

Abstract
Over the past decades, research has defined cAMP as one of the central cellular nodes in sensing and integrating multiple pathways and as a pivotal role player in lung pathophysiology. Obstructive lung disorders, such as chronic obstructive pulmonary disease (COPD), are characterized by a persistent and progressive airflow limitation and by oxidative stress from endogenous and exogenous insults. The extent of airflow obstruction depends on the relative deposition of different constituents of the extracellular matrix, a process related to epithelial‐to‐mesenchymal transition, and which subsequently results in airway fibrosis. Oxidative stress from endogenous and also from exogenous sources causes a profound worsening of COPD. Here we describe how cAMP scaffolds and their different signalosomes in different subcellular compartments may contribute to COPD. Future research will require translational studies to alleviate disease symptoms by pharmacologically targeting the cAMP scaffolds.
Linked Articles
This article is part of a themed section on Adrenoceptors—New Roles for Old Players. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.14/issuetoc
Abbreviations
- AKAP
A‐kinase anchoring protein
- COPD
chronic obstructive pulmonary disease
- ECM
extracellular matrix
- EMT
epithelial‐to‐mesenchymal transition
- Epac
exchange proteins directly activated by cAMP
- ERM
ezrin/radixin/moesin
- IPF
idiopathic pulmonary fibrosis
- ZO‐1
zonula occludens 1
- α‐SMA
α‐smooth muscle actin
1. INTRODUCTION
In this article, we highlight the most recent insights into the signalling pathways regulated by cAMP, one of the most ancient and important second messengers (Billington, Penn, & Hall, 2017). Novel aspects of cAMP scaffolds, which are maintained by a diverse subset of proteins including but not limited to receptors, exchange proteins, PDEs, and A‐kinase anchoring proteins (AKAPs), are also detailed. Our special focus is on epithelial‐to‐mesenchymal transition (EMT) and oxidative stress (Figure 1) in chronic obstructive pulmonary disease (COPD) and how cAMP scaffolds may contribute to alleviation of COPD symptoms and the potential role of these scaffolds in both health and disease conditions.
Figure 1.
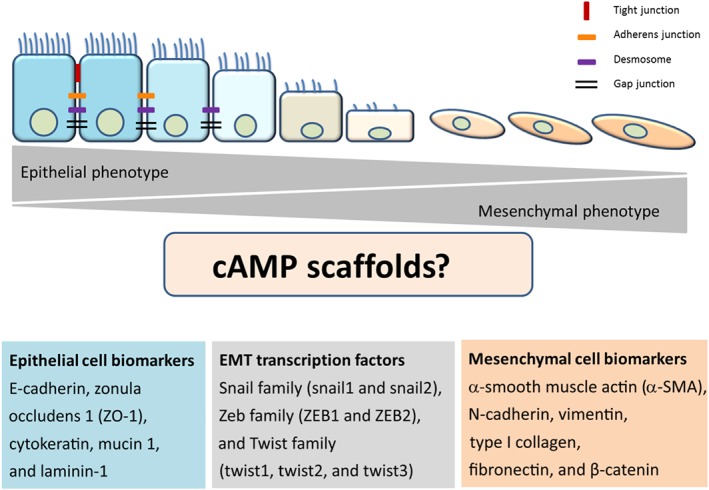
General outline of the epithelial‐to‐mesenchymal transition (EMT) and its potential link to cAMP scaffolds. The epithelial cell layer is maintained by cell–cell contacts through tight and adherens junctions, desmosomes, and gap junctions. The epithelial cell phenotype is identified by some known biomarkers, such as E‐cadherin, zonula occludens 1 (ZO‐1), cytokeratin, mucin 1, and laminin‐1. Transcription factors involved in the EMT process belong to Snail family (snail1 and snail2), Zeb family (ZEB1 and ZEB2), and Twist family (twist1, twist2, and twist3). Mesenchymal cell phenotype is characterized by α‐smooth muscle actin (α‐SMA), N‐cadherin, vimentin, type I collagen, fibronectin, and β‐catenin. For further details, see text
2. EPITHELIAL‐TO‐MESENCHYMAL TRANSITION
The cAMP signalling pathway is one of the many pathways that are implicated in EMT (Bartis, Mise, Mahida, Eickelberg, & Thickett, 2014; Jansen, Gosens, Wieland, & Schmidt, 2018; Jolly, Ware, Gilja, Somarelli, & Levine, 2017; Nieto, 2011). The EMT process comprises the loss of cell–cell junctions (tight junctions, desmosomes, and adherens junctions) and the loss of cell interactions with the basal membrane. EMT also involves the loss of apicobasal polarity, the change in cell shape from cuboidal to fibroblastoid, and the subsequent acquisition of migratory and invasive properties due to a loose organized morphology as demonstrated on a three‐dimensional extracellular matrix (ECM; López‐Novoa & Nieto, 2009; Nieto, 2011; Oldenburger, Poppinga, et al., 2014; Thiery, Acloque, Huang, & Nieto, 2009). In order to characterize the EMT process, biomarkers including the epithelial cell biomarkers E‐cadherin and zonula occludens 1 (ZO‐1) and the mesenchymal cell biomarkers α‐smooth muscle actin (α‐SMA) and β‐catenin are used. Next to biomarkers, transcription factors including family members of Snail, Zeb, and Twist (Figure 1) are also used to characterize the EMT process (Kalluri & Weinberg, 2009; Thiery et al., 2009).
