

Alveolar capillary dysplasia typically presents with neonatal pulmonary hypertension and early mortality. However, there is growing evidence for a subset of disease with atypical late onset and/or prolonged survival. Here we present the variable clinical, genetic, and pathology findings of four such patients.
First described in 1947, alveolar capillary dysplasia with misalignment of pulmonary veins (ACD) is a rare disease that causes neonatal onset pulmonary hypertension (PH).1 ACD is characterized by unique histologic findings on lung biopsy, including reduced number of pulmonary capillaries remote from the alveolar epithelium within thickened alveolar septa, dilated bronchial veins that function as venovenous shunts and course within the same adventitial sheath as pulmonary arteries (previously described as misaligned pulmonary veins), and pulmonary arterial muscularization.2 Although lung biopsy results remain the gold standard for diagnosis, mutations in FOXF1 were identified as pathogenic in 2009 and, to date, FOXF1 is the only unequivocally implicated gene.3,4 Because FOXF1 regulates mesenchymal development, these patients often also have extrapulmonary anomalies including congenital heart, gastrointestinal, and urological defects.3
ACD typically has a fulminant neonatal presentation with complete mortality obviating the opportunity for potential life saving lung transplantation.5 Although the pulmonary arterial bed may be acutely responsive to pulmonary vasodilators, capillary obstruction leads to severe pulmonary edema and respiratory failure. However, there is a subset of patients with an atypical, delayed presentation with prolonged lung transplant-free survival.6–10 The disease pathophysiology for these atypical patients remains poorly understood, but the most widely accepted hypothesis is a “patchy” presentation wherein some areas of the lung are normal and provide an early compensatory mechanism.11 Here we report 4 patients with atypical ACD and widely variable clinical courses, with three harboring FOXF1 mutations and the fourth harboring a genomic deletion predicted to impact FOXF1 expression.
Methods
Clinical histories, results of echocardiography, chest CT, cardiac catheterizations, and genetic testing of all patients with diagnosis of atypical ACD between January 2014 – December 2017 at our institution were abstracted from review of electronic medical records. This study was conducted under Children’s Hospital of Philadelphia institutional review board approved protocols with written informed consent obtained from all parents. Variant pathogenicity was assessed in silico using MutationTaster, VarSome, and PredictSNP2 accessed in June 2018.12–14
Histologic evaluation of open lung biopsy was performed according to institutional protocol. Formalin fixed specimens were sectioned and stained using H&E or CD31 to highlight vascular endothelium. Explanted (patients 1 and 2) or postmortem lungs (patient 4) were further assessed for severity and uniformity of bronchovascular and alveolar anomalies using sections from multiple lung lobes. Radial alveolar count was performed for biopsy and explanted or post-mortem tissue sections and compared with published reference ranges.15
Clinical presentations
Patient 1 was a full term male with hypoxia at birth and a presumed diagnosis of persistent pulmonary hypertension of the newborn (PPHN) given bidirectional shunting across a patent ductus arteriosus (PDA) that responded to a 5-day course of supplemental oxygen. He tolerated spontaneous PDA closure and remained well until presenting with fulminant PH at age three months with suprasystemic pulmonary artery pressure by echocardiogram and pure right to left shunting across a reopened PDA. Chest CT demonstrated nonspecific diffuse ground glass opacities, peribronchial and interlobular septal thickening. ACD was suspected and later confirmed by lung biopsy results at age 4 months (Figure 1, A; available at www.jpeds.com) after he developed severe pulmonary edema with treatment with 40 parts per million (ppm) inhaled nitric oxide (iNO). Subsequent FOXF1 sequencing demonstrated a novel, likely pathogenic (c. 286G>T, p. Val96Leu) mutation. A cardiac catheterization demonstrated severe pulmonary arterial hypertension with a mean pulmonary artery pressure (mPAP) of 43mmHg and an indexed pulmonary vascular resistance (PVRi) of 6.5 indexed Wood units (WU*m2); at that time, the PDA was enlarged and stented to enhance pressure unloading of the right ventricle. Although he developed severe pulmonary edema at his initial presentation, he tolerated a lower dose of iNO (20 ppm) prior to PDA stenting without developing pulmonary edema and was transitioned to intravenous treprostinil following stent placement. By 6 months of age he developed progressive worsening of pulmonary edema treated with diuretics and discontinuation of all pulmonary vasodilators. Following this, he tolerated a slow wean of dopamine and was discharged at eight months of age only on oral diuretics. He required multiple brief readmissions for mild respiratory illnesses, until age 24 months of age when he developed worsening right ventricular dysfunction that responded to dopamine and sildenafil. He remained on these medications until undergoing bilateral lung transplantation at age 26 months. He died 13 months posttransplant (age 39 months) in the setting of chronic rejection and adenovirus and Pseudomonas pneumonia/lung abscess.
