the chloroplast of plants, and apicomplexan parasites, but is absent in animals.1–5 Its structure and mechanism are unique when compared to other ThDP-dependent enzymes.6–9 Its active site is nearly twice the volume of mammalian pyruvate dehydrogenase (PDH) and transketolase (TK), and its domain arrangement places the active site at the interface of two domains within the same monomer rather than at the dimer interface as in PDH and TK.10 Additionally, unlike all other known ThDP-dependent enzymes which display ping-pong kinetics, DXP synthase uses a preferred order random sequential mechanism requiring the formation of a unique Enzyme-LactylThDP-(d-GAP) ternary complex (Scheme 1) prior to release of the first product, CO2.7–9 These mechanistic findings evoke questions about DXP synthase function in bacteria6, and highlight targetable features of DXP synthase that establish a basis for the development of selective chemical probes to study DXP synthase function and to serve as starting points for new antimicrobial agents.11
Scheme 1.

DXP synthase is an essential ThDP-dependent enzyme for bacterial central metabolism. Unique amongst ThDP-dependent enzymes, DXP synthase forms a stable lactyl-ThDP (LThDP) intermediate which requires d-GAP to bind forming a ternary complex before decarboxylation occurs. AlkylAP inhibitors, such as BAP and (R)-14, mimic pyruvate to form reversible covalent phosphono-LThDP intermediates to elicit their inhibitory activity.
We have developed alkylAPs as selective mechanism-based inhibitors of DXP synthase bearing an acetylphosphonate group that mimics pyruvate and a sterically demanding alkyl group that is readily accommodated by the large active site. These inhibitors interact with the ThDP cofactor to form reversible covalent intermediates (phosphono-LThDP).11–13 Our early studies identified butyl acetylphosphonate (BAP) as a promising selective inhibitor with micromolar inhibitory activity and 60-fold selectivity for DXP synthase over PDH and no inhibitory activity against TK.12 Additional studies demonstrated that BAP exhibits growth medium-dependent antibacterial activity against a number of clinically-important bacterial pathogens, with its activity being markedly increased in defined, minimal medium compared to rich medium.13,14 Second generation alkylAPs11 (Scheme 1) were recently developed as bisubstrate analog inhibitors capable of interacting with both the ThDP cofactor and d-GAP binding site. In vitro analyses reveal potent nanomolar inhibition and exquisite selectivity for DXP synthase (>15,000-fold selectivity over PDH) in the best case, further confirming the unique mechanism of this interesting bacterial enzyme and opening new avenues for inhibitor development.
Inhibitor uptake into bacterial cells has emerged as a key barrier in the development of selective DXP synthase inhibitors as potential antibacterial agents.15 Thus, we have sought a strategy to increase the intracellular accumulation of DXP synthase inhibitors to improve their antibacterial activity. Herein, we show that homopropargyl acetylphosphonate (hpAP Ki = 470 ± 40 nM)11, a precursor to second generation bisubstrate analog inhibitors and a more potent inhibitor of DXP synthase than BAP, displays growth medium-dependent activity against E. coli that is similar to BAP. Second generation DXP synthase inhibitors exhibit variable, growth medium-dependent activity and poor correlation to enzyme inhibitory activity with the most potent enzyme inhibitors lacking antibacterial activity altogether. A prodrug strategy inspired by the natural product dehydrophos (Figure 1) was applied to alkylAPs in an effort to increase the efficiency of inhibitor uptake. The peptidic prodrugs take advantage of active transport through the OppA peptide transporter, which is known to be upregulated under nutrient-limited conditions associated with infection16 and has been demonstrated to be required for host colonization, specifically, in uropathogenic E. coli (UPEC).17 Prodrugs were synthesized and evaluated against several Gram-negative bacteria revealing a dramatic increase in antibacterial activity for some DXP synthase inhibitors against several clinically-relevant pathogens. Active prodrugs were shown to behave similarly to the parent alkylAPs with regard to growth medium dependence and ablation of activity by exogenous thiamin (Figure S1), supporting a mechanism of antimicrobial activity involving conversion of enamide prodrugs to active alkylAPs. LC-MS/MS analysis of E. coli cell following prodrug treatment of and use of permease deletion mutants provide direct evidence for the conversion of the enamide prodrug to alkylAP following prodrug uptake by the OppA peptide transporter.
Figure 1. Transport and activation of dehydrophos and alkylAP prodrugs.

The natural product dehydrophos travels to the cytoplasm via the peptide transporter, OppA.18 Once in the cytoplasm it is hydrolyzed an enamine product which then undergoes spontaneous tautomerization and hydrolysis to release MAP. The alkylAP prodrugs are expected to undergo the same transport and activation to reveal potent and selective DXP synthase inhibitors.
Results
Potent alkylAP-mediated inhibition of DXP synthase in vitro does not correlate with antibacterial activity.
BAP was previously identified as a selective DXP synthase inhibitor which possesses antibacterial activity against several Gram-negative pathogens.13,14 Work in our lab has recently demonstrated the application of CuAAC chemistry to expand SAR around the alkylAP scaffold toward enhancing potency and selectivity of inhibition of DXP synthase (Figure 2).11 This new series of alkylAP DXP synthase inhibitors includes bisubstrate analog inhibitors which effectively target the unique Enzyme-LThDP-(d-GAP) ternary complex, resulting in high potency and exquisite selectivity of inhibition of DXP synthase.11 Here, we sought to learn whether this increased inhibition potency leads to enhanced antimicrobial activity. Thus, inhibitors (1 – 21, Figure 2) were evaluated against E. coli MG1655 in liquid culture. Each compound was tested for its ability to halt cell growth and compared to a no inhibitor control to determine fractional growth.
Figure 2. Activity in growth inhibitory assay against MG1655 E. coli.

Antibacterial activity was evaluated in M9-glucose minimal medium. Enzyme inhibition constants (Ki) were previously reported by Bartee and Freel Meyers11. Minimal inhibitory concentration (MIC) is defined as the concentration of inhibitor required to inhibit bacterial growth by 90%.
The alkyne precursor to second generation bisubstrate analog inhibitors, homopropargyl acetylphosphonate (hpAP 1), and alkylAPs 2 – 10 linking uncharged alkyl or aryl groups to the acetylphosphonate through a triazole linker generally exhibit more potent antibacterial activity against E. coli than the charged bisubstrate mimics 11, 13 – 21, despite their generally inferior biochemical inhibitory activity (Figure 2).13 Aromatic inhibitors 7, 8, and 10 exhibit the most potent antibacterial activity with MICs of 4.9, 2.4, and 2.4 μM, respectively. Curiously, the closely related benzyl triazole alkylAP 6 is inactive as an antimicrobial agent under these conditions (MIC > 1250 μM) despite having similar enzyme inhibitory activity to 7, 8, and 10.
Dianionic bisubstrate analogs 11, 13 – 21 generally display weak antibacterial activity or fail to inhibit bacterial growth altogether. The simplest bisubstrate inhibitor 11 and its methylated analogs (S)-13 and (R)-13 exhibit modest growth inhibition (MIC = 313, 156, and 625 μM) while the most potent enzyme inhibitors (R)-14, 16, and 17 (Ki = 90, 110, and 320 nM, respectively) are inactive against E. coli up to 1.25 mM. The N-acyl sulfonamide inhibitors 19 and 20, derived from carboxylate 11, display modest antibacterial activity. Masking the negative charge of carboxylate 11 in the form of methyl ester 12 results in a loss of antibacterial activity suggesting a simple charge-masking prodrug strategy will be ineffective for alkylAPs.
Antibacterial activity does not correlate with enzyme inhibitor activity for these inhibitors (Figure 2). For example, hpAP exhibits 6-fold higher inhibition potency against DXP synthase compared to BAP, but 8-fold lower antibacterial activity against E. coli in liquid culture, and the most potent inhibitors of DXP synthase, (R)-14 and 16, lack antibacterial activity altogether.
Design and synthesis of enamide prodrugs of alkylAPs.
