

The spinocerebellar ataxias (SCAs) are a heterogeneous group of autosomal dominantly inherited progressive ataxia disorders, with a variable clinical presentation. More than 40 gene loci and mutations have been identified so far; however, an expansion of a CAG nucleotide repeat is the cause of the most common SCAs (SCA1, 2, 3, and 6).1 SCA12 is considered rather distinct given that it presents with a characteristic action tremor in the upper limbs, often mistaken for essential tremor. Cerebellar ataxia and other neurological features may occur later in the disease course.2, 3
Here, we report on a patient with genetically confirmed SCA12, who presented with atypical parkinsonism even featuring an abnormal DaTSCAN.
Case
A 76‐year‐old right‐handed man, originally from Punjab, with a 20‐year history of tremor affecting his hands during action, recently sought medical attention because, over the last 3 years, his tremor had worsened and he had become slower in day‐to‐day activities. Moreover, his balance had deteriorated and his speech had become slurred. He also had experienced some mild memory difficulties and some urinary dysfunction, that is, urgency and incontinence at times. He had a remarkable family history, with his mother, 5 of his brothers, and 2 of his sisters being similarly affected by tremor. In 1 sibling, there was also marked speech difficulty, making him barely intelligible. Clinical examination at that time revealed a bilateral rest and action hand and head tremor associated with other parkinsonian signs and eye movements’ abnormalities, namely vertical upgaze restriction (Video 1, Segment 1). Treatment with levodopa 300 mg/day did not yield any benefit. Previous investigations comprised a brain MRI, reported as normal (apart from age‐related atrophy), and a DaTSCAN, which showed reduced uptake in both putamina, particularly on the left (Fig. 1). In the view of the clinical picture, the family history suggesting an autosomal‐dominant disorder and his ethnic origin (Agrawal community), genetic testing for SCA12 was performed and detected a heterozygous pathogenic expansion at the SCA12 locus (56 CAG repeats).
Figure 1.
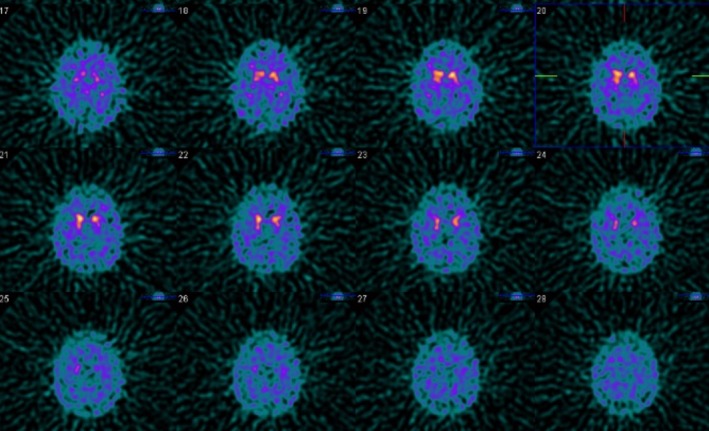
Abnormal DaTSCAN: reduced tracer uptake in both putamina, particularly on the left.
In the last 3 years, the disease has moderately progressed. l‐dopa was stopped because of poor response, and, currently, clinical examination shows more severe extrapyramidal signs compared to before, and in particular generalized bradykinesia, clear supranuclear gaze palsy, and severely impaired postural reflexes, with inability to stand or walk without assistance. He also has dysarthric speech and clear upper limb dysmetria (Video 1, Segment 2). Additionally, the family reported apathy and cognitive dysfunction with deterioration of both attention and memory.