Figure 1; online.
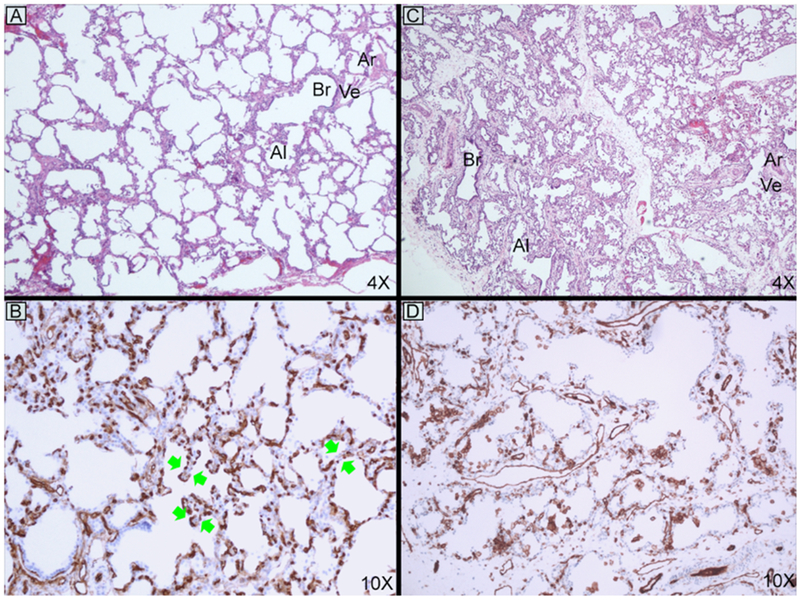
Open lung biopsies with H&E and CD31 staining of patient 1 (A and B) age 4 months and patient 3 (C and D) age 5 weeks. Typical features of ACD are seen in both patients, but are much milder in patient 1. The atypical finding of double-layered capillaries is noted with green double arrows. Abbreviations: alveolus (Al), arteriole (Ar), bronchiole (Br), and vein (Ve).
Patient 2 was a full term infant with tachypnea at birth that was attributed to transient tachypnea of the newborn. An echocardiogram at eight days of age demonstrated severe PH with bowing of the ventricular septum into the left ventricle and right to left shunting across a small PDA. His tachypnea improved with iNO, but ACD was suspected after he developed pulmonary edema on higher doses (>30 ppm). At 6 weeks of age, chest CT and cardiac catheterization were performed on the same day as an open lung biopsy that confirmed ACD (Figure 2, A). The chest CT showed nonspecific dependent atelectasis and scattered air trapping but otherwise normal lung parenchyma. Catheterization revealed pulmonary arterial hypertension (mPAP 34 mmHg and PVRi 8.6 WU*m2) with acute vasoreactivity to oxygen and iNO (mPAP 19 mmHg and PVRi 4.9 WU*m2). FOXF1 sequencing revealed a de novo, novel, and pathogenic FOXF1 mutation (c. 266A>G, p. Tyr89Cys). Clinically, he demonstrated inconsistent response to pulmonary vasodilator therapy with episodes of pulmonary edema. He was discharged to home at age two months on 0.5 L nasal cannula oxygen, but presented two days later in pulmonary hypertensive crisis that recovered with recanalization and stenting of the ductus arteriosus tract. Progressive worsening of his pulmonary vascular disease was evident by gradual widening of the ductal saturation gradient to 20% prior to successful bilateral lung transplantation at 4 months of age with ongoing survival through 25 months.