We hypothesized that the lack of a correlation between enzyme inhibitory activity and antimicrobial activity is due to variable inhibitor uptake and access to the DXP synthase target in bacterial cells. Given the potential importance of these probes to validate DXP synthase as a target and study its function in bacterial pathogens, we sought a strategy to enhance uptake and, thus, antimicrobial activity of our alkylAP inhibitors. Methyl acetylphosphonate (MAP), an indiscriminate inhibitor of ThDP-dependent enzymes, has been known to biochemists for decades19 and to Nature for much longer; Streptomyces luridus produces the natural product dehydrophos, a peptidic prodrug of MAP (Figure 1).20 As a tripeptide mimic, dehydrophos enters bacterial cells through the OppA peptide transporter where it is then hydrolyzed by several intracellular peptidases to yield a phosphono-enamine intermediate which can spontaneously tautomerize to an imine and undergo hydrolysis to release MAP (Figure 1).18 Dehydrophos exhibits growth inhibitory activity against bacteria, including Escherichia coli, Bacillus subtilis, and Salmonella enterica, while MAP was shown to be inactive.18,21
The OppA peptide transporter is of particular interest as it has been implicated in bacterial virulence.16 OppA is the substrate recognition portion of the oligopeptide permease (Opp) transporter complex, an ABC transporter, which spans the inner membrane of many bacteria. Important for the import of short peptides, the Opp transport system is required for colonization by UPEC due to a reliance on amino acids and short peptides present in the bladder and urine to fulfill metabolic needs during infection.17 Exploiting the reliance of UPEC on the Opp system could provide selectivity for pathogenic bacteria over the gut microbiota.
In this work, we have applied a dehydrophos-inspired prodrug strategy to the well-studied, selective DXP synthase inhibitor BAP and the structurally similar hpAP (1), which shows more potent enzyme inhibitory activity against DXP synthase compared to BAP, but less potent antimicrobial activity against E. coli. In addition, we applied this prodrug strategy to the highly potent and selective bisubstrate analogs, (R)-14 and 16, both of which are inactive against E. coli.
The prodrug synthesis reported by Kuemin and van der Donk21 provided an excellent starting point for the development of new enamide prodrugs. Commercially available N-Cbz-l-serine was converted to the N-Boc-Gly-(l-Leu)-phosphonoserine intermediate (22) over several steps, as previously described.21 Phosphonate 22 was then selectively demethylated to produce the monoester triethylammonium salt (23) after purification (Scheme 2). The homopropargyl phosphonoester was installed via the Mitsunobu reaction22 to produce the mixed diester which was immediately subjected to TBAF-mediated desilylation to yield 24. Attempts to synthesize the mixed ester via electrophilic activation of the phosphorus center (e.g., by carbodiimide coupling or by production of the phosphonochloridate via oxalyl chloride) failed, possibly due to steric hinderance of the phosphonic acid. The free alcohol of 24 was activated by reaction with mesyl chloride to give the corresponding mesylate which underwent elimination under basic conditions to yield the enamide 26. Subsequent acid-promoted cleavage of Boc provided the penultimate product (28) which was subjected to another thiophenol-mediated deprotection to selectively remove the methyl ester. The resulting desired zwitterionic prodrug 30 was obtained following C18 reversed phase chromatography. Synthesis of triazole-containing prodrugs of bisubstrate analogs 32 and 33 was achieved through CuAAC23 of alkyne 30 with the corresponding azides (Scheme 3).
Scheme 2.

Synthesis of prodrugs 30 and 31
Scheme 3.

Synthesis of triazole-containing prodrugs
The synthesis of the BAP enamide prodrug 31 was easily accomplished by subjecting 24 to catalytic hydrogenation to generate the butyl ester 25 prior to the generation of the corresponding olefin 27 (Scheme 2). BAP prodrug 31 was accessed from 27 as described above for the synthesis of alkyne 30.
Antimicrobial activity of alkylAP enamide prodrugs.
Prodrugs 30 – 33 were evaluated for bacterial growth inhibitory activity against E. coli grown in M9-glucose. Prodrugs 30 and 31 display a remarkable increase in potency compared to their acetyl phosphonate parent drugs. The MIC decreased by nearly 2000-fold for the hpAP prodrug (30) compared to the parent inhibitor hpAP (1, Table 1). Similarly, a 33-fold increase in potency was observed for the BAP prodrug 31. Interestingly, both of these analogs outperform the natural product dehydrophos (MIC = 680 nM). However, neither prodrugs 32 nor 33 displays enhanced potency compared to bisubstrate analog inhibitors (R)-14 and 16, and thus were not studied further. The additional negative charge and substantial increase in size of (R)-14 and 16 compared to dehydrophos and prodrugs 30 and 31 may reduce affinity for OppA to prevent transport.
Table 1.
Evaluation of Prodrugs against E. coli MG1655.
| Prodrug | MICM9 (μM) | Parenet alkyIAP | MICM9 (μM) | Fold-increase in potency |
|---|---|---|---|---|
| Dehydrophos | 0.61 | MAP | 78 | 128 |
| Pro-hpAP (30) | 0.02 | hpAP (1) | 39 | 1950 |
| Pro-BAP (31) | 0.15 | BAP | 5 | 33 |
| 32 | >1250 | (R)-14 | >1250 | n/a |
| 33 | 1250 | 16 | >1250 | n/a |
To understand how the antibacterial activity of 30 and 31 compare to that of their parent alkylAPs, the prodrugs were further evaluated under exogenous thiamin (100 nM), a condition known to suppress the activity of alkylAPs targeting DXP synthase.13 As expected, supplementation with thiamin rescues the growth of cells treated with 30 or 31, consistent with the behavior of the parent alkylAPs and suggesting that the prodrugs are being processed intracellularly to active alkylAP inhibitors of DXP synthase to exert their antimicrobial activity.
Encouraged by the potent activity observed for 30 and 31 against E. coli MG1655, we sought to examine the potency of these prodrugs against more clinically-relevant Gram-negative bacteria. Similar to the lab strain of E. coli, other bacterial pathogens including Salmonella typhimurium, Enterobacter cloacae, and a clinical strain of E. coli are all susceptible to growth inhibition by the homopropargyl prodrug 30 at sub-micromolar concentrations (Table 2). Notably, only modest antibacterial activity was observed against Klebsiella oxytoca in the presence of 30, and both Pseudomonas species tested were resistant to these prodrugs up to 40 μM. The lack of activity against the more distantly-related Pseudomonas species suggests that differences in active uptake and/or activation likely arising from differences in affinity of prodrugs for OppA or hydrolases, will likely dictate microbial susceptibility to the prodrugs (Figure S2). This suggests that future iterations of enamide alkylAP prodrugs may be tuned for selective uptake by other peptide transporters toward the development of narrow spectrum agents.
Table 2.
Evaluation of hpAP (1), BAP, and their respective prodrugs, 30 and 31, against clinical isolates of Gram-negative bacterial pathogens.
| MIC (μM) |
||||
|---|---|---|---|---|
| Starin | BAP | hpAP (1) | Pro-hpAP (30) | Pro-BAP (31) |
| Escherichia coil (clinical) | 11 | 20 | 0.0375 | 0.3 |
| Escherichia coil (FSR) | 43 | 80 | 0.0375 | 0.15 |
| Salmonella typhimurium | 11 | 20 | 0.01875 | 0.075 |
| Klebsiella oxytoca | 43 | >160 | 20 | >40 |
| Pseudomonas aeruginosa | n/a | >160 | >40 | >40 |
| Pseudomonas fluorescens | 43 | 80 | >40 | >40 |
| Enterobacter cloacae | 688 | >160 | 0.625 | 20 |
OppA is required for transport of alkylAP enamide prodrugs in E. coli.
