

Abstract
To determine the role of reduced dopaminergic transmission for declines of forced versus spontaneous behavior, we used a model of Parkinson’s disease with progressive degeneration of dopamine (DA) neurons, the MitoPark mouse. Mice were subjected to rotarod tests of motor coordination, and open field and cylinder tests for spontaneous locomotor activity and postural axial support. To measure DA release in dorsal striatum and the shell of Nucleus Accumbens (NAc) we used ex vivo fast-scan cyclic voltammetry (FSCV) in 6 to 24 week old mice. To determine decline of DA transporter function we used 18FE-PE2I PET. We show here that FSCV is a sensitive tool to detect evoked DA release dysfunction in MitoPark mice and that electrically evoked DA release is affected earlier in nigrostriatal than mesolimbic DA systems. DA reuptake was also affected more slowly in NAc shell. PET data showed DA uptake to be barely above detection levels in 16 and 20 weeks old MitoPark mice. Rotarod performance was not impaired until mice were 16 weeks old, when evoked DA release in striatum had decreased to ≈ 40% of wild type levels. In contrast, impairment of open field locomotion and rearing began at 10 weeks, in parallel with the initial modest decline of evoked DA release. We conclude that forced behaviors, such as motivation not to fall, can be partially maintained even when DA release is severely compromised, whereas spontaneous behaviors are much more sensitive to impaired DA release, and that presumed secondary non-dopaminergic system alterations do not markedly counteract or aggravate effects of severe impairment of DA release.
Keywords: TFAM, Dopamine, Parkinson’s Disease, Fast Scan Cyclic Voltammetry, PET
Graphical Abstract
We asked how the ability to release dopamine (DA) relates to the ability to carry out spontaneous, versus forced motor activities, taking advantage of a neurotoxin-free transgenic mouse model with degeneration specifically of the DA system. Using ex vivo fast-scan cyclic voltammetry of brain slices we find that impairment of open field locomotion and rearing began already at 10 weeks, in parallel with a modest decline of evoked DA release while rotarod performance was not impaired until mice were 16 weeks old, when evoked DA release in striatum had decreased to 40% of wild type levels. We conclude that forced behaviors, such as motivation not to fall, can be partially maintained even when DA release is severely compromised, whereas spontaneous behaviors are much more sensitive to impaired DA release, and that presumed secondary non-dopaminergic system alterations do not markedly counteract effects of severe impairment of DA release.

Introduction
Despite the identification of genetic causes and risk factors for Parkinson’s disease (Ferreira & Massano 2017, Delamarre & Meissner 2017, Deng et al. 2017), it is striking how much more remains to be understood. It is known that when symptoms first present themselves, degeneration of the mesencephalic dopamine (DA) system is already marked (Braak et al. 2004). However, it remains unclear which symptoms are caused specifically by the degeneration of the ascending dopaminergic systems, versus symptoms that might be contributed by primary neurodegenerative events affecting non-dopaminergic systems and occurring in other areas ranging from the gastrointestinal tract to cortex cerebri. There is a need to study more closely the relationship between the progressive loss of DA innervation and the various symptoms, not the least at early neuropathological stages, to support early diagnosis and future neuroprotective treatments.
One way to approach the specific role of a progressively degenerating DA system is to target this system in mice. This can be accomplished by specific removal of the mitochondrial transcription factor TFAM in midbrain DA neurons, as is the case in the MitoPark mouse (Ekstrand et al. 2007). In this mouse model, impaired mitochondrial function leads to progressive degeneration specifically of the ascending DA systems in an otherwise healthy animal. The model allows for studies of the role of DA degeneration per se, studies of DA neuron pathology, and behavioral effects over an extended time course (Galter et al. 2010, Good et al. 2011, Sterky et al. 2011). In an operant conditioning task based on effort it has also been shown that MitoPark mice show progressive reduction of movement vigor, as the neuronal representation of vigor in dorsal striatum becomes impaired. Without a sufficient dopaminergic input to dorsal striatum, striatal circuity is unable to support adaptation of movement vigor (Panigrahi et al. 2015).
Here we approach the issue of to which extent the DA innervation is functionally impaired prior to overt motor impairments by comparison of spontaneous and forced behaviors and relating these behaviors to the levels of DA release using different stimulation protocols and intensities. Fast-scan cyclic voltammetry was used to characterize stimulation-induced release and reuptake of DA by DA axon terminals ex vivo in two key areas, dorsal striatum and the NAc shell. We find that impaired function of dopaminergic terminals of the nigrostriatal system occurs earlier than those in the mesolimbic DA system. Importantly, we show that decreased release of DA occurs in parallel with impairments of spontaneous locomotion and rearing, while impairment of forced rotarod performance becomes manifest only later. The findings provide additional evidence that a range of motor functions are affected when the primary pathology is restricted only to the DA system (Alexander 2004) and suggest in a relevant animal model that there is an initial period of impaired DA transmission that the system can cope with to maintain performance of forced behaviors, although spontaneous behaviors are affected earlier, being more directly correlated with baseline DA pathway dysfunction.
Material and Methods
Animals.
The breeding scheme for generating MitoPark mice has been described previously (Ekstrand et al. 2007, Galter et al. 2010, Good et al. 2011). Briefly, to generate MitoPark mice, animals on a C57BL6 background, in which the DA transporter (DAT) promoter was used to drive cre-recombinase expression, were crossed with mice in which the Tfam gene had been loxP-flanked. Breeding pairs to generate MitoPark mice were sent to the National Defense Medical Center Taiwan from a colony maintained at the NIDA (NIH) Intramural Program. Male and female 6 – 8 week-old MitoPark mice used in these experiments were heterozygous for DAT-cre expression (DAT/DATcre) and homozygous for the loxP-flanked Tfam gene (TfamloxP/TfamloxP). Age-matched wild type mice were used as controls. In some voltammetric recordings, mice lacking only the DAT-cre construct (DAT/DAT Tfam/Tfamloxp or only the Tfam construct DAT/DATcre Tfam/Tfam) were also used as controls. A total of 72 MitoPark mice (28 male, 44 female) and 27 control (WT) mice (4 male, 23 female) were used. Numbers of animals are summarized in Table 1. We show in figures 5 and 6 that stimulation-induced release of DA in WT mice remains unchanged from 6 to 22 weeks of age (oldest age tested) in both striatum and NAc accumbens shell. It has previously been shown that in striatum of control mice, dopamine, DOPAC, HVA, HCA/DA ratio, norepinephrine, and 5HT are all unchanged between 6 and 40 weeks of age, whereas there is severe progressive decrease of dopamine, DOPAC, and HVA, and an increase of the HVA/DA ratio in MitoPark mice from 6 to 40 weeks (Galter et al 2009). Based in this information, we were able to reduce the number of mice by using controls of only one age in figs. 3, 7 and 8. Breeding pairs for the generation of MitoPark mice are available upon request from the authors.
Table 1.
Numbers, gender and age (weeks) of control and MitoPark mice used.
| I H C |
Behavior | Echem | P E T |
Age (IHC) |
Age (Behavior) |
Age (Echem) |
Age (PET) |
|
|---|---|---|---|---|---|---|---|---|
| WT (Female) | 8 | 8 | 7 | 2 | 32 | 6–24 | 8,12,22 | 14 |
| WT (male) | 4 | 6–24 | ||||||
| MP (Female) | 6 | 8 | 30 | 9 | 8,20 | 6–30 | 6–24 | 12, 16, 20 |
| MP (male) | 6 | 7 | 15 | 8,20 | 6–30 | 6–24 |
Fig. 5. Rotarod performance is maintained in MitoPark mice until 14 weeks of age, despite severe lack of DA release.