Figure 2.
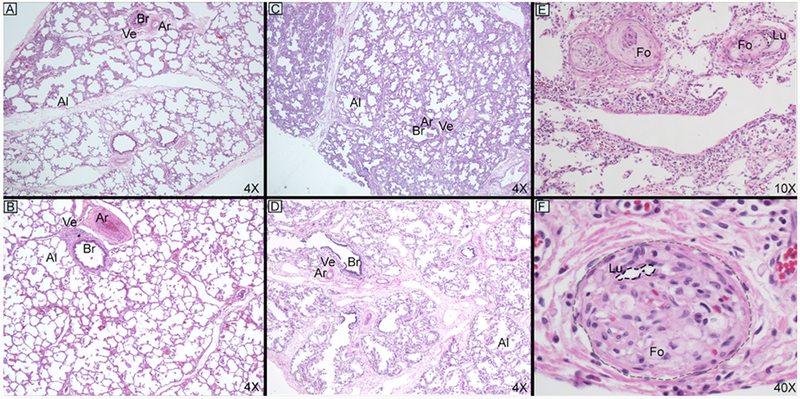
Open lung biopsies and explant or post-mortem with H&E staining for patient 2 (A and B) ages 5 weeks/4months and patient 4 (C – F) ages 3 weeks/10 months. Both patients demonstrate interval alveolar expansion but to a lesser extent than patient 4. Patient 4 also demonstrates a very unusual finding of severe intimal expansion with foamy macrophages in many vessels with near complete obliteration of the arteriolar lumen (E and F, outlined in black dashed lines with margins of vessel outlined in gray dashed line). Abbreviations: alveolus (Al), arteriole (Ar), bronchiole (Br), foamy macrophage (Fo), lumen (Lu), and vein (Ve).
Patient 3 was born full term and was asymptomatic until presenting at three weeks of age with nonspecific emesis, hypoglycemia and metabolic acidosis that responded to rehydration. She had a similar presentation again at five weeks of age. Given the recurrent acidosis, an echocardiogram was performed and demonstrated severe PH with bowing of the ventricular septum into the left ventricle. A chest CT illustrated small, multifocal areas of ground glass opacification in the lung parenchyma and a 1.5 mm PDA. Lung biopsy findings confirmed a diagnosis of ACD (Figure 1, C; available at www.jpeds.com) and cardiac catheterization demonstrated severe pulmonary arterial hypertension (mPAP 63 mmHg and PVRi 15 WU*m2), with a robust response to acute vasodilator testing (mPAP 38 mmHg and PVRi 5.8 WU*m2). She has remained clinically responsive to pulmonary vasodilator therapy (sildenafil, treprostinil) without developing pulmonary edema. FOXF1 sequencing demonstrated a novel likely pathogenic frameshift mutation (c.965delC, p. Pro322fs). She was later discharged at 2 months of age on subcutaneous treprostinil, sildenafil, and diuretics without supplemental oxygen. A subsequent cardiac catheterization at 16 months of age demonstrated significantly improved pulmonary vascular disease (baseline on therapy PVRi 3.6 WU*m2). At the time of this report, she is 26 months old living at home, clinically well, and is not actively listed for transplant.
