

The concept that the immune system has a role in controlling cancer is not a recent one. More than a century ago, the surgeon William Coley hypothesized that postoperative bacterial infections could mobilize a patient’s own resistance to tumour recurrence, and he developed a mixture of heat-killed bacteria for intratumoral injection that occasionally produced durable regressions1. More recently, the elucidation of molecular mechanisms underlying immune regulation has been instrumental in devising strategies to overcome cancer cells’ ability to suppress the immune surveillance that would otherwise protect the host from tumour progression2–4. One approach to activating these antitumour immune responses has been termed ‘checkpoint blockade’ — referring to the use of antibodies that block immune-inhibitory pathways switched on by cancer cells. Five papers published in this issue5–9 reveal a growing list of cancers that respond to checkpoint blockade and describe characteristics of those patients who respond to such therapies.
The immune checkpoints targeted by these therapies serve under normal conditions as molecular brakes, preventing hyperactivity of the T cells of the immune system and, in some cases, preventing autoimmunity10. CTLA-4 and PD-1 are two key cell-surface receptors that, when bound by their ligands, trigger such inhibitory pathways and dampen T-cell activity. In the case of the PD-1 pathway, expression of ligands such as PD-L1 on tumour cells can directly lead to the death of T cells expressing PD-1. Furthermore, engagement of CTLA-4 or PD-1, which are expressed both on T cells and on other immune cells in an inflamed tumour microenvironment, can self-limit the antitumour response. Antibodies that block CTLA-4 (ipilimumab) and PD-1 (pembroli-zumab and nivolumab) have been approved to treat patients, and the clinical responses are often durable, with some patients remaining free from disease progression for many years11–13. But until recently, little efficacy of these treatments has been noted beyond melanoma and renal-cell carcinoma. Furthermore, the precise cellular events triggered by antibody binding and their exact antigenic targets (the molecular structures to which the antibodies bind) remained unclear.
Powles et at.5 (page 558) and Herbst et at.6 (page 563) present results from a phase I clinical trial of MPDL3280A, a monoclonal antibody that blocks the ligand PD-L1. Herbst and colleagues report that the antibody induces therapeutic responses in patients with non-small-cell lung cancer, melanoma, renal-cell carcinoma and other solid tumours — findings that support the known activity of other antibodies that block the PD-1-pathway in some of these diseases11,14–16. Powles and colleagues analyse the effects of this antibody treatment in a larger group of patients with urothelial bladder cancer. Both clinical reports document durable responses in a subset ofpatients, and that the therapy has low toxicity, with only rare high-grade adverse events. These results substantially expand the spectrum of malignancies in which PD-1-pathway blockade has meaningful clinical activity.
Ever since the earliest reports of the effects of PD-1 blockade14,15,17,18, PD-L1 expression by tumour cells has been a focus of studies looking for biomarkers that will predict a therapeutic response. Although it is clear that expression of PD-L1 on tumour cells makes it more likely that the patient will respond to PD1-pathway blockade, this is not a binary, static predictive marker. Herbst et at. and Tumeh et at.7 (page 568) now reveal that it is not solely tumour-cell expression of PD-L1 that can enrich responses to PD-1-pathway blockade, but that expression of PD-L1 on immune cells infiltrating the tumour is also a key predictor of clinical activity (Fig. 1). Tumeh et at. further show, using samples from patients with melanoma that were treated with pembroli- zumab, that a certain set of conditions enables PD-1 blockade to mediate tumour regression. These are the presence of CD8+ T cells (a T-cell subset that directly kills its target cells) and immune cells that express PD-1 and PD-L1 at the tumour margin, together with a T-cell population with less-diverse antigen specificity. Taken together, the findings of these two papers suggest that tumours that have already been recognized by the immune system, and so contain infiltrating immune cells bearing PD-1 and PD-L1, are particularly sensitive to immune-checkpoint blockade.
Figure 1 |. Checkpoint blockade activates antitumour immunity.
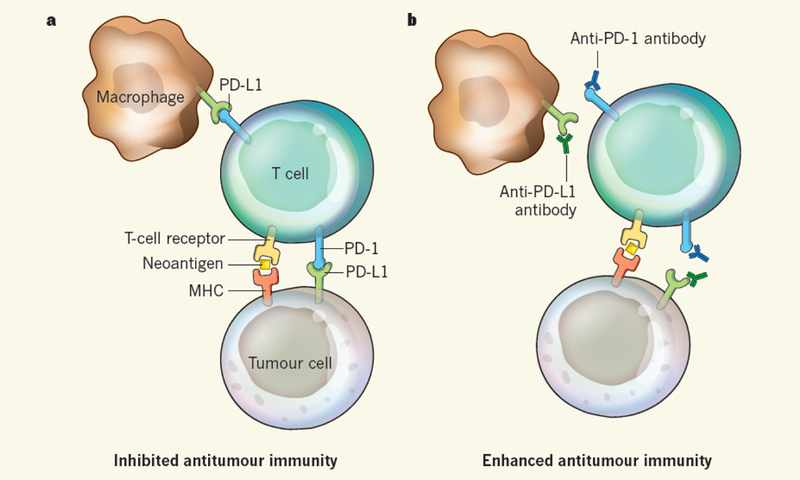
a, Tumour cells express both cancer-driving mutations and ‘passenger’ mutations that cause the expression of neoantigens — ‘new’ molecular structures that, when presented by MHC proteins on the cell surface, are recognized by T cells of the immune system as being foreign, leading to an immune response against the tumour. However, interactions between the receptor PD-1 and its ligand PD-L1, which are expressed on tumour cells, T cells and other immune cells such as macrophages, activate signalling pathways that inhibit T-cell activity and thus inhibit the antitumour immune response. b, Antibodies that block the PD-1 pathway by binding to PD-1 or PD-L1 can reactivate T-cell activity and proliferation, leading to enhanced antitumour immunity.