Peptide uptake is an important and well-studied mechanism used by bacteria to acquire amino acids for cell anabolism and to recycle cell wall components.24 The oligopeptide permease (Opp) system functions to transport peptides, preferentially tri- and tetra-peptides, into the cell in an ATP-dependent manner.25,26 Studies by Cierllo et al.18 demonstrated that in Salmonella enterica, OppA is required for the uptake of dehydrophos and that its intracellular activation to provide MAP is catalyzed by several different peptidases (PepA, B, D, N). To determine which proteins are necessary for the uptake and activation of prodrugs 30 and 31 in E. coli, we measured prodrug activity in several different deletion mutants obtained from the Keio collection27 (Figure 3). Our results show that the ΔoppA deletion mutant is resistant to prodrugs 30 and 31, indicating that OppA is required for transport of 30 and 31 in E. coli, similar to its requirement for dehydrophos transport in S. enterica.18 The deletion of other uptake systems does not affect the growth inhibitory activity of 30 and 31. The activities of the parent acetyl phosphonates (BAP and hpAP) are also unchanged against the Δdpp, Δopp, or Δtpp peptide permease deletion mutants, indicating these transporters are not required for alkylAP uptake. Single knockout mutants of peptidases PepA, PepB, and PepC remain susceptible to prodrugs 30 and 31, suggesting that there is likely redundancy among peptidases that can activate the enamide prodrugs once imported to the cytoplasm, similar to what is observed for activation of dehydrophos in S. enterica.18
Figure 3.

Evaluation of antibacterial activity of BAP, hpAP (1), Pro-hpAP (30), and Pro-BAP (31) in peptide transporter deletion strains. Only the deletion of OppA desensitizes E. coli to prodrugs 30 and 31. The activity of parent alkylAPs (BAP and 1) is not affected by the deletion of peptide transporter tested. Experiments were performed in triplicate and error bars indicate standard error.
Enamide prodrugs are rapidly converted to alkylAP inhibitors of DXP synthase in E. coli.
Toward establishing a mechanism for the dramatic increase in potency of the enamide prodrugs compared to the parent alkylAPs, an LC-MS/MS method was developed to directly observe the intracellular concentrations of both enamide prodrugs and parent alkylAPs. The treatment of E. coli with prodrug results in an immediate observable accumulation of the parent alkylAP and a steady, low concentration of enamide prodrug (Figure 4), suggesting prodrugs are efficiently converted to parent inhibitors upon transport into E. coli. Treatment of E. coli with 5 μM of either parent alkylAP or enamide prodrug reveals a significant increase in the intracellular accumulation of alkylAP with the enamide prodrug compared to direct treatment with BAP or 1 (Figure 5).
Figure 4.

Intracellular concentrations of BAP, hpAP (1), Pro-hpAP (30), and Pro-BAP (31) as measured by LC/MS-MS. MG1655 E. coli were treated with 40 μM of a) 30 or b) 31 and collected over the course of 1h. Cell lysates were analyzed and concentrations of prodrug and corresponding alkylAP were determined using a standard curve (Figure S3, S4). Experiments were performed in duplicate and error bars indicate standard deviation.
Figure 5.

Intracellular concentrations of BAP, hpAP (1), Pro-hpAP (30), and Pro-BAP (31) as measured by LC/MS-MS. MG1655 E. coli were treated with 5 μM of BAP, hpAP (1), Pro-hpAP (30), and Pro-BAP (31) incubated at 37 °C for 5 minutes. Cell lysates were analyzed and concentrations of the corresponding alkylAP (BAP for BAP and Pro-BAP, dark grey, and hpAP for hpAP and Pro-hpAP, light grey) were determined using a standard curve (Figure S5, S6). Experiments were performed in duplicate and error bars indicate standard deviation.
Discussion
The ThDP-dependent enzyme DXP synthase is an essential bacterial enzyme key to the de novo biosynthesis of isoprenoids, ThDP, and PLP. Its involvement in three separate and vital processes in bacterial central metabolism suggest a key role for DXP synthase in bacterial pathogen metabolism during infection. Our insights from mechanistic studies28–30 raise additional questions about other potential functions of this target that require further investigation. To address these interesting biological questions, DXP synthase probes displaying higher penetration into bacterial cells are required. Here, we present the synthesis and evaluation of peptidic prodrugs of alkylAP DXP synthase inhibitors, designed to display enhanced uptake by the OppA permease. The alkylAP prodrugs 30 and 31 exhibit excellent antibacterial activity by a mechanism involving transport and activation of alkylAP prodrugs to the parent alkylAPs with enhanced activity due to increased intracellular accumulation of the active alkylAPs, BAP and hpAP (1). Furthermore, the relative activities of the prodrugs 30 and 31 correlate with the enzyme inhibitory activities of their parent alkylAPs suggesting DXP synthase inhibitor activity is predictive of antibacterial activity if cellular uptake is similar.
Interestingly, extension of the dehydrophos-inspired prodrug strategy to the most potent bisubstrate inhibitors of DXP synthase, 32 and 33, does not afford the same enhancement in antibacterial activity. Dehydrophos and alkylAP analogs studied here enter bacterial cells through the OppA transporter. Early work to determine substrate preferences revealed that most tri- and tetrapeptides are substrates for transport.25,26 OppA is also capable of transporting carboxylic acid esters25, consistent with its transport of phosphonyl monoesters which contain both carboxylate and ester character. Prodrugs 32 and 33 may be too large to be recognized by OppA and likely more closely resemble a penta- or hexapeptide than the preferred tripeptide substrate. Alternatively, the added negative charge distal to the amino terminus could be detrimental to transporter binding. Given the redundancy and broad substrate specificity of bacterial peptidases, it seems unlikely that added bulk and charge introduced with prodrugs 32 and 33 precluded their intracellular activation, however, we cannot exclude this possibility at this time. Future work will explore new peptidic enamide prodrugs of alkylAPs in search of scaffolds which will permit the most potent alkylAP DXP synthase inhibitors to accumulate in bacteria.
The molecular determinants of the transport and accumulation of parent alkylAPs are not fully understood. While simple first generation alkylAPs display similar low micromolar inhibition constants for DXP synthase13, BAP possesses the most potent antibacterial activity. Both longer and shorter chain alkylAPs are apparently unable to reach the same intracellular concentrations resulting in reduced activity. Second generation alkylAPs examined here reveal an apparent preference for uncharged aromatic functional groups as observed with 7 – 8 and 10 however the addition of a charged functional group, present in the most potent DXP synthase inhibitors, appears to abolish antibacterial activity in some cases. In other cases, the presence of a negative charge without added substituents at the bridging methylene to the triazole moiety appears to be tolerated to some extent as evidenced by 11, 13, 19, and 20.
The development of antibacterial agents through the targeted inhibition of DXP synthase has been a goal since the discovery of the enzyme over 20 years ago. Even with the development of selective inhibitors of the enzyme, achieving potent antibacterial activity remains a challenge. While the dehydrophos-inspired strategy reported herein may have limited flexibility for significant structural modifications to the prodrug scaffold, its application to simple alkylAPs can dramatically enhance alkylAP potency. Our results demonstrate the advantage of such strategies for functional studies of this target and the potential of DXP synthase inhibitors to be potent antibacterial agents when intracellular accumulation is achieved. New delivery strategies that are amenable to the most potent DXP synthase inhibitors in the alkylAP class promise to produce extremely potent antibacterial agents.
Experimental
General.
Unless otherwise noted, all reagents were obtained from commercial suppliers and used without further purification. Diisopropylethylamine (iPr2NEt) and dichloromethane (DCM) were distilled after drying over CaH2. Yields of all reactions refer to the purified products. Silica chromatography was carried out in the indicated solvent system using pre-packed silica gel cartridges for use on the Biotage Isolera® Purification System. TLC silica gel 60 F254 plates (Millipore) were used for thin layer chromatography (TLC) and developed with Ninhydrin except where otherwise noted. 1H and 31P NMR spectra were acquired on a Bruker Avance III 500 spectrometer operating at 500 MHz for 1H and 202 MHz for 31P. For 1H NMR, chemical shift values are reported as δ (ppm) relative to CHCl3 at δ 7.27 ppm, MeOH at δ 3.31 ppm, or DMSO at δ 2.50 ppm. 31P chemical shifts are reported relative to triphenylphosphine oxide (TPPO) at δ 0 ppm as an external standard. High resolution mass spectrometry analysis was carried out The Johns Hopkins University School of Medicine, Baltimore, MD using a Thermo Q-exactive Orbitrap mass spectrometer with electrospray ionization.