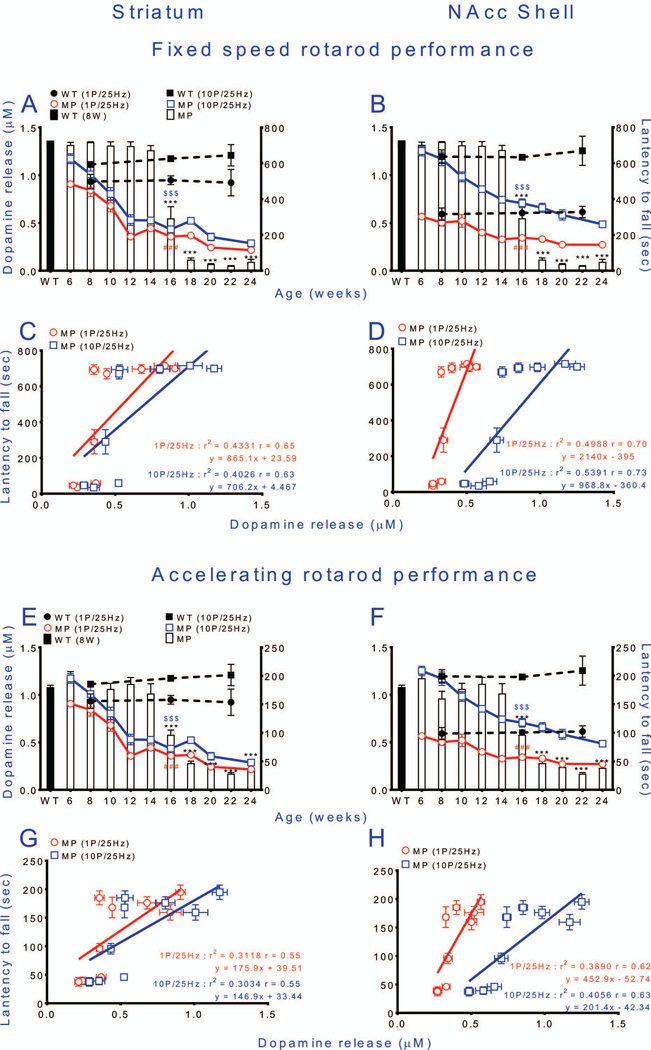
Relationships between tonic (red empty circles) or phasic (blue empty squares) stimulation of DA release (stimulation intensity 10 volt) and fixed (A-D) or accelerating (E-H) speed rotarod performance of MitoPark mice. Corresponding wild type data are shown as dashed black lines for DA release and a black bar for rotarod performance. (A) Tonic and phasic stimulation-induced DA release in striatum decrease before impairment of fixed speed rotarod performance. (B) Tonic and phasic stimulation-induced DA release in NAc shell decrease before impairment of fixed speed rotarod performance. (C) Linear regression shows positive correlation between fixed speed Rotarod performance and amount of DA release in striatum. (D) Linear regression shows positive correlation between fixed speed Rotarod performance and amount of DA release in NAc shell. In E-H the same DA release curves are instead superimposed on performance on the accelerating rotarod and the corresponding linear regression an analyses performed. Mean ± s.e.m. ***P < 0.001, for exact numbers see Table 2.
Fig. 6. Spontaneous locomotion and rearing begin to be impaired as soon as deficits in dopamine release can be detected.
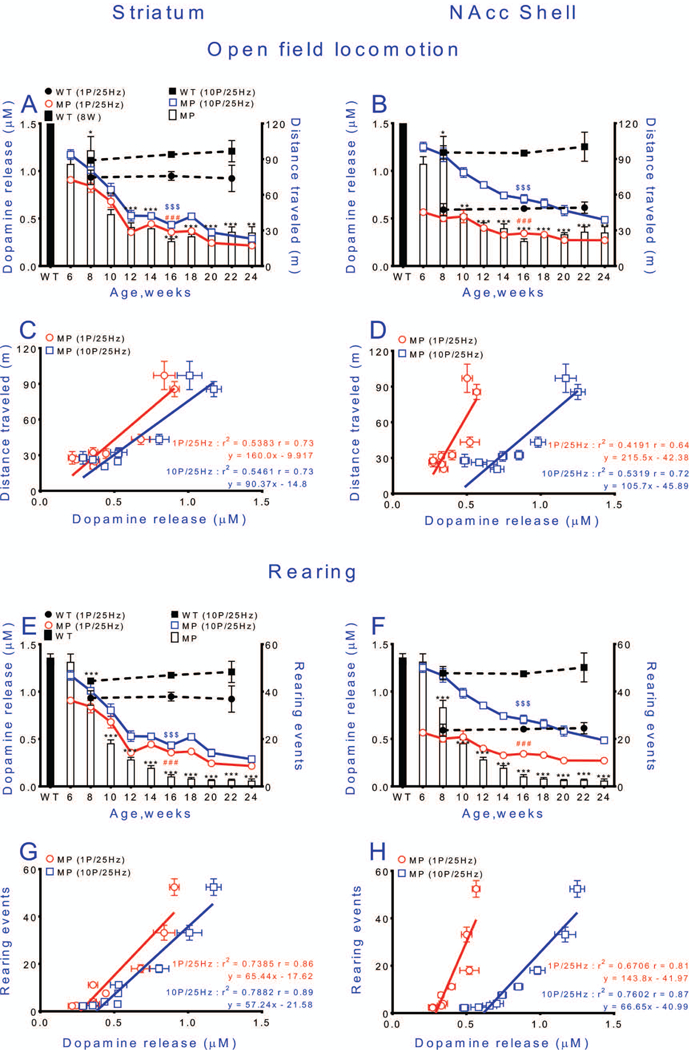
Relationships between stimulation-induced DA levels in striatum or NAc shell and open-field locomotion (distance traveled in 1 h) (A, B) and rearing (cylinder test) (E, F). Relationships between tonic stimulation of DA release (stimulation intensity 10 volts) and locomotion (A, B) of MitoPark mice. Corresponding wild type data are shown as dashed black lines for DA release and a black bar for locomotion or rearing. Correlations between DA release and locomotion and rearing of MitoPark mice are shown in C and D, and G and H, respectively. Mean ± s.e.m. *p < 0.05, **p < 0.01, ***p < 0.001 WT vs. MP; ###p < 0.001 WT (1P/25Hz) vs. MP (1P/25Hz); $ $ $p < 0.001 WT (10P/25Hz) vs. MP (10P/25Hz). For exact statistical data, see Table 2.
Fig. 3. Stimulation-induced dopamine release in response to different stimulation intensities in slices of striatum and NAc shell.
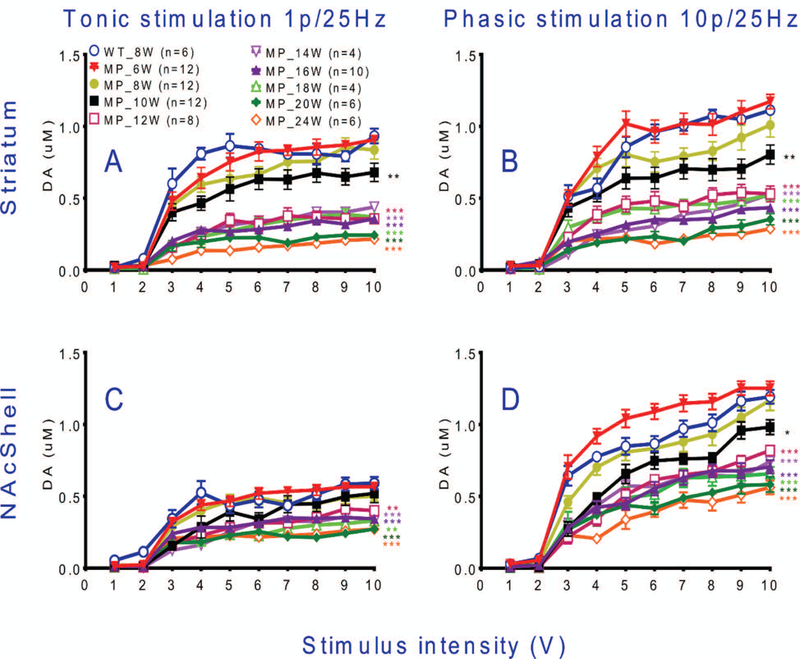
(A) In striatal slices, FSCV data show that tonic DA release is decreased from 10 weeks of age and onwards in MitoPark vs WT mice. (B) A larger suppression of phasic release was found in striatal slices from 10 weeks of age and onwards in MitoPark mice. (C) Tonic DA release was affected significantly in NAc shell of MitoPark mice by 16 weeks of age and (D) phasic DA release declined in the NAc shell of MitoPark mice beginning at 10 weeks of age. Mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, compared to 8 weeks old wild type mice. For detailed statistics see Table 2.
Fig. 7. Dopamine release probability declines with age in MitoPark mice.