Patient 4 was a full term infant who developed respiratory distress on the second day of life that progressively worsened. An echocardiogram at eight days of age demonstrated suprasystemic right ventricular pressure by ventricular septal bowing, and right to left shunting across a trivial PDA. A chest CT identified no abnormalities of lung parenchyma. Initially PPHN was the presumed diagnosis, but ACD was suspected and confirmed by lung biopsy findings at age 4 weeks (Figure 2, C) after he developed pulmonary edema on iNO. Cardiac catheterization demonstrated severe pulmonary arterial hypertension (mPAP 65 mmHg and PVRi 18.4 WU*m2) with positive response to acute vasodilator testing (mPAP 43 mmHg and PVRi 10.7 WU*m2). During the recovery from these procedures, he developed a pleural effusion requiring drainage and he was treated with systemic steroids, sildenafil, bosentan, and subcutaneous treprostinil with a favorable clinical response. He was discharged from the hospital at 3.5 months of age on these therapies with dose adjustments as needed in the setting of recurrent pulmonary edema. He later had a neonatal respiratory distress gene panel (ABCA3, FOXF1, NKX2-1, SFTPB, and SFTPC) performed, which did not identify any pathogenic variants or deletions/duplications. However, a chromosomal microarray demonstrated a 2.35MB 16q23.3q24.1 microdeletion that spanned 50 genes (22 in OMIM database, Table I; available at www.jpeds.com) not including any FOXF1 coding exons, but did include a FOXF1 enhancer and the expressed long non coding RNAs (lncRNAs) LINC010801 and LINC010802 which regulate FOXF1 expression.10 At 7 months of age he was admitted for respiratory distress and increased oxygen requirement. He was listed for lung transplantation, but died on the waitlist at age 10 months from a bradycardic arrest in the setting of a pulmonary hypertensive crisis and bacterial pneumonia.
Table 1.
OMIM genes within genomic deletion identified in patient 4.
| Gene Symbol | Gene Name |
|---|---|
| MLYCD | malonyl-CoA decarboxylase |
| OSGIN1 | oxidative stress induced growth inhibitor 1 |
| SLC38A8 | solute carrier family 38 member 8 |
| MBTPS1 | membrane bound transcription factor peptidase, site 1 |
| DNAAF1 | dynein axonemal assembly factor 1 |
| TAF1C | TATA-box binding protein associated factor, RNA polymerase I subunit C |
| KCNG4 | potassium voltage-gated channel modifier subfamily G member 4 |
| WFDC1 | WAP four-disulfide core domain 1 |
| ATP2C2 | ATPase secretory pathway Ca2+ transporting 2 |
| COTL1 | coactosin like F-actin binding protein 1 |
| USP10 | ubiquitin specific peptidase 10 |
| CRISPLD2 | cysteine rich secretory protein LCCL domain containing 2 |
| ZDHHC7 | zinc finger DHHC-type containing 7 |
| KIAA0513 | KIAA0513 |
| FAM92B | family with sequence similarity 92 member B |
| GSE1 | Gse1 coiled-coil protein |
| GINS2 | GINS complex subunit 2 |
| EMC8 | ER membrane protein complex subunit 8 |
| COX4I1 | cytochrome c oxidase subunit 4I1 |
| IRF8 | interferon regulatory factor 8 |
| LINC01082 | long intergenic non-protein coding RNA 1082 |
| LINC01081 | long intergenic non-protein coding RNA 1081 |
Histopathology
Open lung biopsy results were diagnostic for all 4 patients in this series (Table 2, Figure 1, A and C, and Figure 2, A and C). Histologic features at biopsy and explant or postmortem assessments are summarized in Table 2. There was qualitative variation between the patients in this series and typical ACD, which are summarized below. Abnormal histology findings were most severe in patient 3 and closely resemble typical ACD. Patients 1 and 2 had the mildest histology in this series with generally mild or patchy findings characteristic of ACD. These two patients also had double-layered capillaries directly apposed to the basement membrane, which is uncommon for ACD but can be seen in bronchopulmonary dysplasia or occasionally in early post-natal life.16 The histology findings for Patient 4 were intermediate in severity. At explant (patient 1—age 26 months and patient 2—age 4 months) and postmortem (patient 4—age 10 months), the characteristic features of ACD were much less obvious (Figure 2). The radial alveolar count, an assessment of alveolarization, had normalized for patients 1 and 2 (9.3 and 8.0, respectively) but remained abnormal for patient 4. The most striking finding for patient 4 was marked intimal expansion of many small pulmonary arteries and arterioles comprised of either loose concentric intimal expansion or numerous foamy macrophages incorporated into the intima (Figure 2, E and F). Superimposed on these changes were recent platelet aggregates with very early organization.
Table 2.