Boc-N-Gly-Leu-N-(O-tertbutyldimethylsilyl ether)phosphonoserine dimethyl diester (22).
N-Boc protected glycine (0.689 g, 3.93 mmol) and HATU were suspended in DMF (11 mL) and iPr2NEt (1.15 mL, 6.56 mmol) was added. A solution of H2N- Leu-N-(O-tertbutyldimethylsilyl ether)phosphonoserine dimethyl diester (1.30 g, 3.28 mmol) in DMF (5 mL) was added to the glycine/HATU mixture, and the mixture was stirred at ambient temperature for 16 h. The reaction mixture was diluted with DCM (20 mL) and washed with aqueous citric acid (5% w/v, 30 mL), saturated aqueous NaHCO3 (30 mL), and saturated aqueous NaCl (30 mL). The organic layer was dried over MgSO4, then filtered, and condensed under reduced pressure. The resultant residue was purified by silica flash chromatography (1:0 to 9:1 DCM/MeOH) to yield 22 as a colorless oil (3:2 mixture of diastereomers, 0.984 g, 54% yield). 1H NMR (500 MHz, CDCl3) δ ppm 6.39 – 6.67 (m, 2 H), 5.04 – 5.30 (m, 1 H), 4.49 (br. s., 2 H), 3.94 – 4.09 (m, 2 H), 3.79 – 3.89 (m, 2 H), 3.76 (d, J=10.7 Hz, 6 H), 2.81 (br. s., 6 H), 1.69 – 1.79 (m, 3 H), 1.50 – 1.60 (m, 1 H), 1.46 (s, 9 H), 0.94 (br. s., 6 H), 0.91 (br. s., 9 H), 0.08 (br. s., 6 H). 31P NMR (202 MHz, CDCl3) δ ppm 0.04 (s), −0.08 (s). HRMS (ESI) calcd for C23H49N3O8PSi [M + H]+ 554.3021, found 554.3026.
Boc-N-Gly-Leu-N-(O-tertbutyldimethylsilyl ether)phosphonoserine methyl monoester (23).
To a solution of dimethyl phosphonate 22 (3.00 g, 5.42 mmol) in THF (13.5 mL) was added iPr2NEt (2.84 mL, 16.3 mmol) followed by thiophenol (0.83 mL, 8.13 mmol). The solution was stirred at ambient temperature for 5 days at which point additional iPr2NEt (2.84 mL, 16.3 mmol) and thiophenol (0.83 mL, 8.13 mmol) were added. After overnight stirring, the reaction had reached completion as observed by 31P NMR. The reaction mixture was transferred to a silica plug which was washed sequentially with THF (30 mL), DCM (30 mL), and DCM/MeOH/triethylamine (8:1.9:0.1, 100 mL). The desired product was collected in the DCM/MeOH/TEA elution which was condensed under reduced pressured and then further dried in vacuo to yield monoester 23 as a colorless oil (3:2 mixture of diastereomers, 2.974 g, 86% yield). 1H NMR (500 MHz, MeOD-d4) δ ppm 4.48 – 4.65 (m, 1 H), 4.25 – 4.41 (m, 1 H), 4.03 – 4.14 (m, 1 H), 3.65 – 3.82 (m, 2 H), 3.55 (t, J=10.5 Hz, 3 H), 3.04 – 3.17 (m, 6 H), 1.54 – 1.76 (m, 3 H), 1.45 (s, 9 H), 1.27 (t, J=7.2 Hz, 9 H), 0.93 (t, J=5.7 Hz, 6 H), 0.88 (d, J=5.2 Hz, 9 H), 0.01 – 0.11 (m, 6 H). 31P NMR (202 MHz, METHANOL-d4) δ ppm −7.94 (s), −8.08 (s). HRMS (ESI) calcd for C22H47N3O8PSi [M + H]+ 540.2865, found 540.2869.
Boc-N-Gly-Leu-N-phosphonoserine homopropargyl methyl diester (24).
To a stirred solution of phosphonate monoester 23 (2.97 g, 4.64 mmol) in THF (40 mL), triphenylphosphine (2.43 g, 9.28 mmol) and homopropargyl alcohol (0.70 mL, 9.28 mmol) were added followed by dropwise addition of diisopropyl azodicarboxylate (DIAD, 1.82 mL, 9.28 mmol). After 30 min at ambient temperature, the starting material was consumed as determined by 31P NMR. Solvent was removed under reduced pressure and the resulting residue was purified by silica flash chromatography (1:0 to 9:1 DCM/MeOH) to provide a mixture of mixed diester and TPPO.
The mixture of mixed diester and TPPO was dissolved in THF (30 mL) and treated with tetrabutylammonium fluoride (TBAF, 1.0 M in THF, 9.3 mL, 9.3 mmol). The reaction mixture was stirred at ambient temperature for 30 min, at which point the reaction was quenched by the addition of silica (10 g). Solvent was removed under to reduced pressure and the resulting silica mixture was loaded onto a silica flash column (1:0 to 9:1 DCM/MeOH) and purified to yield 24 as a white solid (mixture of diastereomers, 1.76 g, 85% yield). 1H NMR (500 MHz, CDCl3) δ ppm 7.21 (br. s., 1 H), 6.75 – 6.98 (m, 1 H), 5.35 (br. s., 1 H), 4.52 (br. s., 2 H), 4.09 – 4.26 (m, 2 H), 3.68 – 4.08 (m, 8 H), 2.59 (br. s., 2 H), 2.01 – 2.11 (m, 1 H), 1.69 (br. s., 2 H), 1.59 (d, J=7.7 Hz, 1 H), 1.46 (br. s., 9 H), 0.94 (br. s., 6 H). 31P NMR (202 MHz, CDCl3) δ ppm −1.06 - −1.00 (m), −1.08 (br. s.), −1.18 (s). HRMS (ESI) calcd for C20H37N3O8P [M + H]+ 478.2313, found 478.2314.
Boc-N-Gly-Leu-N-phosphonoserine butyl methyl diester (25).
To a solution of alkyne 24 (0.382 g, 0.8 mmol) in MeOH (10 mL) was added Pd/C (10% w/w, 40 mg). Hydrogen was bubbled through the solution for 2 min to saturate the solution and then the reaction mixture was stirred vigorously overnight at ambient temperature under an atmosphere of H2 maintained by a balloon of H2. After 16 h, the reaction mixture was filtered to remove solids and volatiles were removed under reduced pressure. The resulting residue was purified by silica flash chromatography (1:0 to 9:1 DCM/MeOH) yielding butyl methyl diester 25 as a white solid (1:1:2:2 mixture of diastereomers, 0.219 g, 57% yield). 1H NMR (500 MHz, MeOD-d4) δ ppm 4.44 – 4.58 (m, 2 H), 4.10 (q, J=6.5 Hz, 2 H), 3.82 – 3.90 (m, 1 H), 3.79 (d, J=10.7 Hz, 3 H), 3.74 (br. s., 1 H), 1.57 – 1.78 (m, 5 H), 1.47 (s, 9 H), 1.40 – 1.45 (m, 2 H), 1.19 (m, J=6.1 Hz, 1 H), 0.91 – 1.02 (m, 9 H). 31P NMR (202 MHz, MeOD-d4) δ ppm −0.46 (br. s.), −0.49 (br. s.), −0.70 (s), −0.77 (s). HRMS (ESI) calcd for C20H41N3O8P [M + H]+ 482.2626, found 482.2629.
Boc-N-Gly-Leu-N-phosphonodehydroalanine homopropargyl methyl diester (26).
Alcohol 3 (1.38 g, 2.88 mmol) was dissolved in DCM (29 mL) and cooled to 0 °C. To the solution was added iPr2NEt (2.0 mL, 11.5 mmol) followed by mesyl chloride (0.89 mL, 11.5 mmol). After 10 min at 0 °C, the reaction was quenched by the addition of silica (3.65 g) and volatiles were removed under reduced pressure. The resulting silica mixture was then loaded onto a silica flash column and purified (EtOAc to 9:1 EtOAc/MeOH) to yield the corresponding mesylate which was used immediately in the next reaction.