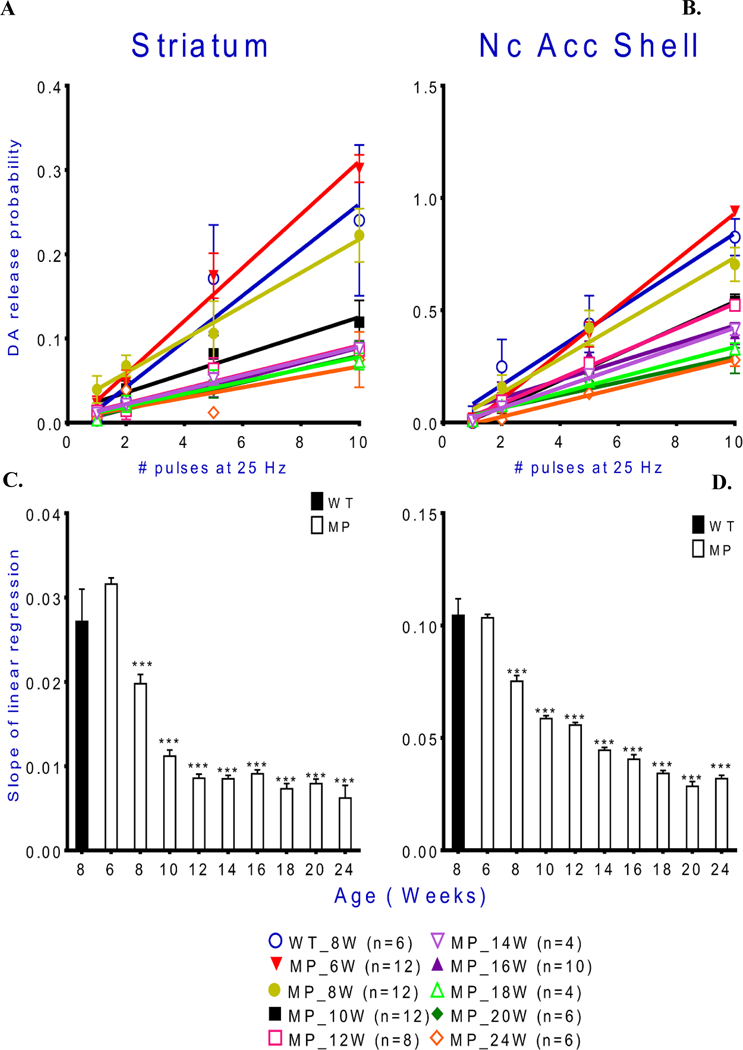
Release probability was calculated as described in the methods section. A decline of release probability with age of MitoPark mice is found in both striatum and NAc shell. Slopes of linear regression lines for tonic DA release in striatum and NAc shell in MitoPark mice decrease with age. Mean ± s.e.m. *p < 0.05, ***p < 0.001 MitoPark versus controls. For exact statistical data, see Table 2.
Fig. 8. Dopamine reuptake efficacy, shown by increasing time for disappearance of FAST-detectable extracellular DA.
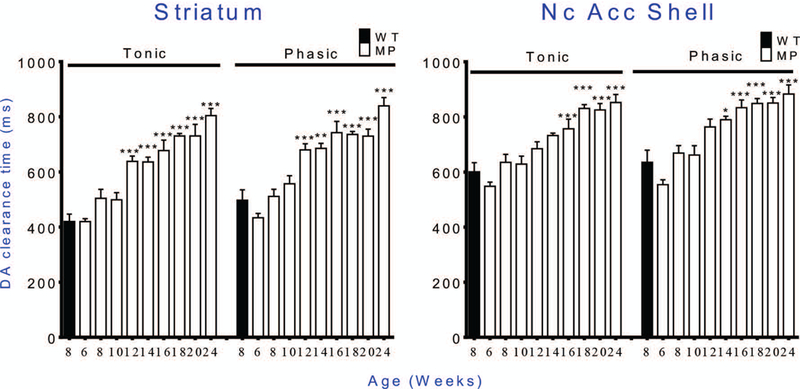
The uptake rate decreases with age in MitoPark mice. Similar patterns of increased time are seen in striatum and Nc. Accumbens Shell, and with either tonic or phasic stimulation. * p < 0.05, ** p < 0.01, *** p < 0.001 WT vs. MP. For statistical data, See Table 2.
Generally, brain tissue from the same animals were used for ex vivo fast-scan cyclic voltammetry (FSCV) in both striatal and NAc brain slices. Genotyping was performed at the Department of Neurological Surgery, Tri-Service General Hospital, National Defense Medical Center, Taipei (NDMC). Animal protocols were approved by the NDMC Animal Care and Use Committee and followed the NDMC Guidelines for the Care and Use of Laboratory Animals (IACUC 17–156 and IACUC 17–261). Times at which behavioral tests and in vitro electrochemistry were performed are summarized in Fig. 1.
Fig. 1. Experimental set-up.
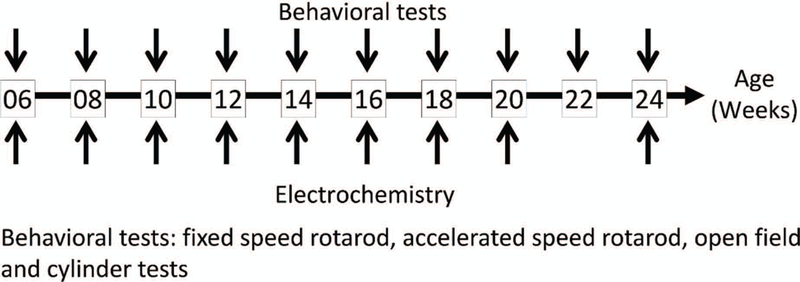
Control and MemoFlex mice were analyzed at 10 (behavior) or 9 (electrochemistry) ages. A set of behavior tests were carried out at 2 consecutive days for each age (upper arrows). Electrochemical analyses of stimulation induced DA release in striatum and Nc Accumbens shell, were analyzed in in ex vivo brain slices at the same ages (except at 22 weeks) (lower arrows). Not shown here, a separate group of mice were subject to PET scans for the dopamine transporter DAT. For numbers of animals, see Table 1. (Total number of animals = 120, n = 20 for immunohistochemistry, n = 27 for behavioral test, n = 52 for electrochemistry and n = 11 for PET scans).
Locomotor activity.
The mice were habituated to a low-noise experimental environment for 1 h and then placed in an open-field chamber (20 cm x 30 cm x 30 cm) in this environment. A digital camera was mounted on the ceiling and filmed the animal while it moved freely for 1 hour. The videos were analyzed by dedicated software (TopScan®, Clever Sys Inc., VA, USA) to detect rodent movements and behaviors based on video-tracking of multiple individual body parts, posture and frequency of movements. The horizontal movements during 1 hour were taken as distance traveled.
Cylinder test.
The cylinder test was performed to evaluate vertical movements and axial set. The mice were habituated to the low-noise experimental environment for 1 h. After habituation, the mice were placed in an open-top, clear plastic cylinder (diameter: 20 cm, height 30 cm) for 5 min and behavior was recorded by a video camera. Rearing was counted from video records. Blinding was not needed.
Rotarod test.
Rotarod tests were performed to evaluate motor coordination and balance. During a training phase, mice were introduced to walking on the rotating rod (47650 Rota-Rod NG, Ugo Basile, Comerio, Italy) one day before being tested. The training was completed when all mice were able to walk forward for 720 s at 15 rpm. Both fixed speed (20 rpm, cut-off time 720 s) and accelerating speed (from 5 rpm to 80 rpm within 240 s) rotarod tests were performed 3 times a day every 2 weeks. The time until the animal fell off the rotating rod was recorded by observers blinded to the mice genotypes. Trials were separated by at least 30 min.
Timing of behavioral tests.
For each time point the behavioral tests were carried out on two consecutive days. Day 1, mice were first habituated to a low-noise experimental environment for 1 h. They were then subjected to the open field test for 1 hr. After a 1 hr pause, they were subjected to the cylinder test for 5 min. Day 2, mice were again habituated to a low-noise experimental environment for 1 h before the Rotarod tests. These started with 3 rounds on the accelerating Rotarod with 20 min between rounds. Then there was a 2 hr break before 3 rounds of tests on a fixed speed rotarod, again with 20 min between rounds.
NAc and striatal brain slice preparation.
Brain slices were prepared as described previously (Chen et al. 2008, Good et al. 2013). After decapitation, the brain was removed and immersed in oxygenated (95%O2/5%CO2) cold cutting solution (in mM: sucrose 194, NaCl 30, KCl 4.5, MgCl2 1, NaH2PO4 1.2, glucose 10, and NaHCO3 26). The tissue blocks containing NAc and striatum, were cut into coronal slices (280 μm) within the chamber filled with cold cutting solution using a tissue slicer (VT 100, Leica). The slices were then transferred to a holding chamber filled with oxygenated artificial CSF solution (aCSF; in mM: NaCl 126, KCl 3, MgCl2 1.5, CaCl2 2.4, NaH2PO4 1.2, glucose 11, NaHCO3 26) at 30°C for 30 min.
Fast scan cyclic voltammetry and dopamine measurements in brain slices.