Summary of histologic findings on initial biopsy and available explant or postmortem assessments.
| Histologic Findings at Diagnosis and Explant or Autopsy | |||||
|---|---|---|---|---|---|
| Diagnostic biopsy | Explant/ Post-mortem | ||||
| Patient | Alveolar Simplification | Capillary features | Dilated Shunt Veins | Arteriolar Muscularization | Lung development and capillarization |
| 1 | Mild and patchy | Most capillaries in a single layer adjacent to epithelium with patchy double-layering | About half of examined BVBs | Moderate | 26 months – Normalized RAC, patchy foci with thickened alveolar septa and misaligned veins |
| 2 | Mild and patchy | Most capillaries in a single layer adjacent to epithelium with patchy double-layering | About half of examined BVBs | Moderate to severe | 4 months – Normalized RAC, rare foci with thickened alveolar septa and patchy misaligned veins |
| 3 | Severe | Frequent to nearly uniform remote capillaries and reduced number | Most BVB | Severe | N/A |
| 4 | Mild but uniform | Most capillaries in a single layer adjacent to epithelium but decreased in density | Most BVB | Moderate | 10 months – Improved alveolarization but abnormal RAC, patchy normal capillaries. Obliteration of arteriolar lumen from intimal expansions and foam cells. |
Abbreviations: bronchovascular bundles (BVBs) and radial alveolar count (RAC)
Genetics
All three of the FOXF1 exonic variants reported here are novel, have not been noted in the gnomAD project and are all predicted to be likely pathogenic or pathogenic according to ACMG classification and in silico tools MutationTaster and PredictSNP2.12–14,17
Discussion
The subset of ACD patients with delayed presentation and/or prolonged survival is incompletely understood. Here we report four cases with presentation of fulminant PH ranging from 8 to 120 days, all surviving beyond the neonatal period. Two patients underwent lung transplantation, one died awaiting transplantation and a fourth remains clinically well with sustained clinical response to pulmonary vasodilators. Three had confirmed FOXF1 mutations and one a genomic deletion affecting lncRNA regulators of FOXF1 (Table 3; available at www.jpeds.com). This expands the literature to ten such cases in addition to at least eight others without reported FOXF1 gene testing.5–11,18
Table 3, online.
Clinical and genetic summaries of all reported atypical ACD/MPV patients with FOXF1 impacting variants.
| Clinical and Genetic Summaries for Patients with Identified Genetic Causes of Atypical ACD/MPV | |||||||
|---|---|---|---|---|---|---|---|
| Age at Px | Preceding Respiratory Symptoms | Acute PVD testing | Clinical PVD Response | Genetic | Extrapulmonary Defects | Outcome | Ref |
| 120 days | PPHN | Not performed | Partial -Responsive to Sildenafil in late course | FOXF1 p.V96L | Duodenal atresia | Transplant – 26 months | Pt 1 |
| 8 days | TTN | PVRi 8.6 to 4.9 WU*m2 | Partial - Intermittent pulmonary edema | FOXF1 p.Y89C | Hirschsprung’s disease | Transplant – 4 months | Pt 2 |
| 35 days | None | PVRi 15.0 to 5.8 WU*m2 | Complete – No pulmonary edema | FOXF1 p.P322fs | Intestinal nonrotation | Transplant-free-survival – 26 months | Pt 3 |
| 8 days | PPHN | PVRi 18.4 to 10.7 WU*m2 | Partial -Intermittent pulmonary edema | 16q23.3q24.1 del including LINC01082 and LINC010801 | Immunodeficiency Hydrocephalus | Death prior to transplant 10 months | Pt 4 |
| 7 months | Respiratory distress at birth | Not reported | Partial – Initial positive clinical response but decompensation at 14 months. | De novo maternal 16q24.1 deletion affecting LINC010802 | None | Transplant – 15 months | 10 |
| 5 months | None | Not reported | Complete – Clinically responsive to iNO, IV and PO PVDs | FOXF1 p.L300fs | None | Transplant-free survival-38 months. | 8 |
| Birth | N/A | Reported positive | Managed with iNO | FOXF1 p.P49Q | Posterior urethral valves | Transplant – 20 months | 11 -Pt 2 |