TGF‐β1 is the best known inducer of EMT (Gonzalez & Medici, 2014; Lamouille, Xu, & Derynck, 2014). TGF‐β1 treatment of rat alveolar epithelial cells increased expression of mesenchymal cell markers, such as α‐SMA, type I collagen, vimentin, and desmin, whereas expression of epithelial markers aquaporin‐5, ZO‐1, and cytokeratin was reduced (Willis et al., 2005). The central role of TGF‐β1 signalling in the process of EMT is supported by its ability to induce its own expression and subsequently lead to an increase in its release following induction by a variety of growth factors and cytokines such as IL‐6 and IL‐8. It is generally believed that these TGF‐β1‐driven, “feedforward” mechanisms act in concert with a distinct subset of external cellular cues to efficiently regulate down‐regulation of epithelial markers and up‐regulation of mesenchymal markers, which are crucial characteristics of EMT (Tan, Olsson, & Moustakas, 2015). A process known as mesenchymal‐epithelial transition (MET) is linked to the transition of primary mesenchymal cells to secondary epithelial cells (Acloque, Adams, Fishwick, Bronner‐Fraser, & Nieto, 2009).
2.1. Classification of the distinct stages of the EMT process
Principally, three different types of EMT have been identified on the basis of their distinct cellular phenotypes and responses (Kalluri & Weinberg, 2009). Type I EMT is primarily linked to epithelial cell phenotypical alterations during gastrulation and embryonic formation, and it is essentially characterized by transition of primitive epithelial cells to primary mesenchymal cells (Kim et al., 2017). Type II EMT is associated with a phenotypical change of secondary epithelial cells to fibroblasts and is stimulated by damage and local inflammation, which occurs primarily in mature tissue during tissue repair (wound healing), tissue regeneration, and organ fibrosis. Type II EMT is characterized by the ability of epithelial cells to migrate into interstitial spaces (Kim et al., 2017; Zeisberg & Neilson, 2009). During tissue repair, inflammatory stimuli such as TGF‐β, TNF‐α, and IL‐1β promote the formation of fibroblasts in a process referred to as fibrosis. Fibrosis is characterized by an excessive deposition of collagens, elastin, tenacin, and other ECM molecules. Persistent inflammation therefore induces fibrosis and permanent organ damage. Furthermore, type II EMT is not confined to epithelial cells but also occurs in endothelial cells, indicating that this process is crucial for tissue repair (Agarwal et al., 2016).
Type III EMT is involved in cancer progression and metastatic processes. Type III EMT is characterized by a phenotypical change of secondary epithelial cells into carcinoma cells with a high degree of migratory and invasive properties and malignant growth subsequently creating a novel tumour nodule (Kim et al., 2017). Tight junctions play an important role in type III EMT; particularly, E‐cadherin down‐regulation leads to a loss of cell–cell adhesion and thus facilitates migration and colonization of cancer cells (Rout‐Pitt, Farrow, Parsons, & Donnelley, 2018; Thiery et al., 2009). A recent study in A549 lung cancer cells demonstrated that the novel TGF‐β1 inhibitor compound 67 inhibited TGF‐β1‐induced down‐regulation of E‐cadherin mRNA and up‐regulation of N‐cadherin mRNA, with findings further confirmed on the protein level by using immunofluorescence (Jeong et al., 2019). The authors also reported that compound 67 reduced transmigration through TGF‐β1‐induced layer of ECM and in turn reduced matrigel invasion (a process commonly referred to as wound healing; Jeong et al., 2019). These findings highlight the importance of inhibiting the TGF‐β1 pathway in EMT and the necessity to comprehensively understand the subtle cellular alterations in the distinct stages of EMT.
3. POTENTIAL ROLE OF EMT IN COPD
As outlined above, EMT plays a vital role during organ fibrosis (Kalluri & Neilson, 2003; Kim et al., 2006; Zeisberg & Neilson, 2009), including pulmonary fibrosis (Chapman, 2011; Kim et al., 2006). In this regard, there is increasing interest in understanding the role of EMT in COPD as well. COPD represents a major global health problem with the ailment estimated to become the third leading cause of death and the fifth leading cause of disability by 2030 (Barnes et al., 2015; Laudette, Zuo, Lezoualc'h, & Schmidt, 2018). Cigarette smoke is implicated as the primary cause of COPD. However, factors like exposure to indoor pollution from biomass fuels and outdoor air pollution including occupational dusts particularly in developing countries also seem to contribute to disease progression (Barnes et al., 2015; Maji, Dikshit, Arora, & Deshpande, 2018; Vogelmeier et al., 2017; Wang et al., 2018).