DBU (0.86 mL, 5.8 mmol) was added to a solution of the corresponding mesylate in anhydrous DCM (12 mL). The reaction mixture was stirred at ambient temperature for 1 h at which point it was quenched by the addition of silica (3.5 g). Volatiles were removed under reduced pressure, and the resulting silica mixture was loaded onto silica flash column and purified (2:8:0 to 0:10:0 to 0:9:1 Hexanes/EtOAc/MeOH) to yield alkene 26 as a white solid (1:1 mixture of diastereomers, 714 mg, 54% yield). 1H NMR (500 MHz, CDCl3) δ ppm 7.84 (br. s., 1 H), 6.61 (br. s., 1 H), 6.64 (d, J=53.0 Hz, 1 H), 5.70 (dd, J=19.5, 7.4 Hz, 1 H), 5.19 (br. s., 1 H), 4.49 (br. s., 1 H), 4.08 – 4.24 (m, 2 H), 3.79 (d, J=11.5 Hz, 3 H), 3.69 – 3.91 (m, 2 H), 2.62 (br. s., 2 H), 2.09 (d, J=13.4 Hz, 1 H), 1.54 – 1.79 (m, 3 H), 1.46 (br. s., 9 H), 0.95 (br. s., 6 H). 31P NMR (202 MHz, CDCl3) δ ppm −12.10 (s), −12.16 (s). HRMS (ESI) calcd for C20H35N3O7P [M + H]+ 460.2207, found 460.2211.
Boc-N-Gly-Leu-N-phosphonodehydroalanine butyl methyl diester (27).
Alcohol 25 (0.219 g, 0.46 mmol) was dissolved in anhydrous DCM (4 .6 mL) and cooled to 0 °C. To the solution was added iPr2NEt (0.32 mL, 1.82 mmol) followed by mesyl chloride (0.14 mL, 1.82 mmol). After 20 min at 0 °C, the reaction was quenched by the addition of silica (2 g) and volatiles were removed under reduced pressure. The resulting silica mixture was then loaded onto a silica flash column and purified (EtOAc to 9:1 EtOAc/MeOH) to yield the corresponding mesylate which was used immediately in the next reaction.
DBU (0.136 mL, 0.91 mmol) was added to a solution of corresponding mesylate in anhydrous DCM (2 mL). The reaction mixture was stirred at ambient temperature for 1h at which point it was quenched by the addition of silica (1 g). Volatiles were removed under reduced pressure and the resulting silica mixture was loaded onto silica flash column and purified (2:8:0 to 0:10:0 to 0:9:1 Hexanes/EtOAc/MeOH) to yield alkene 27 as a white solid (1:1 mixture of diastereomers, 162 mg, 77% yield). 1H NMR (500 MHz, CDCl3) δ ppm 7.83 (br. s., 1 H), 6.68 (br. s., 1 H), 6.60 (br. s., 1 H), 5.61 (dd, J=18.7, 6.4 Hz, 1 H), 5.20 (br. s., 1 H), 4.49 (br. s., 1 H), 3.99 – 4.17 (m, 2 H), 3.83 (q, J=18.1 Hz, 2 H), 3.76 (d, J=11.2 Hz, 3 H), 1.62 – 1.78 (m, 4 H), 1.46 (br. s., 10 H), 1.41 (q, J=7.1 Hz, 2 H), 0.95 (br. s., 9 H). 31P NMR (202 MHz, CDCl3) δ ppm −12.09 (s), −12.12 (s). HRMS (ESI) calcd for C20H39N3O7P [M + H]+ 464.2520, found 464.2523.
H2N-Gly-Leu-N-phosphonodehydroalanine homopropargyl methyl diester -TFA salt (28).
Anisole (0.24 mL, 2.22 mmol) was added to a solution of Boc-protected amine 26 (0.600 g, 1.31 mmol) in DCM (13 mL). TFA (1.42 mL, 18.54 mmoL) was added and the reaction mixture was stirred at ambient temperature for 4 h at which point the starting material was consumed. Toluene (20 mL) was added to the reaction mixture and all volatiles were then removed under reduced pressure. The resulting residue was purified by silica flash chromatography (9:1 DCM/MeOH) yielding the TFA salt amine 28 as a white solid (0.538 g, 87% yield). 1H NMR (500 MHz, MeOD-d4) δ ppm 6.43 (d, J=42.0 Hz, 1 H), 5.82 (d, J=17.9 Hz, 1 H), 4.57 (t, J=7.5 Hz, 1 H), 4.15 (quin, J=7.3 Hz, 2 H), 3.81 (d, J=11.3 Hz, 3 H), 3.70 (s, 2 H), 2.61 (q, J=6.3 Hz, 2 H), 2.37 – 2.43 (m, 1 H), 1.71 (spt, J=6.7 Hz, 1 H), 1.57 – 1.64 (m, 2 H), 0.98 (d, J=6.6 Hz, 3 H), 0.95 (d, J=6.4 Hz, 3 H). 31P NMR (202 MHz, MeOD-d4) δ ppm −11.44 (s). HRMS (ESI) calcd for C15H27N3O5P [M + H]+ 360.1683, found 360.1682.
H2N-Gly-Leu-N-phosphonodehydroalanine butyl methyl diester (29).
A stirred solution of Boc-protected amine 27 (0.160 g, 0.35 mmol) was treated with anisole (63 μL, 0.59 mmol) and TFA (0.38 mL, 11.9 mmol). After stirring at ambient temperature for 4 h, toluene (20 mL) was added to the reaction mixture and all volatiles were removed under reduced pressure. Purification of the resulting residue by silica flash chromatography (9:1 DCM/MeOH) yielded the TFA salt of amine 29 as a white solid (1:1 mixture of diastereomers, 0.133 g, 81% yield). 1H NMR (500 MHz, MeOD-d4) δ ppm 6.45 (dd, J=41.5, 6.9 Hz, 1 H), 5.78 (dd, J=18.0, 8.1 Hz, 1 H), 4.53 – 4.61 (m, 1 H), 4.09 (q, J=6.7 Hz, 2 H), 3.77 (d, J=11.2 Hz, 3 H), 3.66 (s, 2 H), 1.69 (quin, J=7.7 Hz, 4 H), 1.62 (dd, J=14.0, 8.6 Hz, 2 H), 1.43 (sxt, J=7.4 Hz, 2 H), 0.97 (d, J=5.7 Hz, 3 H), 0.95 (d, J=7.1 Hz, 3 H). 31P NMR (202 MHz, MeOD-d4) δ ppm −11.49 (s), −11.53 (s). HRMS (ESI) calcd for C15H31N3O5P [M + H]+ 364.1996, found 364.1994.
H2N-Gly-Leu-N-phosphonodehydroalanine homopropargyl monoester (30).
To a solution of Diester 28 (0.537 g, 1.13 mmol) in DMF (2.2 mL), was added iPr2NEt (1.19 mL, 6.81 mmol) followed by thiophenol (0.35 mL, 3.40 mmol). The solution was stirred at ambient temperature overnight. After 17 h, the reaction appeared to be complete by 31P NMR. Aqueous TFA (0.1% v/v, 1 mL) was added to the reaction mixture resulting in precipitation. The mixture was transferred to Eppendorf tubes and centrifuged to pellet the solid. Supernatant was collected and purified by RP-flash chromatography (solvent A: water, solvent B: acetonitrile; gradient 0–1 CV, 0% B; 1–8 CV, 0 – 60% B; 8 – 11 CV, 100 % B) to yield hpAP prodrug 30 as a white powder (191 mg, 49% yield). 1H NMR (500 MHz, D2O) δ ppm 6.13 (d, J=36.6 Hz, 1 H), 5.66 (d, J=16.2 Hz, 1 H), 4.37 – 4.45 (m, 1 H), 3.83 (s, 2 H), 3.85 (q, J=6.8 Hz, 2 H), 2.47 (td, J=6.2, 2.5 Hz, 2 H), 2.34 (br. s., 1 H), 1.63 (m, J=4.4 Hz, 2 H), 0.91 (d, J=4.2 Hz, 3 H), 0.87 (d, J=4.4 Hz, 3 H). 13C NMR (126 MHz, D2O) δ ppm 173.5 (d, J=7.3 Hz), 167.2 (s), 135.0 (d, J=191.6 Hz), 116.7 (d, J=10.9 Hz), 81.8 (s), 70.6 (s), 63.2 (d, J=5.5 Hz), 53.3 (s), 40.3 (s), 39.7 (s), 24.3 (s), 22.1 (s), 20.6 (s), 20.2 (d, J=7.3 Hz). 31P NMR (202 MHz, D2O) δ ppm −17.63 (s). HRMS (ESI) calcd for C14H25N3O5P [M + H]+ 346.1526, found 346.1525.