| 67 days | Poor feeding | Not reported | Managed with sildenafil and iNO | 16q23.3q24.1 1.5 MB deletion | None | Transplant – 5 months | 11 - Pt 4 |
| 212 days | Tachypnea and mild hypoxia | Reported Positive | Managed with sildenafil and iNO | FOXF1 p.P126L | None | Transplant – 15 months | 11 - Pt 6 |
Abbreviations: alveolar capillary dysplasia/misalignment of pulmonary veins (ACD/MPV), inhaled nitric oxide (iNO), per oral (PO), persistent pulmonary hypertension of the newborn (PPHN), pulmonary hypertension (PH), pulmonary vasodilators (PVD), transient tachypnea of the newborn (TTN), indexed Wood units (WU*m2)
The patients reported here suggest a wide spectrum of severity exists in ACD histopathology and clinical presentation. At initial biopsy, the histopathology for most patients in this series was milder than seen in typical ACD. Interestingly, this was not the case for patient 3, who had the most profound changes and yet the most benign clinical course of the patients reported, with an unusual sustained favorable response to pulmonary vasodilator therapy. We hypothesize that “atypical” patients meet a critical threshold for areas of normal lung parenchyma and/or overall mildness of ACD features allowing for early survival and may be affected by growth of “normal” lung tissue relative to the growth of the diseased areas. The combined pre- and post-capillary obstruction in ACD creates a management paradox as pulmonary vasodilators to treat elevated pulmonary vascular resistance can lead to poorly tolerated pulmonary edema.19 We speculate that areas with thin alveolar walls and normal capillary pattern may be more responsive to pulmonary vasodilators, leading to preferential recruitment in acute management and proliferative expansion of these areas if treatment can be maintained without development of substantial pulmonary edema. This mechanism is supported by a recent case series of six patients with atypical ACD who were treated with pulmonary vasodilators prior to transplantation between four and 20 months of age and had explant pathology demonstrating similar patterns of non-uniform abnormal lung histogenesis.11 However, because of this variability, lung biopsy is not a reliable way to establish likely outcome. We also identified double-layered capillaries in patients 1 and 2, which has not previously been reported in ACD, but can be seen in bronchopulmonary dysplasia and may have reduced the severity of postcapillary obstruction in these patients.
Three patients in this series had mild or transient symptoms at birth, which resulted in preliminary diagnoses of PPHN or transient tachypnea of the newborn. We surmise that the combination of patchy areas of normal lung parenchyma, the relative balance between pre- and post-capillary obstruction in a spectrum of disease severity, and the well-described postnatal transition in pulmonary vascular resistance in normal arterioles may explain this slower disease progression. However, it should also be noted that in an autopsy series of patients with a presumptive diagnosis of PPHN all six neonatal mortalities had features of ACD on histology.20 Thus, mild initial symptoms may be common in typical and atypical ACD, and the overall incidence may be under recognized.
The post-mortem lung histology for patient 4 demonstrated findings not previously reported in any patient with ACD, consisting of obliteration of some arteriolar lumina from loose intimal expansion, foamy macrophages incorporated into the intima, and early platelet organization. Although the luminal foamy macrophages have never been reported, the early platelet organization can be seen in chronic, severe pulmonary arterial hypertension such as that seen with unrestrictive left to right shunts from uncorrected congenital heart defects.21 It is possible this finding represents an aspect of end stage disease that few patients with ACD reach given its natural history.