Sohal et al. (2010) reported on fragmentation of the reticular basement membrane in large airways from endobronchial biopsies of smokers, and the findings positively correlated with the subjects' smoking history. Similar observations were reported from another study by comparing current smokers and ex‐smokers suffering from COPD, with healthy non‐smokers (Soltani et al., 2010). Intriguingly, using immunohistochemical staining for bronchial biopsy sections, it was demonstrated that the fibroblast protein marker S100A4 was significantly increased in cells within the reticular basement membrane clefts of smokers and COPD patients as compared with never‐smoking control subjects (Sohal et al., 2010). This finding was further confirmed with S100A4 and vimentin double staining, thereby indicating an active EMT process in the large airway of CODP patients. These findings were strongly correlated with cigarette smoke exposure (Sohal et al., 2010). In addition, Wang, Wang, Zhang, Zhang, and Xiao (2013) assessed the expression of the epithelial marker E‐cadherin and the mesenchymal marker vimentin in small airway epithelium from non‐smokers, smokers, non‐smokers with COPD, and smokers with COPD. Compared with non‐smokers, a dramatic increase of vimentin positive cells was detected in the small airway epithelium from smokers and COPD subjects, together with a marked decrease of E‐cadherin, thereby suggesting an active EMT process in small airway epithelium during the pathogenesis of cigarette smoke‐induced COPD (Wang et al., 2013). Even though EMT was found to be active in both large and small airways from COPD patients with chronic airflow limitation, EMT observed in small airways was uniformly less than that in large airways, thereby implying different mechanisms of EMT in small airways as compared with large airways (Mahmood et al., 2015). In small airways, it was considered as type II EMT (profibrotic, see above) rather than as type III EMT (malignancy associated, see above). This distinction was primarily based on the virtual lack of hypervascularization as studied by staining for type IV collagen (Mahmood et al., 2015).
The presence of EMT in COPD has been further identified in in vitro cell models. Compared with primary human bronchial epithelial cells from healthy controls, cells from COPD patients showed an up‐regulation of mesenchymal markers (α‐SMA, collagen type I, vimentin, and NOX4) and a down‐regulation of epithelial markers (E‐cadherin, ZO‐1, KRT5, and KRT18), suggesting that EMT was significantly increased in COPD patients (Milara, Peiró, Serrano, & Cortijo, 2013). Additionally, it was demonstrated that cigarette smoke activated the EMT process in isolated primary epithelial cells (Milara et al., 2013; Milara et al., 2014; Wang et al., 2013), findings which are in line with studies in epithelial cell lines such as A549 and BEAS‐2B (Eurlings et al., 2014; Shen et al., 2014). Indeed, BEAS‐2B cells, primary normal human bronchial epithelial cells and (rat) alveolar cells, were able to undergo EMT primarily induced by TGF‐β1, indicating that at least these lung epithelial cells retained the potential to transform into mesenchymal cells (Kamitani et al., 2011; Molloy et al., 2008; Willis et al., 2005).
EMT can be induced by environmental stresses or factors such as ROS. ROS have been implicated in COPD progression and exacerbations known as episodes of acute worsening of disease symptoms (Antus & Kardos, 2015; Bernardo, Bozinovski, & Vlahos, 2015; Kirkham & Barnes, 2013). Milara et al. (2013) demonstrated that preincubation of differentiated primary human bronchial epithelial cells with the antioxidants N‐acetyl‐l‐cysteine and apocynin inhibited cigarette smoke‐induced up‐regulation of mesenchymal markers (α‐SMA, vimentin, and collagen type I) and down‐regulation of epithelial markers (E‐cadherin, ZO‐1, KRT5, and KRT18) both mRNA and protein. These findings were further confirmed in a knockout mouse model of the transcription factor Nrf2 , a key regulator in the antioxidant defence system known to protect against oxidative stress (Zhou et al., 2016). Compared with wild‐type mice instilled with bleomycin, vimentin, α‐SMA, and collagen were further and significantly augmented in Nrf2‐knockout mice, whereas E‐cadherin protein was reduced, albeit not significantly (Zhou et al., 2016).
Another important process that is involved in EMT in COPD but far from being completely understood is the interaction between fibroblasts and epithelial cells. To gain more mechanistic insights, a recent study investigated the potential of conditioned medium derived from normal human lung fibroblasts and COPD human lung fibroblasts in inducing EMT in normal human bronchial epithelial cells and COPD human bronchial epithelial cells (Nishioka et al., 2015). Exposure of both normal and COPD human bronchial epithelial cells to conditioned medium from normal human lung fibroblasts induced vimentin mRNA, whereas N‐cadherin mRNA increased only in COPD human bronchial epithelial cells, indicating that COPD human bronchial epithelial cells had partly undergone EMT (Nishioka et al., 2015). In addition, normal human bronchial epithelial cells exposed to COPD human lung fibroblasts‐conditioned medium showed up‐regulation of E‐cadherin, N‐cadherin, and vimentin protein, thereby confirming that COPD human lung fibroblasts‐conditioned medium promoted EMT in normal human bronchial epithelial cells (Nishioka et al., 2015). The results from Nishioka and colleagues revealed that the interaction between fibroblasts and epithelial cells plays a crucial role in the EMT process in COPD, and future studies ought to unravel the molecular nature of this interaction.