H2N-Gly-Leu-N-phosphonodehydroalanine butyl monoester (31).
To a solution of diester 29 (0.133 g, 0.28 mmol) in DMF (1 mL), was added iPr2NEt (0.29 mL, 1.7 mmol) followed by thiophenol (86 μL, 0.84 mmol). The solution was stirred at ambient temperature overnight. After 17 h, the reaction appeared to be complete by 31P NMR. Aqueous TFA (0.1% v/v, 1 mL) was added to the reaction mixture resulting in precipitation. The mixture was transferred to Eppendorf tubes and centrifuged to pellet the solid. Supernant was collected and purified by RP-flash chromatography (solvent A: water, solvent B: acetonitrile; gradient 0–1 CV, 0% B; 1–8 CV, 0 – 60% B; 8 – 11 CV, 100 % B) to yield BAP prodrug 31 as a white powder (49 mg, 51% yield). 1H NMR (500 MHz, D2O) δ ppm 6.13 (d, J=35.5 Hz, 1 H), 5.63 (d, J=15.9 Hz, 1 H), 4.34 – 4.48 (m, 1 H), 3.81 (s, 2 H), 3.75 (q, J=6.4 Hz, 2 H), 1.63 (m, J=4.9 Hz, 3 H), 1.52 (quin, J=7.0 Hz, 2 H), 1.25 – 1.34 (m, 2 H), 0.91 (d, J=4.6 Hz, 3 H), 0.87 (d, J=4.4 Hz, 3 H), 0.84 (t, J=7.4 Hz, 3 H). 13C NMR (126 MHz, D2O) δ ppm 173.5 (d, J=7.3 Hz), 167.4 (s), 135.1 (d, J=188.0 Hz), 116.2 (d, J=10.9 Hz), 65.4 (d, J=5.4 Hz), 53.4 (s), 40.4 (s), 39.8 (s), 31.9 (d, J=6.4 Hz), 24.3 (s), 22.1 (s), 20.6 (s), 18.3 (s), 12.9 (s). 31P NMR (202 MHz, D2O) δ ppm −17.39 (s). HRMS (ESI) calcd for C14H29N3O5P [M + H]+ 350.1839, found 350.1842.
d-PheTrAP Prodrug (32).
To a suspension of alkyne 30 (20 mg, 58 μmol) in t-butanol (0.5 mL) and water (0.2 mL) was added (R)-2-azido-2-benzyl acetic acid (22 mg, 0.116 mmol), Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, 29 μL, 0.1 M in DMSO, 2.9 μmol), and CuSO4 (5.8 μmol, 0.5 M in water, 2.9 μmol) followed by sodium ascorbate (23.2 μL, 0.5 M, 11.6 μmol). The reaction mixture was stirred at ambient temperature overnight. Volatiles were removed under reduced pressured and the resulting residue was dissolved in water (0.5 mL) and purified by RP-flash chromatography (solvent A: water, solvent B: acetonitrile; gradient 0–1 CV, 0% B; 1–8 CV, 0 – 60% B; 8 – 11 CV, 100 % B) to yield d-PheTrAP prodrug 32 as a pale yellow foam (14 mg, 46% yield). 1H NMR (500 MHz, MeOD-d4) δ ppm 7.98 (br. s., 1 H), 7.13 – 7.25 (m, 3 H), 7.09 (d, J=7.1 Hz, 2 H), 6.27 (d, J=35.2 Hz, 1 H), 5.54 (s, 1 H), 5.47 (d, J=13.4 Hz, 1 H), 4.41 (dd, J=9.9, 3.8 Hz, 2 H), 3.95 (s, 2 H), 3.80 (dd, J=28.8, 16.0 Hz, 2 H), 3.62 (d, J=11.6 Hz, 1 H), 3.47 (t, J=10.4 Hz, 1 H), 2.94 (s, 2 H), 1.61 – 1.75 (m, 2 H), 1.52 – 1.61 (m, 1 H), 0.94 (d, J=5.8 Hz, 3 H), 0.89 (d, J=5.7 Hz, 3 H). 31P NMR (202 MHz, D2O:pyridine-d5, 9:1) δ ppm −17.54 (s). HRMS (ESI) calcd for C23H34N6O7P [M + H]+ 537.2221, found 537.2224.
o-phenol-TrAP Prodrug (33).
To a suspension of alkyne 30 (20 mg, 58 μmol) in t-butanol (0.5 mL) and water (0.2 mL) was added 2-azidophenol (39 mg, 0.29 mmol), Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, 29 μL, 0.1 M in DMSO, 2.9 μmol), and CuSO4 (5.8 μmol, 0.5 M in water, 2.9 μmol) followed by sodium ascorbate (23.2 μL, 0.5 M, 11.6 μmol). The reaction mixture was stirred at ambient temperature overnight. Volatiles were removed under reduced pressured and the resulting residue was dissolved in water (0.5 mL) and purified by RP-flash chromatography (solvent A: water, solvent B: acetonitrile; gradient 0–1 CV, 0% B; 1–8 CV, 0 – 60% B; 8 – 11 CV, 100 % B) to yield o-phenol prodrug 33 as a pale yellow foam (19 mg, 65% yield). 1H NMR (500 MHz, D2O) δ ppm 8.16 (s, 1 H), 7.48 (d, J=7.9 Hz, 1 H), 7.41 (t, J=7.8 Hz, 1 H), 7.11 (d, J=8.3 Hz, 1 H), 7.07 (t, J=7.7 Hz, 1 H), 6.08 (d, J=36.3 Hz, 1 H), 5.55 (d, J=16.3 Hz, 1 H), 4.28 (dd, J=9.5, 5.0 Hz, 1 H), 4.08 (q, J=5.7 Hz, 2 H), 3.79 (s, 2 H), 3.06 (t, J=5.9 Hz, 2 H), 1.46 – 1.59 (m, 2 H), 1.37 – 1.45 (m, 1 H), 0.79 (d, J=6.3 Hz, 3 H), 0.76 (d, J=6.1 Hz, 3 H).31P NMR (202 MHz, D2O) δ ppm −17.41 (s). HRMS (ESI) calcd for C20H30N6O6P [M + H]+ 481.1959, found 481.1965.
Evaluation of antibacterial activity of DXP synthase inhibitors and prodrugs.
MG1655 E. coli K-12
Inhibitors were evaluated as previously described by Sanders et al.13 Briefly, using aseptic techniques, 3 isolated colonies were selected from a plate containing ATCC MG1655 E. coli K-12, inoculated into 5 mL of M9-glucose minimal medium and grown to saturation overnight with shaking at 37 °C. The saturated culture was diluted 50-fold into fresh M9-glucose medium and grown to exponential phase (OD600 = 0.4). The exponential phase cell culture was diluted 1:1000 into M9-glucose medium to yield the experimental inoculum which was mixed 1:1 with M9-glucose medium containing the antimicrobial agent at 2× the desired concentration. To confirm consistency between experiments, colony counts of the experimental inoculum were verified by dilution and enumeration on CAMHB agar for 16 h at 37 °C. The final concentration of bacteria in each well was approximately 105 CFU/mL in a final volume of 200 μL. The 96-well plates were incubated at 37 °C for 16 h with periodic shaking. Fractional growth was determined at 16 h relative to a no drug control. Experiments were performed in triplicate.