FSCV recording was performed as described previously (Chen et al. 2017, Cho et al. 2006). Slices were transferred into a chamber (0.5ml, 31–33°C) filled with aCSF with a perfusion rate of 2ml/min. Carbon fibers (7 μm diameter; Goodfellow Corp., Oakdale, PA, USA) were lowered to a depth of 100 μm into striatum or NAc under stereoscopic observation. Pipettes containing the carbon fibers were filled with a solution of 4M K-acetate/150 mM KCl. The carbon fiber was positioned between the separated tips of a bipolar stimulating electrode (FHC Inc., Bowdoin, ME, USA) using a stereo microscope. Voltammetric scans, stimulus wave form generation timing, and data collection were performed using A/D boards (PCI 6052E and PCI-6711E, National Instruments, Austin, TX, USA) and custom LabView-based software (TarHeel CV, courtesy of Drs. Joseph Cheer and Michael Heien, University of North Carolina). The potential of the carbon fiber was driven from −0.4 to 1.0 V and back to −0.4 V using a triangular waveform (400 V/s scan rate, 7 ms duration) applied every 100 ms. A 5-s (50-scan) control period was initially applied to the carbon fiber. The stimulation intensity was defined as voltage levels (from 1 to 10 volts in input/output curves) instead of current as used previously (Chen et al. 2017). DA peak oxidation currents were digitally subtracted from those obtained during the peak of the response following electrical stimulation. All signals used in the statistical analyses matched the expected voltammetric profile for DA (Kawagoe et al. 1993). The DA release evoked by a single pulse (tonic release) represent both the underlying release per pulse and the re-uptake by DAT (Wightman & Zimmerman 1990). The level of tonic release depends upon the DA terminal density and the rate of DA uptake by DAT. The signals were converted to DA concentrations using a calibration performed for each electrode with a 1-μM DA standard solution. To assess the capacity of axon terminals to release DA during stimulation, 2 types of voltammetric signals were obtained at each recording site by using a single pulse (for tonic) or 10 pulses (for phasic) delivered at 25 Hz under different stimulation intensities (from 1 to 10 volts). After tonic and phasic DA signals were obtained with different stimulation intensities at each site, these values were summed and averaged for plotting input/output (I/O) curves.
DA uptake was assessed by fitting a single exponential function to the signal decay using a least-squares minimization algorithm: Y(t) = A-t/τ, where A = peak signal amplitude (nA), t = time (ms), and Y = signal amplitude at any given t (WinWCP; Dr. John Dempster, Strathclyde Institute for Biomedical Sciences, Glasgow, UK; http://spider.science.strath.ac.uk). A tau (τ) value was obtained for each recording site by averaging all time constants obtained from each DA signal generated during the input-output curves (stimulus intensity vs. DA signal). The first-order rate constant (k or 1/τ) obtained using this approach provides an index of the efficiency (Vmax/Km) of DA clearance mediated primarily by the DA transporter (DAT) (Chen et al. 2008, Sabeti et al. 2002).
Release probability of DA was calculated after 3 signals at each site had been averaged. To assess the capacity of axon terminals to release DA; the difference between the peak DA signal obtained immediately after burst stimulation or single pulses was determined as DAnp - DA1p, where DAnp is the amplitude of the voltammetric signal for 2, 5, and 10 pulses, and DA1p is the amplitude of the voltammetric signal obtained following a single electrical pulse. The data were fit to a linear regression model (y=mx+b; Prism 5.02; GraphPad, San Diego, CA, USA), where the slope m represents the relative change in DA concentration per pulse (Chen et al. 2017, Chen et al. 2008).
[18F]FE-PE2I PET scan imaging for Dopamine Transporter function.
Using an automatic synthesizer system (GE TRACERlab FX2 N), radioactive 18F nuclei were produced by a cyclotron. After separation, the tosylethyl-PE2I precursor was dissolved in anhydrous dimethyl sulfoxide, and reacted with k[18F]/ Kryptofix2.2.2 at −140° for five minutes (Fig. 2), followed by purification using high performance liquid chromatography with a C18 column, to obtain the PET ligand, [18F]FE-PE2I.
Fig. 2. Synthesis of 18F-labeled DA-transporter ligand [18F]FE-PE2I.

A tosylethyl-PE2I precursor was dissolved in anhydrous dimethyl sulfoxide, and reacted with k[18F]/ Kryptofix2.2.2 at 140° for five minutes, followed by purification using high performance liquid chromatography.
A separate set of MitoPark mice aged 12, 16 and 20 weeks were used for PET scans. Tail vein injections of 0.3 mCi [18F]FE-PE2I were delivered during anesthesia (5% isoflurane/oxygen mixture for induction, 2% for maintenance). Each injected animal was first kept in a radiation prevention box for 20 minutes and then transferred to a PET scanner for static image scans with the energy window being set from 250 to 700 KeV. 3D images of the brain were thus captured ≈ 20 minutes after ligand injection. Images were smoothened using a Gaussian algorithm. Scans from a control animal was used as reference for volumes of interest (VOI). The specific uptake ratio (SUR) was calculated as SUR = (VOI of striatum)- (VOI of cerebellum)/VOI of cerebellum.
Statistics.
Statistical analyses of data for DA release input/output curves and behavioral tests were performed using a two-way analysis of variance (ANOVA) followed by a Bonferroni post hoc test for multiple comparisons. Statistical analyses of data for DA reuptake and slope differences between phasic and tonic releases were performed using an unpaired t-test. All statistical tests were two-tailed and were performed using appropriate software (GraphPad Prism 5.02, GraphPad Scientific, San Diego, CA, USA). No animals were excluded, no inclusion or exclusion criteria were determined, no outliers were excluded, tests for normality were not performed. A p-value < 0.05 using a two-tailed test was considered significant. Statistical levels are indicated in the figures and exact numbers are found in Table 2.
Table 2:
Statistics details
| Figure | Statistics | F (DFn, DFd) | P value | Statistics details | |
|---|---|---|---|---|---|
| Figure 3 | A | Two-way ANOVA | F81, 1449 = 4.571 | P < 0.001 | **P < 0.01, ***P < 0.001 MP compared to 8 weeks old WT mice |
| B | Two-way ANOVA | F81, 1448 = 5.030 | P < 0.001 | **P < 0.01, ***P < 0.001 MP compared to 8 weeks old WT mice | |
| C | Two-way ANOVA | F81, 1364 = 2.127 | P < 0.001 | **P < 0.01, ***P < 0.001 MP compared to 8 weeks old WT mice | |
| D | Two-way ANOVA | F81, 1442 = 4.213 | P < 0.001 | *P < 0.05, ***P < 0.001 MP compared to 8 weeks old WT mice | |
| Figure 4 | A | One-way ANOVA | F9, 143 = 25.62 | P < 0.001 | *P < 0.05, ***P < 0.001 MP compared to 8 weeks old WT mice |
| B | One-way ANOVA | F9, 143 = 26.99 | P < 0.001 | **P < 0.01, ***P < 0.001 MP compared to 8 weeks old wWT mice | |
| C | One-way ANOVA | F9, 134 = 9.883 | P < 0.001 | *P < 0.05, **P < 0.01, ***P < 0.001 MP compared to 8 weeks old WT mice | |
| D | One-way ANOVA | F9, 140 = 23.03 | P < 0.001 | ***P < 0.001 MP compared to 8 weeks old WT mice | |
| Figure 5 | A | Two-way ANOVA | F1, 59 = 8.524 | P < 0.001 | ### p < 0.001 WT (1P/25Hz) vs. MP (1P/25Hz) |
| Two-way ANOVA | F1, 56 = 10.56 | P =0.002 | $ $ $ p < 0.001 WT (10P/25Hz) vs. MP (10P/25Hz) | ||
| One-way ANOVA | F10, 140 = 122.4 | P < 0.001 | *** p < 0.001 WT vs. MP | ||
| B | Two-way ANOVA | F1, 53 = 3.138 | P = 0.082 | ### p < 0.001 WT (1P/25Hz) vs. MP (1P/25Hz) | |
| Two-way ANOVA | F1, 56 = 6.862 | P = 0.011 | $ $ $ p < 0.001 WT (10P/25Hz) vs. MP (10P/25Hz) | ||
| One-way ANOVA | F10, 140 = 122.4 | P < 0.001 | *** p < 0.001 WT vs. MP | ||