As key effector cells during fibrosis, the accumulation of myofibroblasts may occur as a consequence of transition of resident fibroblasts to myofibroblasts, as a transition of airway smooth muscle cells to myofibroblasts, as a transition of epithelial cells to fibroblasts (and subsequently to myofibroblasts), or as a consequence of the recruitment of circulating fibroblastic stem cells (Karvonen et al., 2013; Milara et al., 2013; Scotton & Chambers, 2007). TGF‐β1 induced the transition of primary pulmonary fibroblasts from individuals with COPD to myofibroblasts, a process positively correlated with the severity of COPD (Baarsma et al., 2011). In addition, Karvonen et al. (2013) reported recently that (α‐SMA‐positive) myofibroblasts were variably localized in lungs from non‐smokers, smokers without COPD, and smokers with COPD, further suggesting that this cell type is linked to both lung regeneration and COPD development. Further investigation, however, is necessary in order to establish the extent to which EMT‐derived myofibroblasts contribute to fibrosis in COPD as current findings strongly indicate that distinct stages of EMT are active in airways of COPD patients and that cigarette smoke exposure and oxidative stress may play vital roles during this process.
4. COMPARTMENTALIZATION OF cAMP
Research on cyclic nucleotides was initiated early in 1953 by Earl Sutherland (Berthet, Rall, & Sutherland, 1957). For the past 60 years, the importance of cyclic nucleotides has been elucidated, resulting in more than five distinguished Nobel Prizes (Beavo & Brunton, 2002), including one that was awarded in 2012 to Robert J. Lefkowitz and Brian K. Kobilka for their contribution in unravelling the molecular topography of the β2‐adrenoceptor (Chung et al., 2011; Lefkowitz, Roth, & Pastan, 1970; Lefkowitz, Roth, Pricer, & Pastan, 1970; Rasmussen, Choi, et al., 2011; Rasmussen, DeVree, et al., 2011), strongly pointing to the importance of fundamental research to unravel the distinct molecular mechanisms underlying the signalling properties of cAMP.
Compartmentalization, a key feature of cAMP signalling, allows extracellular signals to propagate into the cells along defined and specific pathways within the network. Stimulation of prototypical Gs‐protein‐coupled receptors, such as the β2‐adrenoceptor and distinct prostanoid receptors, leads to the activation of adenylyl cyclases (ACs), which catalyse the synthesis of cAMP from ATP. cAMP is able to exert such diverse signalling properties by activating distinct effectors, which include PKA (Taylor, Knighton, Zheng, Ten Eyck, & Sowadski, 1992), the exchange proteins directly activated by cAMP (Epacs; Schmidt, Dekker, & Maarsingh, 2013), cyclic nucleotide‐gated ion channels (Biel & Michalakis, 2009; Kaupp & Seifert, 2002), and the most recently defined novel class of three‐pass transmembrane popeye domain‐containing proteins, which bind cAMP with a high affinity (Schindler & Brand, 2016). The intracellular concentration of cAMP is spatially and temporally controlled by PDEs, a superfamily of metallohydrases that hydrolyse cAMP to AMP and thereby terminate its signalling properties (Omori & Kotera, 2007). Additionally, the AKAPs, a group of structurally diverse proteins localized at specific subcellular sites, play a critical role in maintaining subcellular cAMP compartmentalization by generation of spatially discrete signalling complexes that create local gradients of cAMP (Beene & Scott, 2007; Skroblin, Grossmann, Schäfer, Rosenthal, & Klussmann, 2010). In addition to anchoring cAMP effectors such as PKA to distinct subcellular complexes, AKAPs also bind PDEs, phosphatases (which terminate phosphorylation), thereby contributing to spatial regulation of local and specific cAMP cellular processes.
4.1. Players of cAMP compartmentalization: AKAPs, PKA, and Epac
Members of the AKAPs family bind to the regulatory subunits of PKA and target PKA to discrete sites or macromolecular complexes, thereby playing a central role in the regulation of cAMP compartmentalization. In addition, compartmentalization of cAMP signalling by AKAP proteins plays a central role in pathological cellular responses, primarily due to the fact that the expression level of AKAPs is subject to profound changes under disease conditions (Poppinga, Muñoz‐Llancao, González‐Billault, & Schmidt, 2014; Tröger, Moutty, Skroblin, & Klussmann, 2012). It was reported recently that the mRNA of both AKAP5 and AKAP12 was reduced in the lung tissue of COPD patients, compared with lung tissue from controls (Poppinga et al., 2014). Similar findings were obtained in primary airway smooth muscle cells exposed to cigarette smoke (Poppinga et al., 2015). Because both AKAP5 and AKAP12 have been implicated in the recycling of β2‐adrenoceptors (see Poppinga et al., 2014), alterations in their expression profile under disease pressure (as shown in lung tissue from COPD patients) may alter the mode of action of β2‐adrenoceptor agonists. Such findings should be envisioned in the context of the current treatment regime of COPD, which relies mainly on bronchodilator therapy (β2‐adrenoceptor agonists, anticholinergics, and theophylline) and on PDE4 inhibitors used in concert with either corticosteroid or bronchodilator treatment especially in COPD patients with a high risk of exacerbations (Barnes et al., 2015; Maji et al., 2018; Vogelmeier et al., 2017; Wang et al., 2018). This hypothesis, however, still remains to be elucidated in additional COPD patient cohorts and/or additional experimental models of COPD. Previously, studies focused on the role of AKAPs in the airway epithelium. Exposure of human bronchial epithelial 16HBE14o− cells to cigarette smoke extract reduced the epithelial barrier, a process accompanied by a reduction of E‐cadherin and AKAP9 both of which colocalize at the cell membrane. Interestingly, E‐cadherin, but not AKAP9, protein expression was reduced in lung tissue from COPD patients, compared with controls. However, AKAP9 mRNA expression was decreased in primary bronchial epithelial cells from current smokers as compared with non‐smokers or ex‐smokers (Oldenburger, Poppinga, et al., 2014). The results pointed to a divergence between AKAP9 protein expression and mRNA expression potentially reflecting functional differences in the effects of E‐cadherin and AKAP9 on distinct EMT phenotypes. Taken together, the findings outlined above point to an alteration of the expression profile and subsequent change in function of some members of the AKAP family in experimental models of COPD.