For thiamin rescue experiments, antibacterial activity was determined as above with a key exception. Rather than diluting the exponential culture 1000-fold into fresh M9-glucose medium, it was diluted 10-fold into fresh M9-glucose medium prior to addition to M9-glucose medium containing 200 nM thiamin (2× the desired concentration). This results in a 107 CFU/mL starting inoculum and 100 nM thiamin in each well.
Gram-negative bacteria clinical isolates
Inhibitors were evaluated as previously described by Sanders et al.13 Clinical isolates of all pathogens were from an in-house strain library maintained at Northern Arizona University.31 All microbial manipulation of pathogenic bacteria was conducted in a certified biosafety level 2 laboratory while following all associated safety protocols. All clinical isolates were streaked onto CAMHB agar and allowed to grow for 12 h at 37 °C. Using aseptic technique, colonies were inoculated into CAMHB (5 mL) and grown to saturation overnight. Following overnight growth, bacteria were subcultured into M9-glucose medium (approximately 5 μL per 5 mL of fresh medium), grown to saturation at 37 °C, and then used in subsequent broth microdilution assays. Experiments were performed essentially as described above, and experimental inoculum for all strains was verified by dilution and enumeration to be approximately 2 × 108 CFU/mL. To maintain retention of the pJLA-Fsr plasmid by the cells involving the E. coli-Fsr expression strain, experiments were performed in the presence of kanamycin (70 μg/ mL). At a minimum, experiments were performed in triplicate.
LC-MS/MS method to determine intracellular inhibitor concentrations.
E. coli samples were treated with inhibitor and prepared for LC-MS/MS analysis as described by Sanders et al.15 Samples were analyzed with an LC-MS/MS system comprised of a Waters NanoAcquity UPLC and a TSQ Vantage Triple Quadripole (Thermo Scientific). The liquid chromatography separation was performed on a Waters Acquity UPLC HSS C18 column (1.0 × 50 mm, 1.8 μm) with mobile phase A (1 mM acetic acid in water or 5 mM dimethylhexylammonium acetate) and mobile phase B (acetonitrile). The flow rate was 50 μL min−1. The autosampler temperature was set at 10 °C. The injection volume was 3 μL. The LC gradient was as follows: 0 – 1 min, 2% B; 1 – 7 min, 0 to 100% B; 7 – 9 min, 100% B; 9 – 10.1 min, 100 to 2% B; 10.1 – 12 min, 2% B.
BAP was detected using selective reaction monitoring (SRM) follow the transition of 179 m/z to 63 m/z. The instrument settings were as follows: negative ion mode; scan width – 0.5 m/z; scan time – 1.000 s; collision energy - 18 V; collision gas pressure – 1.6 mTorr; Spray Voltage −4000 V; Sheath Gas 30; Ion Sweep Gas – 0; Aux Gas Pressure – 0; Capillary Temperature 237 °C; S-Lens RF Amplitude – 133; Declustering Voltage – 0.
hpAP (1) was detected using selective reaction monitoring (SRM) follow the transition of 175 m/z to 63 m/z. The instrument settings were as follows: negative ion mode; scan width – 0.5 m/z; scan time – 1.000 s; collision energy - 48 V; collision gas pressure – 1.6 mTorr; Spray Voltage −2000 V; Sheath Gas 40; Ion Sweep Gas – 0; Aux Gas Pressure – 0; Capillary Temperature 240 ˚C; S-Lens RF Amplitude – 133; Declustering Voltage – 0.
Prodrug 30 was detected using selective reaction monitoring (SRM) follow the transition of 346.15 m/z to 143.12 m/z. The instrument settings were as follows: positive ion mode; scan width – 0.1 m/z; scan time – 1.000 s; collision energy - 20 V; collision gas pressure – 1.0 mTorr; Spray Voltage 3000 V; Sheath Gas 0; Ion Sweep Gas – 0; Aux Gas Pressure – 0; Capillary Temperature 270 ˚C; S-Lens RF Amplitude – 133; Declustering Voltage – 10.
Prodrug 31 was detected using selective reaction monitoring (SRM) follow the transition of 350.18 m/z to 143.12 m/z. The instrument settings were as follows: positive ion mode scan width – 0.1 m/z; scan time – 1.000 s; collision energy - 20 V; collision gas pressure – 1.0 mTorr; Spray Voltage 3000 V; Sheath Gas 0; Ion Sweep Gas – 0; Aux Gas Pressure – 0; Capillary Temperature 270 ˚C; S-Lens RF Amplitude – 133; Declustering Voltage – 10.
Supplementary Material
Acknowledgements
We thank Carley Heck and Namandje Bumpus for help with high resolution mass spectrometry analyses for compound characterization. This work was supported by funding from the National Institutes of Health (GM084998 for C.L.F.M., S. S., and D.B., T32 GM08018901 for D.B., and T32 GM007445 for S.S). We also wish to acknowledge support from the Johns Hopkins University School of Medicine, Institute for Basic Biomedical Sciences. We thank the National Science Foundation (CHE-1412648) and the NAU Office of the Vice President for Research for funds supporting M.J.H., J.L.A., and A.T.K. Support was also provided to A.T.K. through NIH/NIMHD RCMI U54MD012388 (Baldwin/Stearns-MPI).
Footnotes
Associated Content
The Supporting Information is available free of charge on the ACS Publication website http://pubs.acs.org/.
Evaluation of antibacterial activity of 30 and 31 in the presence of exogenous thiamin, phylogenetic analysis of peptide transporters of Gram-negative bacteria tested for antibacterial activity, LC-MS/MS standard curves and representative ion chromatograms, and NMR spectra for compounds 22 – 33.