Discussion
SCA12 is caused by the expansion of CAG repeats in the protein phosphatase 2 regulatory subunit Bβ (PPP2R2B) gene at position 32 on the long arm of chromosome (5q32), with 43 CAG repeats considered as the diagnostic threshold.2 Age at onset ranges from 8 to 56 years, but the disease usually manifests in the fourth decade of life. The highest prevalence of SCA12 is in India, where it is the second‐most common SCA.4 The presence of a common founder for SCA12 in India has been demonstrated, and, interestingly, most of the Indian cases stem from the Agrawal community.5 Yet, rarely, SCA12 mutations do occur also in other populations.2 Clinically, it is characterized by an unusual tremor and a possible later onset of dystonia, subtle parkinsonian features, gait ataxia, and cognitive decline.3, 4, 6 Brain imaging typically reveals both cerebral and cerebellar atrophy with relative sparing of brainstem, thalamus, and basal ganglia.7 Nevertheless, absence of atrophic changes has been reported in genetically confirmed cases.2
To the best of our knowledge, this is the first case of SCA12 presenting with atypical parkinsonism and featuring an abnormal DaTSCAN. The phenotype with a supranuclear vertical gaze palsy and postural instability resembled progressive supranuclear palsy (PSP), although the prominent tremor affecting not only the hands, but also the head would be unusual for classic PSP. Signs typically observed in PSP have not been detected on MRI, but considering that motor deficit correlates with the degree of midbrain atrophy in PSP, we cannot exclude that they would have appeared later in the course of the disease.
Whether the presence of parkinsonism in this SCA12 case might be coincidental remains an open question; however, we do not believe this is the case. Parkinsonian features may be common in SCA12,3 and, apart from SCA6, which gives rise to a pure cerebellar syndrome, the other SCAs are multisystemic disorders characterized by a variety of non‐cerebellar symptoms, including movement disorders.8 Furthermore, substantia nigra degeneration has been proven in SCA2 and SCA3 patients,9 in which parkinsonian features evolve rarely, but, when present, can be l‐dopa responsive.8 Interestingly, regardless of the nigrostriatal involvement, some SCAs present with parkinsonian features and others do not. It has been proposed that lesions in the motor territory of the subthalamic nucleus may prevent the manifestation of parkinsonism in SCA2 and SCA3,9 and also that cerebellar dysfunction may counteract the manifestation of parkinsonism.10
In summary, here we presented a patient with SCA12 mutation manifesting with a PSP‐like phenotype. Presence of a prominent and atypical tremor, the strong family history, and the ethnic origin have guided us to the genetic diagnosis. However, the final diagnosis has not been pathologically confirmed. Interestingly, a SCA12 case with Creutzfeldt‐Jakob disease (CJD) pathology has been reported,11 but whether the presence of the two was coincidental is uncertain. Nevertheless, a link between CJD and SCA12 might be the protein phosphatase, PP2A, of which the SCA12 gene encodes for a specific subunit and that may play a part in tau phosphorylation. We therefore cannot exclude that SCA12 could predispose to the occurrence of tau pathology.
This report might give new insight on this rare condition and help in the differential diagnosis of atypical parkinsonism chameleons and mimics, given that it may suggest SCA12 for the differential diagnosis of “atypical atypical parkinsonism” with PSP phenotype.12
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Manuscript: A. Writing of the First Draft, B. Review and Critique.
A.L.: 1A, 1B, 1C, 2A
C.D.G.: 1C, 2B
E.M.: 1C, 2B
B.B.: 2B
F.B.: 2B
K.P.B.: 1A, 2B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. The patient has given written and informed consent for online publication of his videos.
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: B.B. is supported by an EAN research fellowship and by the Bosch Foundation. F.B. is supported by a Baasch‐Medicus stipend. K.P.B. serves on the editorial boards of Movement Disorders and Therapeutic Advances in Neurological Disorders; receives royalties from the publication of Oxford Specialist Handbook of Parkinson's Disease and Other Movement Disorders (Oxford University Press, 2008); received speaker honoraria and travel from GlaxoSmithKline, Ipsen, Merz Pharmaceuticals LLC, Novartis, and Sun Pharmaceutical Industries Ltd; received personal compensation for scientific advisory board for GSK and Boehringer Ingelheim; and received research support from Ipsen and from the Halley Stewart Trust through Dystonia Society UK, and the Wellcome Trust MRC strategic neurodegenerative disease initiative award (Ref. number WT089698), a grant from the Dystonia Coalition, and a grant from Parkinson's UK (Ref. number G‐1009).