The major cAMP effectors are PKA and Epac, which can act in concert or alone in several physiological processes. The guanine nucleotide exchange factor Epac family consists primarily of two members, Epac1 (cAMP‐GEF‐I) and Epac2 (cAMP‐GEF‐II), which are able to activate Ras‐like small GTPases. Epac proteins are known to regulate numerous biological responses, including but not limited to inflammation, cell proliferation, remodelling, and barrier functions (Grandoch, Roscioni, & Schmidt, 2010; Insel et al., 2012; Robichaux & Cheng, 2018; Schmidt et al., 2013). Exposure of airway smooth muscle cells to cigarette smoke extract reduced the protein expression of Epac1, but not Epac2, a process involving miRNA‐7. In addition, miRNA‐7 was increased in bronchial smooth muscle of COPD stage II patients isolated by laser dissection, as compared with controls (Oldenburger, van Basten, et al., 2014). In lung tissue from COPD patients, Epac1 protein expression was also reduced. The loss of Epac1 was associated with a higher degree of neutrophilic inflammation measured by an increase in NF‐κB‐dependent production of IL‐8 (Oldenburger et al., 2012). In Epac1‐deficient mice, higher levels of TGF‐β1 (mRNA), collagen I, and fibronectin (both mRNA and protein levels) were observed (Oldenburger, Timens, et al., 2014). In line with these findings, it has been reported that binding of Epac1 to the activated TGF‐β1 type I receptor subsequently decreased the phosphorylation of Smad2 and Smad2‐dependent transcription (Conrotto, Yakymovych, Yakymovych, & Souchelnytskyi, 2007), raising the possibility that Epac1 may potentially inhibit the production of collagen by binding to the TGF‐β1 type I receptor. Given the fact that cigarette smoke is one of the main inducers of COPD (Barnes et al., 2015; Maji et al., 2018; Vogelmeier et al., 2017; Wang et al., 2018), the findings outlined above point to a link between COPD and an impaired Epac1 signalling. As it is generally accepted that local inflammation contributes to TGF‐β1‐induced EMT, impaired Epac1 signalling may not only cause a higher deposition of ECM but may also worsen the process of chronic inflammation. Because Epac2 seems to act in a proinflammatory manner in lung tissue (Oldenburger, Timens, et al., 2014), Epac1 and Epac2 may cooperatively regulate the process of EMT induced by TGF‐β1. The development of Epac1‐ and Epac2‐specific inhibitors from 2012 onwards continues to foster research in this area (Parnell, Palmer, & Yarwood, 2015; Robichaux & Cheng, 2018; Schmidt et al., 2013). Studies in airway smooth muscle cells exposed to TGF‐β1 induced the de novo synthesis of ECM components, such as collagen types I, III, and IV, and fibronectin (Lambers et al., 2014). Treatment with long‐acting β2‐adrenoceptor agonists (formoterol and salmeterol) exclusively prevented the TGF‐β1‐induced de novo synthesis of a distinct subset of ECM components, specifically type I and type III collagen. Using the P‐site AC inhibitor 2′‐5′‐dideoxyadenosine, the authors reported on the intriguing finding that the de novo synthesis of ECM components distinctly depends on cAMP, suggesting that specific cAMP scaffolds are most likely operational in these structural lung cells (Lambers et al., 2014). It will therefore be of interest in the future to study the extent to which distinct ACs (Halls & Cooper, 2017), acting in concert with a distinct subset of PDEs, PKA, and Epac, may contribute to the differential regulation of ECM deposition. Even though such cAMP microdomains have been extensively studied in the cardiovascular system (Laudette et al., 2018; Musheshe, Schmidt, & Zaccolo, 2018), they still have to be defined in more detail in the pulmonary system.
4.2. Players of cAMP compartmentalization: PDE
The superfamily of PDEs comprises 11 family members and at least 21 isoforms with different splice variants (Page & Spina, 2012). PDEs hydrolyse cyclic nucleotides (cAMP and cGMP) to their respective inactive 5′‐monophosphates within subcellular microdomains, thereby leading to an organization of cyclic nucleotides signalling in time and space. PDE4, PDE7, and PDE8 are specific for cAMP, and particularly, PDE4 is the most widely studied PDE isozyme being evidently linked to compartmentalization of cAMP and thus to cellular signalling and adaptation (Conti et al., 2003; Manganiello, 2002). Oral administration of the PDE4 inhibitor roflumilast (1 or 5 mg·kg−1) prevented lung parenchyma destruction induced by cigarette smoke exposure for 5 days per week for 7 months in mice (Martorana, Beume, Lucattelli, Wollin, & Lungarella, 2005; Figure 2). These results point to PDE4 inhibition as a potential player in the prevention of lung tissue remodelling in experimental models of COPD. Also, these findings suggest that roflumilast acted by inhibiting the activation and recruitment of macrophages, which in turn prevented parenchymal destruction induced by chronic cigarette smoke exposure through a reduced release of metalloproteases from macrophages. In concert with these findings, Martorana et al. (2005) observed a reduced macrophage density in mice treated with roflumilast. Persistent inflammation is linked to a distinct subset of EMT; therefore, the prevention of inflammation by the PDE4 inhibitor might be of benefit for different phenotypes of EMT.