| C | Linear regression |
F 1, 122 = 93.2 | P < 0.0001 | Equation Y = 865.1*X + 23.59 R2 = 0.4331, R = 0.65 (1P/25Hz) |
|
| Linear regression |
F 1, 94 = 53.82 | P < 0.0001 | Equation Y = 671.9*X + 30.49 R2 = 0.3641, R = 0.63 (10P/25Hz) |
||
| D | Linear regression |
F 1, 122 = 121.4 | P < 0.0001 | Equation Y = 2140*X − 395.0 R2 = 0.4988, R = 0.7 (1P/25Hz) |
|
| Linear regression |
F 1, 122 = 142.7 | P < 0.0001 | Equation Y = 968.8*X − 360.4 R2 = 0.5391, R = 0.73 (10P/25Hz) |
||
| E | Two-way ANOVA | F1, 59 = 8.524 | P < 0.001 | ### p < 0.001 WT (1P/25Hz) vs. MP (1P/25Hz) | |
| Two-way ANOVA | F1, 56 = 10.56 | P =0.002 | $ $ $ p < 0.001 WT (10P/25Hz) vs. MP (10P/25Hz) | ||
| One-way ANOVA | F10, 139 = 32.57 | P < 0.001 | *** p < 0.001 WT vs. MP | ||
| F | Two-way ANOVA | F1, 53 = 3.138 | P = 0.082 | ### p < 0.001 WT (1P/25Hz) vs. MP (1P/25Hz) | |
| Two-way ANOVA | F1, 56 = 6.862 | P = 0.011 | $ $ $ p < 0.001 WT (10P/25Hz) vs. MP (10P/25Hz) | ||
| One-way ANOVA | F10, 139 = 32.57 | P < 0.001 | *** p < 0.001 WT vs. MP | ||
| G | Linear regression |
F 1, 122 = 55.28 | P < 0.0001 | Equation Y = 175.9*X + 39.51 R2 = 0.3118, R = 0.55 (1P/25Hz) |
|
| Linear regression |
F 1, 122 = 53.14 | P < 0.0001 | Equation Y = 146.9*X + 33.44 R2 = 0.3034, R = 0.55 (10P/25Hz) |
||
| H | Linear regression |
F 1, 122 = 77.66 | P < 0.0001 | Equation Y = 452.9*X − 52.74 R2 = 0.3890, R = 0.62 (1P/25Hz) |
|
| Linear regression |
F 1, 122 = 83.26 | P < 0.0001 | Equation Y = 201.4*X − 42.34 R2 = 0.4056, R = 0.63 (10P/25Hz) |
||
| Figure 6 | A | Two-way ANOVA | F1, 59 = 8.524 | P < 0.001 | ### p < 0.001 WT (1P/25Hz) vs. MP (1P/25Hz) |
| Two-way ANOVA | F1, 56 = 10.56 | P =0.002 | $ $ $ p < 0.001 WT (10P/25Hz) vs. MP (10P/25Hz) | ||
| One-way ANOVA | F10, 140 = 25.04 | P < 0.001 | * p < 0.05, ** p < 0.01, *** p < 0.001 WT vs. MP | ||
| B | Two-way ANOVA | F1, 53 = 3.138 | P = 0.082 | ### p < 0.001 WT (1P/25Hz) vs. MP (1P/25Hz) | |
| Two-way ANOVA | F1, 56 = 6.862 | P = 0.011 | $ $ $ p < 0.001 WT (10P/25Hz) vs. MP (10P/25Hz) | ||
| One-way ANOVA | F10, 140 = 25.04 | P < 0.001 | * p < 0.05, ** p < 0.01, *** p < 0.001 WT vs. MP | ||
| C | Linear I regression |
F 1, 122 = 142.2 | P < 0.0001 | Equation Y = 106.0*X − 9.917 R2 = 0.5383, R = 0.73 (1P/25Hz) |
|
| Linear regression |
F 1, 122 = 146.8 | P < 0.0001 | Equation Y = 90.37*X − 14.80 R2 = 0.5461, R = 0.73 (10P/25Hz) |
||
| D | Linear regression |
F 1, 122 = 88.01 | P < 0.0001 | Equation Y = 215.5*X − 42.38 R2 = 0.4191, R = 0.64 (1P/25Hz) |
|
| Linear regression |
F 1, 122 = 138.6 | P < 0.0001 | Equation Y = 105.7*X − 45.89 R2 = 0.5319, R = 0.72 (10P/25Hz) |
||
| E | Two-way ANOVA | F1, 53 = 3.138 | P = 0.082 | ### p < 0.001 WT (1P/25Hz) vs. MP (1P/25Hz) | |
| Two-way ANOVA | F1, 56 = 10.56 | P =0.002 | $ $ $ p < 0.001 WT (10P/25Hz) vs. MP (10P/25Hz) | ||
| One-way ANOVA | F10, 140 = 91.89 | P < 0.001 | *** p < 0.001 WT vs. MP | ||
| F | Two-way ANOVA | F1, 53 = 3.138 | P = 0.082 | ### p < 0.001 WT (1P/25Hz) vs. MP (1P/25Hz) | |
| Two-way ANOVA | F1, 56 = 6.862 | P = 0.011 | $ $ $ p < 0.001 WT (10P/25Hz) vs. MP (10P/25Hz) | ||
| One-way ANOVA | F10, 140 = 91.89 | P < 0.001 | *** p < 0.001 WT vs. MP | ||
| G | Linear regression |
F 1, 122 = 344.6 | II P < 0.0001 | Equation Y = 65.44*X − 17.62 R2 = 0.7385, R = 0.86 (1P/25Hz) |
|
| Linear regression |
F 1, 122 = 453.9 | III P < 0.0001 | Equation Y = 57.24*X − 21.58 R2 = 0.7882, R = 0.89 (10P/25Hz) |
||
| H | Linear regression |
F 1, 122 = 248.7 | IV P < 0.0001 | Equation Y = 143.8*X − 41.97 R2 = 0.6709, R = 0.81 (1P/25Hz) |
|
| Linear regression |
F 1, 122 = 386.8 | P < 0.0001 | Equation Y = 66.65*X − 40.99 R2 = 0.7602, R = 0.87 (10P/25Hz) |
||
| Figure 7 | C | One-way ANOVA | F9, 66 = 80.17 | P < 0.001 | *** p < 0.001 WT vs. MP |
| D | One-way ANOVA | F9, 66 = 134.7 | P < 0.001 | *** p < 0.001 WT vs. MP | |
| Figure 8 | A | One-way ANOVA | F9, 141 = 17.56 | P < 0.001 | Tonic: *** p < 0.001 WT vs. MP |
| One-way ANOVA | F9, 145 = 18.49 | P < 0.001 | Phasic: ** p < 0.01, *** p < 0.001 WT vs. MP | ||
| B | One-way ANOVA | F9, 133 = 12.52 | P < 0.001 | Tonic: ** p < 0.01, *** p < 0.001 WT vs. MP | |
| One-way ANOVA | F9, 133 = 14.11 | P < 0.001 | Phasic: * p < 0.05, ** p < 0.01, *** p < 0.001 WT vs. MP | ||
| Figure5-1 | A | Two-way ANOVA | F9, 242 = 72.32 | P < 0.001 | *** p < 0.001 WT vs. MP |
| B | Two-way ANOVA | F9, 234 = 13.16 | P < 0.001 | ** p < 0.01, *** p < 0.001 WT vs. MP | |
| C | Two-way ANOVA | F8, 208 = 12.25 | P < 0.001 | * p < 0.05, *** p < 0.001 WT vs. MP | |
| D | Two-way ANOVA | F9, 235 = 9.380 | P < 0.001 | ** p < 0.01, *** p < 0.001 WT vs. MP | |
ANOVA followed by a Bonferroni post hoc test for multiple comparisons
Results
Tonic and phasic stimulation-evoked dopamine release in striatum and NAc shell in MitoPark mice at 9 different ages reveals details of functional losses.
We used ex vivo brain slice FSCV measures of DA release evoked by different stimulations to generate input/output (I/O) curves in striatum and the shell part of NAc. Our technique is similar to that used e.g. by Zhang et al (Zhang 2012) ex vivo to monitor DA release in adult and ageing mice, and Bergstrom and Garris to monitor DA release in vivo after unilateral 6OHDA lesions (Bergstrom 2003). Because recordings were carried out ex vivo in slices (rather than in vivo) recordings could be carried out with very high anatomical precision and reproducibility between animals not only for dorsal striatum, but also with high precision for the shell of NAc. Moreover, the recordings were free from the effects of anesthesia seen in in vivo recordings. The input/output curves showed that tonic stimulation required a 3 volt stimulus to cause robust DA release which leveled off at 5 volts, whereas phasic stimulation caused a larger release of DA in striatum, and also markedly in NAc shell with increasing stimuli up to 10 volts in controls and younger MitoPark mice (Fig 3).
Tonic stimulation showed that striatal DA release curves were decreased from 10 weeks of age in MitoPark mice, compared to WT mice (Fig. 3A). A stronger suppression of striatal tonic stimulation-induced releases was found from 12 weeks of age in MitoPark mice (Fig. 3A). Using the stronger phasic stimulation protocol we found no impairment in 6 week old MitoPark mice, a tendency at 8 weeks and clear, increasingly severe effects in the input/output curves from 10 to 24 weeks of age (Fig 3B). In agreement with previous studies (Ekstrand et al. 2007) the DA nerve terminals in striatum, as identified by tyrosine hydroxylase (TH) immunoreactivity, were almost completely lost in 20 week old MitoPark mice (data not shown).
Dopaminergic transmission in the shell of nucleus accumbens was also surveyed by ex vivo FSCV. We found that tonic DA release was significantly affected in MitoPark animals from 12 weeks of age (Fig. 3C) while phasic DA release in the NAc shell portion appeared to be affected from 10 weeks of age (Fig. 3D). Phasic stimulation in the NAc shell portion showed differences between MitoPark mice of different ages more clearly than tonic stimulation. The stronger phasic stimulation led to a markedly larger release of DA from NAc shell, which increased with voltage increases from 3 to 10 volts. Interestingly, there was a tendency for the phasic stimulation-induced DA release to be somewhat larger in 6 weeks old MitoPark mice than in control mice. This may relate to the fact that behavioral activities (locomotion and rearing) are also somewhat increased in 6 week old MitoPark mice (Ekstrand & Galter 2009, Galter et al. 2010), perhaps due to DA release from degenerating DA terminals and/or loss of reuptake sites.
Time course of declining evoked DA release differs between striatum and NAc shell.