The molecular mechanisms for non-uniform abnormal development in atypical ACD are poorly understood. It is possible that these specific mutations result in less disrupted FOXF1 protein expression or activity. In our cohort, we identified a frameshift mutation, two missense mutations that lie in the DNA binding domain, and a genomic deletion that included LINC010801 and LINC010802, which have been previously implicated in both FOXF1 regulation and ACD.10 In contrast to our patient 4 who had an intermediate early clinical course, in that prior report, a patient with neonatal mortality from ACD had loss of both lncRNAs, and prolonged survival was observed in another patient with loss of LINC010802 when the lung-specific LINC010801 remained intact. Because FOXF1 is a dosage-sensitive protein expressed from a gene that exhibits incomplete paternal imprinting in the lung with about 35% paternal allele expression, high level lung mosaicism or paternally inherited variants may also be contributors.22,23 Mosaic heterozygous FOXF1 p.H89Q mutations were previously reported in an asymptomatic mother with 29% expression in peripheral blood compared with 56% in one of her multiple affected children.24 Mosaic lung expression was not tested in that family, but it is possible that patchy areas of abnormal lung parenchyma in atypical ACD may align with mosaic mutant expression. Finally, FOXF1 variants in ACD are predominantly identified on the maternal allele.4,25 In contrast, in reports of FOXF1 variants occurring on the paternal allele there is a wide spectrum of disease from no discernible lung pathology to ACD with prolonged survival or ACD with neonatal mortality.18,26
In conclusion, this series supports that delayed presentation, prolonged survival, and variable response to pulmonary vasodilators may be attributable to patchy lung involvement at birth and postnatal expansion of normal parenchyma. Although most atypical ACD patients, including those in our series, have milder findings on diagnostic biopsy, it remains difficult to predict which patients will have a mild course or clinically respond to pulmonary vasodilator therapy based on lung biopsy histology or specifics of genetic testing.11 This report reinforces the importance of considering ACD in any infant presenting with PH, especially in the setting of vasodilator-induced pulmonary edema, and the utility of early genetic testing.
Supplementary Material
Acknowledgments
Supported by NIH (5T32HL007915 [to JJ.E.], 2T32GM008638-21 [to C.M.], K12-HD043245, K08-HL140129), and Parker B. Francis Foundation Fellowship (to D.F.). The authors declare no conflicts of interest.
Abbreviations:
- ACD
Alveolar capillary dysplasia
- PVRi
indexed pulmonary vascular resistance
- mPAP
mean pulmonary artery pressure
- PPHN
persistent pulmonary hypertension of the newborn
- PCWP
pulmonary capillary wedge pressure
- PH
pulmonary hypertension
- ppm
parts per million
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Portions of this study were presented at the CHOP Cardiology conference, 2018.
Data sharing: After manuscript is accepted for publication, we will submit these variants to dbSNP and DECIPHER.
References
- 1.Mac MH. Congenital alveolar dysplasia of the lungs. Bull New Engl Med Cent. 1947; 9: 48. [PubMed] [Google Scholar]
- 2.Eulmesekian P, Cutz E, Parvez B, Bohn D, Adatia I. Alveolar capillary dysplasia: a six-year single center experience. J Perinat Med. 2005; 33: 347–52. [DOI] [PubMed] [Google Scholar]
- 3.Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009; 84: 780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szafranski P, Gambin T, Dharmadhikari AV, Akdemir KC, Jhangiani SN, Schuette J, et al. Pathogenetics of alveolar capillary dysplasia with misalignment of pulmonary veins. Hum Genet. 2016; 135 : 569–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michalsky MP, Arca MJ, Groenman F, Hammond S, Tibboel D, Caniano DA. Alveolar capillary dysplasia: a logical approach to a fatal disease. J Pediatr Surg. 2005; 40: 1100–5. [DOI] [PubMed] [Google Scholar]
- 6.Abdallah HI, Karmazin N, Marks LA. Late presentation of misalignment of lung vessels with alveolar capillary dysplasia. Crit Care Med. 1993; 21: 628–30. [DOI] [PubMed] [Google Scholar]