Figure 2.

The role of cAMP scaffolds in TGF‐β‐induced epithelial‐to‐mesenchymal transition (EMT) in the lung. cAMP, which localizes in specific subcellular microdomains, modulates the activities of downstream effectors PKA and Epacs. PDEs, central players in spatio‐temporal dynamics, hydrolyse cAMP and prevent it from diffusing to other compartments. Expression of PDE4 and PDE8 mRNA expression is significantly up‐regulated by TGF‐β1. A‐kinase anchoring proteins (AKAPs) are a group of scaffolding proteins with the ability to associate with PKA via a short α‐helical structure. Ezrin is associated with PGE2‐induced β‐catenin transcription. AKAP9 plays a crucial role in E‐cadherin maintenance. AKAP13, known to act as a guanine nucleotide exchange factor for RhoA, may be able to promote αvβ6 integrin‐mediated TGF‐β activation in response to epithelial injury. As main inducing factors, cigarette smoke and air pollution are able to modulate cAMP scaffolds in the lung structural cells. For further details, see text. β2‐AR, β2‐adrenoceptor; EP2, prostanoid EP2 receptor
5. cAMP COMPARTMENTALIZATION IN EMT
The process of EMT is driven by a complex regulatory network beyond the transcriptional level, which integrates epigenetics, alternative splicing, protein stability, and most importantly subcellular localization (Jolly et al., 2017; Nieto, 2011). Several lines of evidence indicate that cAMP—a central player in compartmentalized signalling—acts as a potential novel pharmaceutical target in EMT (Kolosionek et al., 2009; Milara et al., 2014; Table 1).
Table 1.
The role of cAMP compartmentalization during the process of EMT
| Protein family | Subfamily/isoform | Effect in EMT‐linked process | Reference |
|---|---|---|---|
| AKAP | Ezrin | Morphological changes, actin filament remodelling, and cell migration and invasion | Chen et al. (2014) |
| Increased metastatic potential | Huang et al. (2010), Jansen et al. (2016), and Li et al. (2012) | ||
| Actin stress fibre assembly and morphological transition | Haynes, Srivastava, Madson, Wittmann, and Barber (2011) | ||
| AKAP9 | Cancer development and metastasis of cancers | Frank et al. (2008), Kabbarah et al. (2010), and Truong et al. (2010) | |
| Ezrin/radixin/moesin | Actin cytoskeleton remodelling | Tsukita and Yonemura (1999) | |
| AKAP13 | Increased expression in idiopathic pulmonary fibrosis | Allen et al. (2017) | |
| PDE | PDE4A, PDE4D, and PDE8A | Increased mRNA expression after TGF‐β1 exposure | Kolosionek et al. (2009) |
| PDE4 | PDE4 inhibition restores epithelial marker and inhibits mesenchymal markers and prevention of EMT induced by cigarette smoke | Kolosionek et al. (2009) and Milara et al. (2014) | |
| PKA and EPAC | Epac1 | Increased RNA expression in PGE2‐induced EMT | Jansen et al. (2016) |
| PKA | PKA‐selective cAMP agonist reduces α‐SMA elevation by TGF‐β1 | Insel et al. (2012) |
Note: AKAP: A‐kinase anchoring protein; EMT: epithelial‐to‐mesenchymal transition; α‐SMA: α‐smooth muscle actin.
5.1. The role of AKAP proteins in EMT
The membrane–cytoskeleton linker AKAP, ezrin, plays a crucial role in cell migration and invasion by regulating the assembly of cytoskeleton elements to promote cytoskeletal reorganization and cellular phenotypical alterations (Chen et al., 2014; Elliott, Meens, SenGupta, Louvard, & Arpin, 2005; Ohtani et al., 1999). In human alveolar epithelial cells, ezrin is highly associated with morphological changes, actin filament remodelling, and regulation of cell migration and invasion in EMT induced by TGF‐β1 (Chen et al., 2014; Figure 2). Moreover, in tumour‐related studies, overexpression of ezrin enhanced the metastatic potential, whereas the knockdown of ezrin inhibited cell migration and invasion (Huang et al., 2010; Jansen et al., 2016; Li et al., 2012). Interestingly, ezrin altered its intracellular localization from the apical membrane to the cytoplasm in lung cancers, thereby indicating that subcellular compartmentalization of ezrin is subject to alterations during cancer progression (Li et al., 2012). In asthma, ezrin protein expression was significantly decreased in exhaled breath condensate and serum from asthma patients as compared with normal subjects (Jia et al., 2018). These findings were further confirmed in IL‐13‐stimulated human bronchial epithelial 16HBE cells and ovalbumin‐treated allergic mouse model (Jia et al., 2018), indicating that ezrin is most likely involved in the pathogenesis of asthma.