To compare the magnitude of the evoked DA release deficits in the nigrostriatal and mesocortical limbic systems, the timelines for FSCV data for tonic and phasic release evoked by a fixed stimulation intensity (10 volts) in striatum and the NAc shell portion in MitoPark mice were compared. Tonic and phasic release decreased significantly from 10 weeks in striatum (Figs. 4A,B) in MitoPark mice. A similar decrease was significant 2 weeks later in NAc shell with both the tonic and the phasic stimulation protocols (Figs. 4C,D). Using phasic stimulation, the marginally significant decrease noted in 10 week old mice shown with the input-output curves (Fig. 3D, two-way ANOVA), was not seen in Fig. 4D (one-way ANOVA) although the mean 10 week level was lower than at 8 weeks and higher than at 12 weeks. Taken together, this indicates that impaired release of DA may begin around 10 weeks also in NAc shell. The phasic DA releases in NAc shell decreased more slowly than in striatum. The overall marked decrease of induced DA release between 10 and 12 weeks of age suggests a critical time during which the lack of TFAM leads to severe energy failure in DA neurons. Representative evoked DA release and cyclic voltammetry traces of declining evoked DA signals in striatum and NAc shell of WT and MitoPark mice at different ages are also shown (Figs. 4E,F).
Fig. 4. Stimulation-induced dopamine release in striatum declines gradually with age in MitoPark mice.
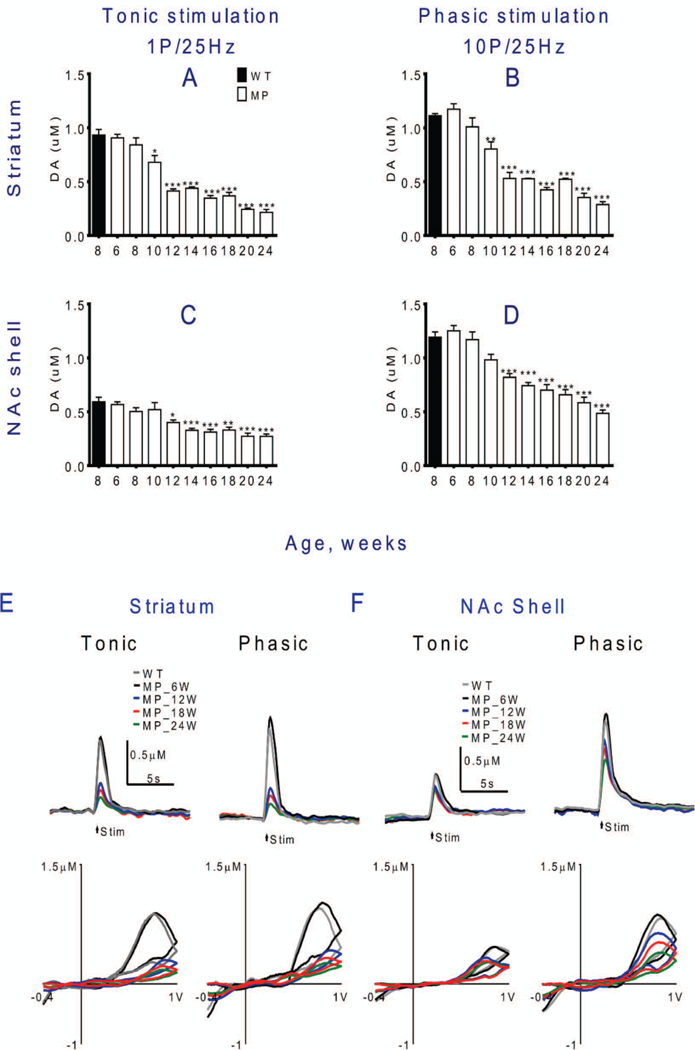
(A) In striatum, tonic and (B) phasic stimulation-induced DA release are both decreased significantly by 10 weeks of age (stim. intensity 10 V). (C) In NAc shell, the decline of tonic release was not significant until 12 weeks of age. (D) Phasic DA release also decreased more slowly in NAc than in striatum, with significant decline only at 12 weeks and later. Representative IT and CV traces of evoked DA signals in (E) striatal slices and (F) NAc shell slices at each stage in MitoPark and WT mice. Mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, compared to 8 weeks old wild type mice. For statistical details see Table 2.
Rotarod performance is upheld in MitoPark mice until 14 weeks of age, despite severe lack of DA release.
To detect coordination and balance functions, MitoPark and control WT mice were subjected to rotarod tests with both fixed and accelerating speeds. MitoPark mice performed as well as wild type mice up to 14 weeks of age on the fixed, as well as the accelerating speed tests. At 16 weeks of age, MitoPark mice were severely impaired in both these tests, and the impairment further increased from 16 to18 weeks (fixed speed: Fig 5A; accelerating speed: Fig. 5E). Comparing the DA release with the rotarod test, it was obvious that decline of striatal DA release, elicited by either tonic or phasic stimulation, precedes the shortening of latency to fall off the rotarod (fixed speed: Fig 5A, accelerating speed Fig. 5E). Linear regression analysis showed that both tonic and phasic DA release were nevertheless modestly positively correlated to latency to fall in either fixed speed or accelerated rotarod tests (Fig. 5C,G).
In the NAc shell, the decrease in tonic DA release occurred later than in striatum and the decline curve was shallow. The decline of phasic DA release in MitoPark mice was almost linearly correlated with animal age. Hence impairment of DA release in NAc shell also preceded impairment of fixed and accelerating rotarod performance (Fig. 5B,F). There was also a positive correlation between phasic DA release in NAc shell and latency to fall, but not between tonic release and latency to fall in this brain area (Fig. 5D,H).
Spontaneous locomotion and rearing decline when deficits in dopamine release are first detected.
We determined overall spontaneous motor activity and rearing, using open field movement and cylinder tests, to examine the relationship between these behaviors and declines in evoked DA release in MitoPark mice. Striatal DA release and distance covered during open field running both decreased as MitoPark mice aged (Fig. 6A). We found that the decline in DA release in response to either tonic or phasic stimulation paralleled the reduced running distances as animals aged. Linear regression analysis showed a good correlation between DA release reduction and impaired movement (Fig. 6C). Cylinder test data showed that rearing in MitoPark mice also decreased as animals aged, and in parallel with the decreased DA release following tonic and phasic stimulation in striatum (Fig. 6E). A strong correlation was found between the decline of striatal DA release and decreased rearing in MitoPark mice (Fig. 6G).
DA release in the shell part of NAc declined gradually and more slowly than in striatum (Fig. 6B) with a correlation with overall locomotor activity only with phasic release (Fig. 6D). The decline of tonic stimulation-induced DA release was small in NAc shell and occurred later than in striatum (Fig. 6B,F). Both tonic and phasic stimulation-induced decline of DA release correlated with the decreased number of rearings in the cylinder test (Fig. 6F,H).
Dopamine release probability and clearance decline with age in MitoPark mice.
DA release probabilities were expressed as the slope of the linear regression line (see methods for calculations; Fig. 7A,B). The values of the slopes in striatum were higher than those in NAc shell portion, which may have resulted from significant frequency augmentation in striatum (Rice & Cragg 2004, Zhang & Sulzer 2004). Both striatal and NAc shell slopes decreased as mice aged, and rapidly so in striatum (Fig. 7C). The decrease in release probability in NAc shell was more gradual (Fig. 7D).
The DA clearance rates were significantly prolonged in MitoPark mice, and occurred earlier in striatum, compared with the NAc shell. The clearance rate decreased by 12 weeks of age in striatum (Fig. 8A) while a significant decrease could not be found before 16 weeks of age in the NAc shell portion (Fig. 8B).
PET imaging demonstrates a marked decrease of DA transporter binding in MitoPark mice.
PET scans with [18F]FE-PE2I, a radioligand used to image the DA transporter (DAT), showed decreased binding in right and left striata in 12 week old MitoPark mice and very low binding in 16 and 20 week age old MitoPark mice (Fig. 9). These observations provide an independent measure of the progressive loss of striatal dopaminergic nerve terminals in MitoPark mice, confirming the loss of DA neurons in our mouse model in which TFAM is deleted in DAT-expressing neurons.
Fig. 9. PET shows decrease of dopamine transporter in MitoPark striatum.