- 7.Ahmed S, Ackerman V, Faught P, Langston C. Profound hypoxemia and pulmonary hypertension in a 7-month-old infant: late presentation of alveolar capillary dysplasia. Pediatr Crit Care Med. 2008; 9: e43–6. [DOI] [PubMed] [Google Scholar]
- 8.Ito Y, Akimoto T, Cho K, Yamada M, Tanino M, Dobata T, et al. A late presenter and long-term survivor of alveolar capillary dysplasia with misalignment of the pulmonary veins. Eur J Pediatr. 2015; 174: 1123–6. [DOI] [PubMed] [Google Scholar]
- 9.Shankar V, Haque A, Johnson J, Pietsch J. Late presentation of alveolar capillary dysplasia in an infant. Pediatr Crit Care Med. 2006; 7: 177–9. [DOI] [PubMed] [Google Scholar]
- 10.Szafranski P, Dharmadhikari AV, Wambach JA, Towe CT, White FV, Grady RM, et al. Two deletions overlapping a distant FOXF1 enhancer unravel the role of lncRNA LINC01081 in etiology of alveolar capillary dysplasia with misalignment of pulmonary veins. Am J Med Genet A. 2014; 164A: 2013–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Towe CT, White FV, Grady RM, Sweet SC, Eghtesady P, Wegner DJ, et al. Infants with Atypical Presentations of Alveolar Capillary Dysplasia with Misalignment of the Pulmonary Veins Who Underwent Bilateral Lung Transplantation. J Pediatr. 2018;194: 158–64 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bendl J, Musil M, Stourac J, Zendulka J, Damborsky J, Brezovsky J. PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions. PLoS Comput Biol. 2016; 12: e1004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014; 11: 361–2. [DOI] [PubMed] [Google Scholar]
- 14.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooney TP, Thurlbeck WM. The radial alveolar count method of Emery and Mithal: a reappraisal 1--postnatal lung growth. Thorax. 1982; 37: 572–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker CD, Alvira CM. Disrupted lung development and bronchopulmonary dysplasia: opportunities for lung repair and regeneration. Curr Opin Pediatr. 2014; 26: 306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The Genome Aggregation Database (gnomAD) [Available from: http://gnomad.broadinstitute.org.
- 18.Reiter J, Szafranski P, Breuer O, Perles Z, Dagan T, Stankiewicz P, et al. Variable phenotypic presentation of a novel FOXF1 missense mutation in a single family. Pediatr Pulmonol. 2016; 51: 921–7. [DOI] [PubMed] [Google Scholar]
- 19.Steinhorn RH, Cox PN, Fineman JR, Finer NN, Rosenberg EM, Silver MM, et al. Inhaled nitric oxide enhances oxygenation but not survival in infants with alveolar capillary dysplasia. J Pediatr. 1997; 130: 417–22. [DOI] [PubMed] [Google Scholar]
- 20.Tibballs J, Chow CW. Incidence of alveolar capillary dysplasia in severe idiopathic persistent pulmonary hypertension of the newborn. J Paediatr Child Health. 2002; 38: 397–400. [DOI] [PubMed] [Google Scholar]
- 21.Dishop MK. Diagnostic Pathology of Diffuse Lung Disease in Children. Pediatr Allergy Immunol Pulmonol. 2010; 23: 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szafranski P, Dharmadhikari AV, Brosens E, Gurha P, Kolodziejska KE, Zhishuo O, et al. Small noncoding differentially methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder. Genome Res. 2013; 23: 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dharmadhikari AV, Sun JJ, Gogolewski K, Carofino BL, Ustiyan V, Hill M, et al. Lethal lung hypoplasia and vascular defects in mice with conditional Foxf1 overexpression. Biol Open. 2016; 5: 1595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luk HM, Tang T, Choy KW, Tong MF, Wong OK, Lo FM. Maternal somatic mosaicism of FOXF1 mutation causes recurrent alveolar capillary dysplasia with misalignment of pulmonary veins in siblings. Am J Med Genet A. 2016; 170: 1942–4. [DOI] [PubMed] [Google Scholar]
- 25.Alsina Casanova M, Monteagudo-Sanchez A, Rodiguez Guerineau L, Court F, Gazquez Serrano I, Martorell L, et al. Maternal mutations of FOXF1 cause alveolar capillary dysplasia despite not being imprinted. Hum Mutat. 2017;38:615–20. [DOI] [PubMed] [Google Scholar]
- 26.Sen P, Gerychova R, Janku P, Jezova M, Valaskova I, Navarro C, et al. A familial case of alveolar capillary dysplasia with misalignment of pulmonary veins supports paternal imprinting of FOXF1 in human. Eur J Hum Genet. 2013;21:474–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.