Twhe actin‐binding proteins, ezrin, radixin and moesin (ERM) are known to organize the cortical cytoskeleton by linking filamentous actin to the apical membrane of cells (Neisch & Fehon, 2011; Schmidt et al., 2013; Tsukita & Yonemura, 1999). Studies in mouse mammary gland epithelial cells showed that although TGF‐β1 up‐regulated the expression of moesin, ezrin expression was down‐regulated, whereas that of radixin remained unchanged (Haynes et al., 2011). Additionally, cells, whose moesin expression was suppressed by short hairpin RNA, had dramatically fewer actin stress fibres, and the bundled filaments were thinner and shorter, compared with those in control cells, further indicating that moesin promoted actin stress fibre assembly and morphological transition during TGF‐β1‐induced EMT (Haynes et al., 2011). The distinct changes in the expression of ERM proteins during the initial stages of TGF‐β1‐induced EMT suggest that ERM proteins may differentially contribute to distinct EMT phenotypes. However, the function of ERM proteins is yet to be elucidated in lung epithelial cells.
It has been reported previously that AKAP9 is involved in the development and metastasis of cancers such as breast cancer, lung cancer, and melanomas (Frank et al., 2008; Kabbarah et al., 2010; Truong et al., 2010). Although further evidence has to be provided that AKAP9 plays a key role in regulating EMT in the lung, findings in colorectal cancer Lovo cells exposed to TGF‐β1 indicated that knockdown of AKAP9 expression using short hairpin RNA restored E‐cadherin expression and in contrast attenuated N‐cadherin and vimentin expression, suggesting that AKAP9 may play an important role in TGF‐β1‐induced EMT (Hu et al., 2016; Figure 2).
Recently, Allen et al. (2017) studied 2,760 patients with idiopathic pulmonary fibrosis (IPF) and 8,561 controls, and they identified a novel genome‐wide significant association of variant rs62025270 of AKAP13 as a susceptibility gene for IPF. Using immunohistochemical staining, it was reported that AKAP13 protein was primarily expressed in bronchial epithelium and alveolar type 1 and 2 cells in control lung tissue, whereas in lung tissue from IPF patients, high AKAP13 expression was detected in fibrotic regions. Additionally, a substantial higher AKAP13 expression was observed in alveoli of patients with IPF as compared with controls (Allen et al., 2017). Likewise, AKAP13 mRNA expression was 1.42 times higher in lung tissue from patients with IPF, compared with controls (Allen et al., 2017). It has been suggested that AKAP13, known to act as a guanine nucleotide exchange factor for RhoA, might be able to promote αvβ6 integrin‐mediated TGF‐β activation in response to epithelial injury, suggesting that AKAP13 may be associated with the pathogenesis of the EMT process (Diviani, Soderling, & Scott, 2001; Jansen et al., 2018; Jenkins et al., 2006; Majumdar, Seasholtz, Buckmaster, Toksoz, & Brown, 1999; Xu et al., 2009). In addition, AKAP13 seems to target the prostaglandin endoperoxide synthase and might provide a molecular link between the cAMP and the EMT process (Table 1).
5.2. The role of PDE family members in EMT
Although it has been reported that PDE1–PDE8 subtypes are highly expressed in lung epithelial cells (Fuhrmann et al., 1999; Haddad et al., 2002; Page & Spina, 2012; Zuo et al., 2018), the precise role of the distinct PDEs in the diverse functions of epithelial cells is still unclear. In A549 cells, TGF‐β1 treatment resulted in a significant increase in gene expression of PDE4A, PDE4D, and PDE8A, whereas the gene expression of PDE1A, PDE3A, and PDE7B was decreased (Kolosionek et al., 2009). Among the up‐regulated PDE isoforms, PDE4D showed the most prominent increase in mRNA (Kolosionek et al., 2009). Additionally, the PDE4‐specific inhibitor rolipram restored the expression of the epithelial marker E‐cadherin and abolished up‐regulation of the mesenchymal markers fibronectin and collagen I (both mRNA and protein) by TGF‐β1. These findings were further confirmed by using siRNA targeting PDE4A and PDE4D (Kolosionek et al., 2009). In another separate study, Milara et al. (2015, 2014) reported that roflumilast N‐oxide, the active metabolite of the PDE4 inhibitor roflumilast, prevented cigarette smoke‐induced EMT in differentiated human bronchial epithelial cells by restoration of the loss of intracellular cAMP after cigarette smoke exposure (Figure 2). These studies emphasized the importance of PDE4 inhibition in blocking the EMT process induced by either TGF‐β1 or cigarette smoke. Additional investigation is needed however to further characterize the distinct role of other PDE family members in EMT phenotypes in the lung.