References
- (1).Eisenreich W; Bacher A; Arigoni D; Rohdich F Biosynthesis of Isoprenoids via the Non-Mevalonate Pathway. Cell. Mol. Life Sci 2004, 61 (12), 1401–1426, DOI 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Broers STJ About the Early Steps of Biosynthesis of Isoprenoids in Escherichia Coli. PhD Dissertation No. 10978, Swiss Federal Institute of Technology, Zurich, PhD Dissertation no. 10978, Swiss Federal Institute of Technology, Zurich, 1994. [Google Scholar]
- (3).Rohmer M; Knani M; Simonin P; Sutter B; Sahm H Isoprenoid Biosynthesis in Bacteria: A Novel Pathway for the Early Steps Leading to Isopentenyl Diphosphate. Biochem. J. 1993, 295 (Pt 2) (2), 517–524, DOI 10.1042/BJ2950517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Schwarz MK Terpene Biosynthesis in Ginkgo Biloba: A Surprising Story. PhD Dissertation No. 10951, Swiss Federal Institute of Technology, Zurich, PhD Dissertation no. 10951, Swiss Federal Institute of Technology, Zurich, 1994. [Google Scholar]
- (5).Jomaa H; Wiesner J; Sanderbrand S; Altincicek B; Weidemeyer C; Hintz M; Türbachova I; Eberl M; Zeidler J; Lichtenthaler HK; et al. Inhibitors of the Nonmevalonate Pathway of Isoprenoid Biosynthesis as Antimalarial Drugs. Science 1999, 285 (5433), 1573–1576, DOI 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- (6).Bartee D; Freel Meyers CL Toward Understanding the Chemistry and Biology of 1-Deoxy-d-Xylulose 5-Phosphate (DXP) Synthase: A Unique Antimicrobial Target at the Heart of Bacterial Metabolism. Acc. Chem. Res 2018, 51 (10), 2546–2555, DOI 10.1021/acs.accounts.8b00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Patel H; Nemeria NS; Brammer LA; Freel Meyers CL; Jordan F Observation of Thiamin-Bound Intermediates and Microscopic Rate Constants for Their Interconversion on 1-Deoxy-d-Xylulose 5-Phosphate Synthase: 600-Fold Rate Acceleration of Pyruvate Decarboxylation by d-Glyceraldehyde-3-Phosphate. J. Am. Chem. Soc 2012, 134 (44), 18374–18379, DOI 10.1021/ja307315u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Brammer Basta LA; Patel H; Kakalis L; Jordan F; Freel Meyers CL Defining Critical Residues for Substrate Binding to 1-Deoxy-d-Xylulose 5-Phosphate Synthase - Active Site Substitutions Stabilize the Predecarboxylation Intermediate C2α-Lactylthiamin Diphosphate. FEBS J. 2014, 281 (12), 2820–2837, DOI 10.1111/febs.12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Brammer LA; Smith JM; Wade H; Meyers CF 1-Deoxy-d-Xylulose 5-Phosphate Synthase Catalyzes a Novel Random Sequential Mechanism. J. Biol. Chem 2011, 286 (42), 36522–36531, DOI 10.1074/jbc.M111.259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Xiang S; Usunow G; Lange G; Busch M; Tong L Crystal Structure of 1-Deoxy-d-Xylulose 5-Phosphate Synthase, a Crucial Enzyme for Isoprenoids Biosynthesis. J. Biol. Chem 2007, 282 (4), 2676–2682, DOI 10.1074/jbc.M610235200. [DOI] [PubMed] [Google Scholar]
- (11).Bartee D; Freel Meyers CL Targeting the Unique Mechanism of Bacterial 1-Deoxy-d-Xylulose 5-Phosphate (DXP) Synthase. Biochemistry 2018, 57 (29), 4349–4356, DOI 10.1021/acs.biochem.8b00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Smith JM; Vierling RJ; Meyers CF Selective Inhibition of E. Coli1-Deoxy-d-Xylulose-5-Phosphate Synthase by Acetylphosphonates. Medchemcomm 2012, 3 (1), 65–67, DOI 10.1039/C1MD00233C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sanders S; Vierling RJ; Bartee D; DeColli AA; Harrison MJ; Aklinski JL; Koppisch AT; Freel Meyers CL Challenges and Hallmarks of Establishing Alkylacetylphosphonates as Probes of Bacterial 1-Deoxy-d-Xylulose 5-Phosphate Synthase. ACS Infect. Dis 2017, 3 (7), 467–478, DOI 10.1021/acsinfecdis.6b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Smith JM; Warrington NV; Vierling RJ; Kuhn ML; Anderson WF; Koppisch AT; Freel Meyers CL Targeting DXP Synthase in Human Pathogens: Enzyme Inhibition and Antimicrobial Activity of Butylacetylphosphonate. J. Antibiot. (Tokyo). 2014, 67 (1), 77–83, DOI 10.1038/ja.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sanders S; Bartee D; Harrison MJ; Phillips PD; Koppisch AT; Freel Meyers CL Growth Medium-Dependent Antimicrobial Activity of Early Stage MEP Pathway Inhibitors. PLoS One 2018, 13 (5), e0197638, DOI 10.1371/journal.pone.0197638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Garai P; Chandra K; Chakravortty D Bacterial Peptide Transporters: Messengers of Nutrition to Virulence. Virulence 2017, 8 (3), 297–309, DOI 10.1080/21505594.2016.1221025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Alteri CJ; Smith SN; Mobley HLT Fitness of Escherichia Coli during Urinary Tract Infection Requires Gluconeogenesis and the TCA Cycle. PLoS Pathog. 2009, 5 (5), e1000448, DOI 10.1371/journal.ppat.1000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Circello BT; Miller CG; Lee J-H; van der Donk WA; Metcalf WW The Antibiotic Dehydrophos Is Converted to a Toxic Pyruvate Analog by Peptide Bond Cleavage in Salmonella Enterica. Antimicrob. Agents Chemother 2011, 55 (7), 3357–3362, DOI 10.1128/AAC.01483-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).O’Brien TA; Kluger R; Pike DC; Gennis RB Phosphonate Analogues of Pyruvate. Probes of Substrate Binding to Pyruvate Oxidase and Other Thiamin Pyrophosphate-Dependent Decarboxylases. Biochim. Biophys. Acta - Enzymol. 1980, 613 (1), 10–17, DOI 10.1016/0005-2744(80)90186-2. [DOI] [PubMed] [Google Scholar]
- (20).Whitteck JT; Ni W; Griffin BM; Eliot AC; Thomas PM; Kelleher NL; Metcalf WW; van der Donk WA Reassignment of the Structure of the Antibiotic A53868 Reveals an Unusual Amino Dehydrophosphonic Acid. Angew. Chemie Int. Ed 2007, 46 (47), 9089–9092, DOI 10.1002/anie.200703810. [DOI] [PubMed] [Google Scholar]
- (21).Kuemin M; van der Donk WA Structure–activity Relationships of the Phosphonate Antibiotic Dehydrophos. Chem. Commun 2010, 46 (41), 7694, DOI 10.1039/c0cc02958k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Campbell DA The Synthesis of Phosphonate Esters; an Extension of the Mitsunobu Reaction. J. Org. Chem 1992, 57 (23), 6331–6335, DOI 10.1021/jo00049a051. [DOI] [Google Scholar]
- (23).Chan TR; Hilgraf R; Sharpless KB; Fokin VV Polytriazoles as Copper(I)-Stabilizing Ligands in Catalysis. Org. Lett 2004, 6 (17), 2853–2855, DOI 10.1021/OL0493094. [DOI] [PubMed] [Google Scholar]
- (24).Dik DA; Fisher JF; Mobashery S Cell-Wall Recycling of the Gram-Negative Bacteria and the Nexus to Antibiotic Resistance. Chem. Rev 2018, 118 (12), 5952–5984, DOI 10.1021/acs.chemrev.8b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Guyer CA; Morgan DG; Staros JV Binding Specificity of the Periplasmic Oligopeptide-Binding Protein from Escherichia Coli. J. Bacteriol 1986, 168 (2), 775–779, DOI 10.1128/JB.168.2.775-779.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tame JR; Murshudov GN; Dodson EJ; Neil TK; Dodson GG; Higgins CF; Wilkinson AJ The Structural Basis of Sequence-Independent Peptide Binding by OppA Protein. Science 1994, 264 (5165), 1578–1581, DOI 10.1126/SCIENCE.8202710. [DOI] [PubMed] [Google Scholar]
- (27).Baba T; Ara T; Hasegawa M; Takai Y; Okumura Y; Baba M; Datsenko KA; Tomita M; Wanner BL; Mori H Construction of Escherichia Coli K-12 in-Frame, Single-Gene Knockout Mutants: The Keio Collection. Mol. Syst. Biol 2006, 2 (1), 2006.0008, DOI 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).DeColli AA; Nemeria NS; Majumdar A; Gerfen GJ; Jordan F; Freel Meyers CL Oxidative Decarboxylation of Pyruvate by 1-Deoxy-d-Xyulose 5-Phosphate Synthase, a Central Metabolic Enzyme in Bacteria. J. Biol. Chem 2018, 293 (28), 10857–10869, DOI 10.1074/jbc.RA118.001980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Brammer LA; Meyers CF Revealing Substrate Promiscuity of 1-Deoxy-d-Xylulose 5-Phosphate Synthase. Org. Lett 2009, 11 (20), 4748–4751, DOI 10.1021/ol901961q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Morris F; Vierling R; Boucher L; Bosch J; Freel Meyers CL DXP Synthase-Catalyzed C-N Bond Formation: Nitroso Substrate Specificity Studies Guide Selective Inhibitor Design. ChemBioChem 2013, 14 (11), 1309–1315, DOI 10.1002/cbic.201300187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Leid JG; Ditto AJ; Knapp A; Shah PN; Wright BD; Blust R; Christensen L; Clemons CB; Wilber JP; Young GW; et al. In Vitro Antimicrobial Studies of Silver Carbene Complexes: Activity of Free and Nanoparticle Carbene Formulations against Clinical Isolates of Pathogenic Bacteria. J. Antimicrob. Chemother 2012, 67 (1), 138–148, DOI 10.1093/jac/dkr408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.