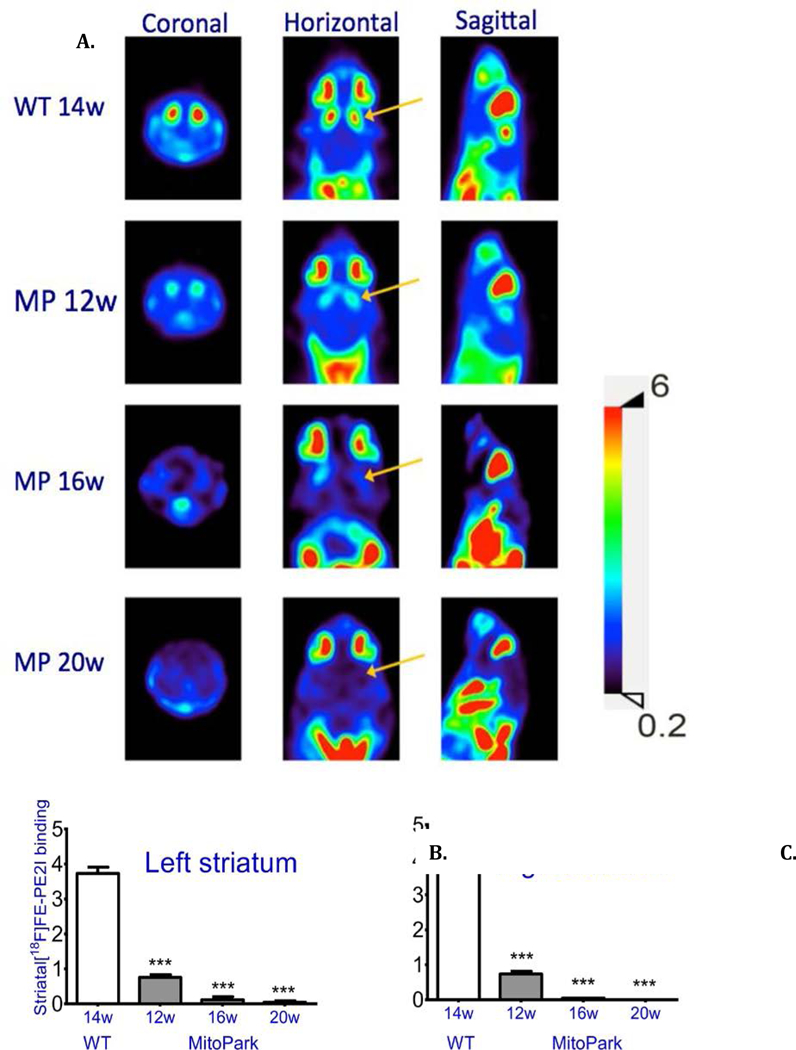
Top: DA transporter ligand binding in a WT and three MitoPark (MP) mice at different ages is shown in coronal, horizontal and sagittal planes. Bottom: Quantification of ligand binding in striatum on the left and right side. The signal in cerebellum was used as a basal background level. Mean +/− SEM; ***P < 0.001. WT n=2, MP 12 w n=3, MP 16 w n=3, MP 20w n= 3.
Discussion
Parkinson’s disease pathophysiology includes motor and non-motor deficits (Beitz 2014, Kalia & Lang 2015), the major histopathology being degeneration of the ascending DA systems from substantia nigra and the ventral tegmental area in mesencephalon to caudate, putamen and accumbens, and to cortex cerebri and limbic structures, respectively. The loss of DA in patients with Parkinson’s disease reaches 95% in putamen and 80% in the caudate nucleus (Hornykiewicz 2001). In the latter the loss is uneven with < 10% remaining in the rostral part and ≈ 25% remaining in the caudal part (Cheng et al. 2010). As DA neurons die, the remaining neurons increase DA synthesis and are able to compensate for the loss of DA neurons in humans until ≈80% of the striatal DA has been lost (Hornykiewicz 2001, Marsden 1990). Since this occurs in putamen, but not always in the caudate nucleus, it is assumed that the motor deficits are coupled to DA loss in putamen, while the caudate loss of DA may impact cognition and other non-motor symptoms (Hornykiewicz 2001). Importantly, loss of those DA neurons in mesencephalon that provide extrastriatal innervation, including cortex cerebri, may also contribute to Parkinson symptoms (Rommelfanger & Wichmann 2010); (Smith & Villalba 2008).
In MitoPark mice, lack of the mitochondrial transcription factor TFAM in DA neurons leads to dysfunctional mitochondria, deficient respiratory chain function, and progressive cell death (Ekstrand et al. 2007). All DA neurons that express DAT are affected, including both those projecting to striatal and those projecting to extrastriatal regions of the brain. There is also a modest loss of DA neurons in the olfactory bulb of MitoPark mice, while the tubero-infundibular DA neurons, which do not have DAT, are spared. Thus the DA loss in the mouse model is somewhat similar to that in Parkinson patients.
The time course of pathology in MitoPark mice (Ekstrand et al 2007, Galter et al 2009, Good et al 2011 and Li et al 2013), including a marked decline of function and increased loss of DA cells at around 16 weeks, is similar to the time course previously observed in “MILON” mice, in which TFAM was deleted in forebrain neurons, as driven by the CaMKII promoter coupled to Cre recombinase (Sorensen et al. 2001). This suggests a time after which DA and non-DA neurons, respectively, with mitochondrial dysfunction and impaired oxidative phosphorylation, can no longer be compensated for by increased glycolysis and/or glial interactions. It has also been shown that anterograde axonal transport of mitochondria from the mesencephalic DA cell bodies is impaired in MitoPark mice, leading to a decreased supply of mitochondria in the target areas (Sterky et al. 2011). Thus energy-dependent processes become severely compromised not only in cell bodies but also in dopaminergic terminals, leading to their degeneration in striatum and NAc.
FSCV is a sensitive tool to measure evoked DA release in the brain (Jones et al. 1994, Budygin et al. 2000). Here, we compared impairment of evoked release of DA to two types of behavior, forced and voluntary, in MitoPark mice of different ages. As expected, release of DA was stronger with the phasic, compared with the tonic, evoked stimulation protocol. Both forms of evoked DA release declined with age in striatum and the shell of NAc. Interestingly, this decline of releasable DA was parallel to the decline of the two spontaneous behaviors, voluntary open field locomotion and rearing, beginning at 10 weeks of age. Individual DA neurons in MitoPark mice are probably impaired longer before they die, than DA neurons damaged by mechanical lesions, 6OHDA or MPTP. We showed that release of DA as well as the characteristic pacemaker activity is impaired already by 6–8 weeks in MitoPark DA neurons (Good et al 2011), which we suggest is the reason for to the early impairment of spontaneous behavior. For spontaneous low-stress activities, the DA system is presumably not called upon to increase activity to the same extent as when mice are challenged by higher-stress activities, such as rotarod performance. Thus, as long as the DA system operates at a “resting state” level, as during low-stress open field activity and rearing tests, less DA would be released in MitoPark than in control mice. Less resting state DA signaling may lower the motivation and/or locomotor control, explaining the early dampening effects on spontaneous open field exploration activity. In contrast, performance on the rotarod, which is stressful and demanding, presumably activates the DA system and allows sufficient amounts of DA to be released for the MitoPark mice to perform at control mouse levels for several weeks longer before becoming hampered by the progressive degeneration of the DA neurons.
In contrast to early decline of spontaneous behaviors, MitoPark mice managed to maintain normal forced behavior for at least 6 additional weeks as tested by ability to stay on a rod with constant or accelerating rotational speed. We speculate that the mice make increased efforts not to fall, including activating remaining DA systems more strongly, just as phasic stimulation has stronger effects than tonic stimulation. The fact that there comes a time between 15 and 16 weeks of age, when the denervation of the DA system is so severe that rotarod performance also becomes impaired, lends further support to the idea that the better performance during the previous weeks is due to increased activation of remaining DA terminals, rather than to non-dopaminergic mechanisms, and that there is a low DA release level below which forced behaviors begin to decline. Linear regression analysis (Fig.5 C, D and G, H), indicates that both tonic (red) and phasic (blue) release is correlated with the behavioral results. Moreover, in Fig.6C, D, G and H there is a stronger correlation with higher r values (064~0.89), which indicates that the impairments in tonic (1P/25Hz) and phasic (10P/25Hz) DA release and deficits in spontaneous behaviors can be considered correlated. These data indicate that spontaneous behaviors are declining in parallel with the decline of DA (Fig.6), while it is not until weeks later that DA releases are insufficient also for forced behaviors (Fig.5). This is akin to the well-known human situation, when patients with Parkinson’s disease are able to perform demanding motor activities only when the motivation and sensory input are sufficiently strong (Ko et al. 2013, Narayanan et al. 2013). Our methods thus appear sensitive enough to detect disturbed behavior at a time when release impairment is modest.
In models of Parkinson’s disease where genes known to cause or increase risk of Parkinson’s disease in humans are deleted or mutated in mice, other systems than the CNS DA neurons are also affected, as indicated by the specific expression patterns of the targeted genes. The severe behavioral disturbances found at later stages in MitoPark mice occur when only the DA system has been specifically targeted. We have previously demonstrated that the standard Parkinson drug levodopa has strong beneficial effects in MitoPark mice, but no effects are seen in control mice (Galter et al 2009). We conclude that loss of DA release is sufficient for these motor impairments and that secondary alterations on non-dopaminergic systems, such as DA receptor supersensitivity that occurs in the DA-denervated striatum (Ungerstedt 1971) are unable to usefully compensate, even though the degeneration here is a slowly progressive process. Our findings are relevant for the question if neuropathology of non-dopaminergic circuitry is secondary to the degeneration of DA neurons or a specific additional pathology seen in some, but not all forms of Parkinson’s disease.