The contributions from Yadav et at.8 (page 572) and Gubin et at.9 (page 577) add another dimension by suggesting that ‘passenger’ mutations — cancer-cell mutations that do not directly contribute to cancer initiation and progression — play a key part in tumour immunity. Although it is increasingly evident that the new antigens generated by such mutations are targeted by antitumour T cells, identifying which ofthese neo-antigens are functionally important has been a challenge. Yadav et al. sequenced the exomes (the protein-coding regions of the genome) of two mouse tumour-cell lines and compared these with the reference mouse exome to predict candidate neo-antigens in the tumour cells. In parallel, they identified which ofthe neo-antigens could potentially elicit immune responses by isolating those that bind to major histocompatibility complex (MHC) proteins, which present antigens to T cells, and then analysing the bound peptides by mass spectrometry.
Surprisingly, this process identified only a few candidate neo-antigens, but these were highly immunogenic in vivo (that is, they provoked a strong immune response) and were found to be encoded by genes that are unlikely to directly contribute to cancer development, confirming that changes in immunogenicity can result from passenger mutations (Fig. 1). The approach presented in this report is a key advance for the discovery of immunogenic antigens and is applicable to many experimental systems. However, it remains to be seen whether the low numbers of neo-antigens discovered reflects an inherently limited sensitivity of the approach or whether the number of MHC-presented neo-antigens is indeed low.
A previous paper from the research group of Gubin et al. showed that a mutant spectrin-β2 protein was responsible for the strong immunogenicity of tumours in a particular mouse model19. Now, Gubin and colleagues find that tumours from the same model that become resistant to immune-mediated rejection have lost this neo-antigen. They go on to show that treatment with anti-PD-1 and/or anti-CTLA-4 antibodies enabled the mice to again reject these tumours. Using a similar approach to that of Yadav et al., the authors identify two mutations, in the Alg8 and Lama4 genes, that created neo-antigens mediating these effects. Vaccinating mice with these antigens induced tumour rejection at a level comparable to that of checkpoint-blockade therapy, convincingly demonstrating that tumour neo-antigens are potent functional targets of this therapy. This work also corroborates recent findings from another group20.
These five papers, together with other recent studies, support the hypothesis that immune responses to tumour-specific mutations are central to both natural antitumour immunity and to the antitumour activity generated by checkpoint-blockade therapy. In another twist to the story, a paper21 just published reports that, in patients with melanoma treated with ipilimumab, specific neo-antigens in the tumour are associated with a favourable clinical response. Intriguingly, these antigens bear a striking similarity to immunogenic antigens derived from bacteria and viruses, suggesting, perhaps, that Coley was on to something.
Contributor Information
JEDD D. WOLCHOK, Department of Medicine and the Ludwig Center, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA.
TIMOTHY A. CHAN, Department of Radiation Oncology and the Human Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center.
References
- 1.Coley WB Proc. R. Soc. Med. 3, 1–48 (1910). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunn GP, Bruce AT, Ikeda H, Old LJ & Schreiber RD Nature Immunol. 3, 991–998 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Koebel CM et al. Nature 450, 903–907 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Shankaran V et al. Nature 410, 1107–1111 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Powles T et al. Nature 515, 558–562 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Herbst RS et al. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tumeh PC et al. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yadav M et al. Nature 515, 572–576 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Gubin MM et al. Nature 515, 577–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Page DB, Postow MA, Callahan MK, Allison JP & Wolchok JD Annu. Rev. Med. 65, 185–202 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Robert C et al. Lancet 384, 1109–1117 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Topalian SL et al. J. Clin. Oncol. 32, 1020–1030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolchok JD et al. Ann. Oncol. 24, 2174–2180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brahmer JR et al. N. Engl. J. Med. 366, 2455–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Topalian SL et al. N. Engl. J. Med. 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamid O et al. N. Engl. J. Med. 369, 134–144 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taube JM et al. Sci. Transl. Med. 4, 127ra37 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brahmer JR et al. J. Clin. Oncol. 28, 3167–3175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsushita H et al. Nature 482, 400–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan F et al. J. Exp. Med. 211, 2231–2248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snyder A et al. N. Engl. J. Med. 10.1056/NEJMoa1406498 (2014). [DOI] [Google Scholar]