Supporting information
Video S1. Segment 1 shows the patient at age 76. Vertical gaze palsy and slow and limited saccades, much more pronounced in the vertical than the horizontal plane; hypomimia; nodding head tremor and chin tremor; high‐amplitude bilateral rest, postural and kinetic tremor in the upper limbs; possible bradykinesia, more on the left than the right; slow and short stepped gait with freezing on turning; and impaired postural reflexes evidenced by a positive pull test. Segment 2 shows the patient 3 years later. The vertical gaze palsy has become more prominent; slow and limited horizontal saccades initiated by head thrust; hypomimia with reduced blinking rate; jaw tremor; generalized bradykinesia; bilateral arm tremor at rest, but also with a severe, not re‐emergent postural and kinetic component; dysmetria; short stepped gait and severely impaired postural reflexes; and festination with mild camptocormia.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Diallo A, Jacobi H, Cook A, et al. Survival in patients with spinocerebellar ataxia types 1, 2, 3, and 6 (EUROSCA): a longitudinal cohort study. Lancet Neurol 2018;17:327–334. [DOI] [PubMed] [Google Scholar]
- 2. Srivastava AK, Takkar A, Garg A, Faruq M. Clinical behaviour of spinocerebellar ataxia type 12 and intermediate length abnormal CAG repeats in PPP2R2B. Brain 2017;140:27–36. [DOI] [PubMed] [Google Scholar]
- 3. Choudhury S, Chatterjee S, Chatterjee K, et al. Clinical characterization of genetically dagnosed cases of spinocerebellar ataxia type 12 from India. Mov Disord Clin Pract 2018;5:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Srivastava AK, Choudhry S, Gopinath MS, et al. Molecular and clinical correlation in five Indian families with spinocerebellar ataxia 12. Ann Neurol 2001;50:796–800. [DOI] [PubMed] [Google Scholar]
- 5. Bahl S, Virdi K, Mittal U, et al. Evidence of a common founder for SCA12 in the Indian population. Ann Hum Genet 2005;69(Pt 5):528–534. [DOI] [PubMed] [Google Scholar]
- 6. Holmes SE, Hearn EO, Ross CA, Margolis RL. SCA12: an unusual mutation leads to an unusual spinocerebellar ataxia. Brain Res Bull 2001;56:397–403. [DOI] [PubMed] [Google Scholar]
- 7. Holmes SE, O'Hearn E, Margolis RL. Why is SCA12 different from other SCAs? Cytogenet Genome Res 2003;100:189–197. [DOI] [PubMed] [Google Scholar]
- 8. van Gaalen J, Giunti P, van de Warrenburg BP. Movement disorders in spinocerebellar ataxias. Mov Disord 2011;26:792–800. [DOI] [PubMed] [Google Scholar]
- 9. Schols L, Reimold M, Seidel K, et al. No parkinsonism in SCA2 and SCA3 despite severe neurodegeneration of the dopaminergic substantia nigra. Brain 2015;138(Pt 11):3316–3326. [DOI] [PubMed] [Google Scholar]
- 10. Haugarvoll K, Bindoff LA, Tzoulis C. Nigrostriatal denervation sine parkinsonism. Brain 2016;139(Pt 4):e25. [DOI] [PubMed] [Google Scholar]
- 11. Hellenbroich Y, Schulz‐Schaeffer W, Nitschke MF, et al. Coincidence of a large SCA12 repeat allele with a case of Creutzfeld‐Jacob disease. J Neurol Neurosurg Psychiatry 2004;75:937–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stamelou M, Quinn NP, Bhatia KP. “Atypical” atypical parkinsonism: new genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy‐a diagnostic guide. Mov Disord 2013;28:1184–1199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Segment 1 shows the patient at age 76. Vertical gaze palsy and slow and limited saccades, much more pronounced in the vertical than the horizontal plane; hypomimia; nodding head tremor and chin tremor; high‐amplitude bilateral rest, postural and kinetic tremor in the upper limbs; possible bradykinesia, more on the left than the right; slow and short stepped gait with freezing on turning; and impaired postural reflexes evidenced by a positive pull test. Segment 2 shows the patient 3 years later. The vertical gaze palsy has become more prominent; slow and limited horizontal saccades initiated by head thrust; hypomimia with reduced blinking rate; jaw tremor; generalized bradykinesia; bilateral arm tremor at rest, but also with a severe, not re‐emergent postural and kinetic component; dysmetria; short stepped gait and severely impaired postural reflexes; and festination with mild camptocormia.