In summary, in the context of Parkinsonian behavior, the present work uses a mouse Parkinson model targeting mitochondria in DA neurons, leading to progressive degeneration of ascending DA systems. We show how spontaneous behavior declines early, while strong motivation overcomes DA deficiency as long as the denervation is partial, and that this is accomplished by using the remaining DA system, rather than other compensatory mechanisms.
Acknowledgements
Supported by National Science Council of Taiwan (MOST105–2314-B-016–001-MY3), Tri-Service General Hospital of Taiwan (TSGH-C107–068, TSGH-C107–071, TSGH-C107–070), National Taiwan Defense Medical Center (MAB-107–022), US National Institutes of Health (NS094152), ERC Advanced Investigator grant (322744 LO), Swedish Research Council (K2012–62x-03185–42-4; LO), StratNeuro (LO), and Swedish Brain Foundation (LO).
Abbreviations:
- τ
Tau
- aCSF
artificial CSF
- ANOVA
analysis of variance
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CNS
Central Nervous System
- CV
Current vs. Voltage
- DA
Dopamine
- DAT
DA transporter
- ERC
European Research Council
- FSCV
Fast-Scan Cyclic Voltammetry
- I/O
Input/Output
- IT
Intensity Time
- MILON
Mitochondrial Late-Onset Neurodegeneration
- MP
MitoPark
- NAc
Nucleus Accumbens
- NDMC
National Defense Medical Center
- PET
Positron Emission Tomography
- RPM
Revolution(s) Per Minute
- SUR
Specific Uptake Ratio
- TFAM
Transcription Factor A, Mitochondrial
- TH
Tyrosine Hydroxylase
- VOI
Volumes of Interest
- WT
Wild Type
Footnotes
Conflict of interest
YHC, THH, TTK, JHK, KHM, KHM, EYKH, YCC, and BJH declare no competing financial interests. LO is a co-owner of a company that owns commercial rights to the MitoPark mouse.
--Human subjects --
Involves human subjects:
If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if it is a Review or Editorial, skip complete sentence => if No, include a statement: “ARRIVE guidelines were not followed for the following reason:”
(edit phrasing to form a complete sentence as necessary).
=> if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
References
- Alexander GE (2004) Biology of Parkinson’s disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues in clinical neuroscience, 6, 259–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitz JM (2014) Parkinson’s disease: a review. Frontiers in bioscience (Scholar edition), 6, 65–74. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H and Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell and tissue research, 318, 121–134. [DOI] [PubMed] [Google Scholar]
- Budygin EA, Kilpatrick MR, Gainetdinov RR and Wightman RM (2000) Correlation between behavior and extracellular dopamine levels in rat striatum: comparison of microdialysis and fast-scan cyclic voltammetry. Neuroscience letters, 281, 9–12. [DOI] [PubMed] [Google Scholar]
- Chen YH, Harvey BK, Hoffman AF, Wang Y, Chiang YH and Lupica CR (2008) MPTP-induced deficits in striatal synaptic plasticity are prevented by glial cell line-derived neurotrophic factor expressed via an adeno-associated viral vector. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 22, 261–275. [DOI] [PubMed] [Google Scholar]
- Chen YH, Huang EY, Kuo TT, Hoffer BJ, Miller J, Chou YC and Chiang YH (2017) Dopamine release in the nucleus accumbens is altered following traumatic brain injury. Neuroscience, 348, 180–190. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM and Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Annals of neurology, 67, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Reddy SP and Kleeberger SR (2006) Nrf2 defends the lung from oxidative stress. Antioxidants & redox signaling, 8, 76–87. [DOI] [PubMed] [Google Scholar]
- Delamarre A and Meissner WG (2017) Epidemiology, environmental risk factors and genetics of Parkinson’s disease. Presse medicale, 46, 175–181. [DOI] [PubMed] [Google Scholar]
- Deng H, Wang P and Jankovic J (2017) The genetics of Parkinson disease. Ageing research reviews, 42, 72–85. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI and Galter D (2009) The MitoPark Mouse - an animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism & related disorders, 15 Suppl 3, S185–188. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D et al. (2007) Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proceedings of the National Academy of Sciences of the United States of America, 104, 1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira M and Massano J (2017) An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta neurologica Scandinavica, 135, 273–284. [DOI] [PubMed] [Google Scholar]
- Galter D, Pernold K, Yoshitake T, Lindqvist E, Hoffer B, Kehr J, Larsson NG and Olson L (2010) MitoPark mice mirror the slow progression of key symptoms and L-DOPA response in Parkinson’s disease. Genes, brain, and behavior, 9, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good CH, Hoffman AF, Hoffer BJ et al. (2011) Impaired nigrostriatal function precedes behavioral deficits in a genetic mitochondrial model of Parkinson’s disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 25, 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good CH, Wang H, Chen YH, Mejias-Aponte CA, Hoffman AF and Lupica CR (2013) Dopamine D4 receptor excitation of lateral habenula neurons via multiple cellular mechanisms. The Journal of neuroscience : the official journal of the Society for Neuroscience, 33, 16853–16864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornykiewicz O (2001) Chemical neuroanatomy of the basal ganglia--normal and in Parkinson’s disease. Journal of chemical neuroanatomy, 22, 3–12. [DOI] [PubMed] [Google Scholar]
- Jones SR, Mickelson GE, Collins LB, Kawagoe KT and Wightman RM (1994) Interference by pH and Ca2+ ions during measurements of catecholamine release in slices of rat amygdala with fast-scan cyclic voltammetry. Journal of neuroscience methods, 52, 1–10. [DOI] [PubMed] [Google Scholar]
- Kalia LV and Lang AE (2015) Parkinson’s disease. Lancet, 386, 896–912. [DOI] [PubMed] [Google Scholar]
- Kawagoe KT, Zimmerman JB and Wightman RM (1993) Principles of voltammetry and microelectrode surface states. Journal of neuroscience methods, 48, 225–240. [DOI] [PubMed] [Google Scholar]
- Ko JH, Antonelli F, Monchi O, Ray N, Rusjan P, Houle S, Lang AE, Christopher L and Strafella AP (2013) Prefrontal dopaminergic receptor abnormalities and executive functions in Parkinson’s disease. Human brain mapping, 34, 1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden CD (1990) Parkinson’s disease. Lancet, 335, 948–952. [DOI] [PubMed] [Google Scholar]
- Narayanan NS, Rodnitzky RL and Uc EY (2013) Prefrontal dopamine signaling and cognitive symptoms of Parkinson’s disease. Reviews in the neurosciences, 24, 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi B, Martin KA, Li Y, Graves AR, Vollmer A, Olson L, Mensh BD, Karpova AY and Dudman JT (2015) Dopamine Is Required for the Neural Representation and Control of Movement Vigor. Cell, 162, 1418–1430. [DOI] [PubMed] [Google Scholar]
- Rice ME and Cragg SJ (2004) Nicotine amplifies reward-related dopamine signals in striatum. Nature neuroscience, 7, 583–584. [DOI] [PubMed] [Google Scholar]
- Rommelfanger KS and Wichmann T (2010) Extrastriatal dopaminergic circuits of the Basal Ganglia. Frontiers in neuroanatomy, 4, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti J, Adams CE, Burmeister J, Gerhardt GA and Zahniser NR (2002) Kinetic analysis of striatal clearance of exogenous dopamine recorded by chronoamperometry in freely-moving rats. Journal of neuroscience methods, 121, 41–52. [DOI] [PubMed] [Google Scholar]
- Smith Y and Villalba R (2008) Striatal and extrastriatal dopamine in the basal ganglia: an overview of its anatomical organization in normal and Parkinsonian brains. Movement disorders : official journal of the Movement Disorder Society, 23 Suppl 3, S534–547. [DOI] [PubMed] [Google Scholar]
- Sorensen L, Ekstrand M, Silva JP, Lindqvist E, Xu B, Rustin P, Olson L and Larsson NG (2001) Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. The Journal of neuroscience : the official journal of the Society for Neuroscience, 21, 8082–8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterky FH, Lee S, Wibom R, Olson L and Larsson NG (2011) Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proceedings of the National Academy of Sciences of the United States of America, 108, 12937–12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerstedt U (1971) Postsynaptic supersensitivity after 6-hydroxy-dopamine induced degeneration of the nigro-striatal dopamine system. Acta physiologica Scandinavica. Supplementum, 367, 69–93. [DOI] [PubMed] [Google Scholar]
- Wightman RM and Zimmerman JB (1990) Control of dopamine extracellular concentration in rat striatum by impulse flow and uptake. Brain research. Brain research reviews, 15, 135–144. [DOI] [PubMed] [Google Scholar]
- Zhang H and Sulzer D (2004) Frequency-dependent modulation of dopamine release by nicotine. Nature neuroscience, 7, 581–582. [DOI] [PubMed] [Google Scholar]