5.3. The role of PKA and Epac in EMT
PKA and Epac are two of the best known downstream effectors of cAMP. There are two Epac isoforms, Epac1 and Epac2, which have distinct tissue expression patterns (Schmidt et al., 2013). Studies have demonstrated that gene expression of Epac1 but not Epac2 was increased by PGE2 in lung epithelial A549 cells (Jansen et al., 2016), thereby emphasizing that Epac1 was involved in PGE2‐induced EMT. It has been shown that PGE2 is able to induce EMT and enhance cell migration by augmenting ZEB1 and suppressing E‐cadherin expression in non‐small cell lung carcinoma, which is associated with stabilization of β‐catenin and activation of β‐catenin‐dependent transcription (Dohadwala et al., 2006; Singh & Katiyar, 2013; Zhang et al., 2014). More importantly, cotreatment of A549 cells with the Epac1 inhibitor CE3F4 or down‐regulation of Epac1 expression with siRNA fully abolished the induction of cell migration by PGE2. Moreover, ezrin knockdown prevented PGE2‐induced β‐catenin transcriptional activity, indicating that the scaffold protein ezrin acts as a physical link between β‐catenin and Epac1 (Jansen et al., 2016).
Interestingly, another PG. PGD2, has been reported to inhibit TGF‐β1‐induced EMT in MDCK cells. In addition, PKA inhibition by H89 was able to block the inhibitory effect of AC activator forskolin on TGF‐β1‐induced EMT, whereas inhibition of endogenous cAMP activity via H89 had no effect of PGD2‐induced inhibition of EMT (Zhang, Dong, & Yang, 2006). In additional studies in MDCK cells, i 8‐Me‐cAMP—a cAMP derivative that selectively activates Epac but not N6‐cAMP—a PKA‐selective cAMP agonist, blunted the TGF‐β1‐induced up‐regulation of α‐SMA. In contrast, both 8‐Me‐cAMP and N6‐cAMP reversed the TGF‐β1‐induced E‐cadherin down‐regulation (Insel et al., 2012). Taken together, these data indicate that both PKA and Epacs are involved in EMT, although their particular contribution may differ depending on distinct cAMP pools that are activated.
6. OUTLOOK AND FUTURE PERSPECTIVES
As one of the best known second messengers, cAMP transmits the information carried by hormones, neurotransmitters, and other extracellular signals into the intracellular environment (Berthet et al., 1957; Musheshe et al., 2018). Despite a wide array of information being unravelled about the cAMP signalling pathway, however, a number of outstanding questions still remain. It is still not clear how a simple molecule such as cAMP coordinates such a wide range of physiological and pathophysiological processes; why cAMP accumulation induced by stimulation of different Gs‐protein‐coupled receptors evoke distinct cell type‐specific responses; why β2‐adrenoceptor agonists cause airway smooth muscle relaxation but have no clinically relevant effects on airways inflammation or airway remodelling; why PDE3 inhibitors are able to relax airway smooth muscle, yet they are not anti‐inflammatory; why PDE4 inhibitors act as anti‐inflammatory drugs but have no acute bronchodilator effects; and lastly, why theoretically PDE4 inhibitor roflumilast provides clinically relevant effects in patients with COPD, yet the development of various inhalable PDE4 inhibitors has been halted due to lack of efficacy. These outstanding questions need to be addressed urgently to further enhance understanding of cAMP research in the lung. Comprehensive understanding of the spatio‐temporal dynamics of cAMP compartments will provide a platform for unravelling the distinct cAMP signalling properties and for screening of novel therapeutic agentss with higher efficacies and less side effects for the treatment of obstructive lung diseases.
As one of the main bronchodilator therapies, β2‐adrenoceptor agonists augment the anti‐inflammatory effects of corticosteroids in obstructive lung diseases (Barnes et al., 2015; Maji et al., 2018; Vogelmeier et al., 2017; Wang et al., 2018). Even though it has been shown that the β2‐adrenoceptor agonists inhibited cytokine release in vitro (Bosmann et al., 2012; Hallsworth, Twort, Lee, & Hirst, 2001; Poppinga et al., 2015), evidence for their anti‐inflammatory properties in vivo is still lacking (Giembycz & Maurice, 2014; Giembycz & Newton, 2006). Potential explanations for lacking evidence in vivo may be due to the development of β2‐adrenoceptor desensitization, followed by receptor internalization (Charlton, 2009; Dekkers, Racké, & Schmidt, 2013; Giembycz & Newton, 2006) and due to the fact that the β2‐adrenoceptor agonists induced biased signalling via β‐arrestin‐2, subsequently leading to airway hyperresponsiveness and inflammation (Nguyen et al., 2017; Walker, Penn, Hanania, Dickey, & Bond, 2011). Another unresolved issue that needed to be addressed in the field of compartmentalized cAMP signalling was the controversial effects of PDE3 inhibition on airway hyperresponsiveness and inflammation. It has been proven that PDE4 inhibition effectively reduces activation and recruitment of inflammatory cells and reduces the release of various cytokines and the production of ROS. This characteristic of PDE4 inhibitors is most likely due to the fact that PDE4 is widely expressed in inflammatory and immune cells (Barber et al., 2004; Engels, Fichtel, & Lübbert, 1994). Despite PDE3 being present in T lymphocytes, it has a limited impact on T‐cell proliferation and cytokine production (Giembycz, Corrigan, Seybold, Newton, & Barnes, 1996). Recently, Beute et al. (2018) studied the role of PDE3A and PDE3B in inflammation using an acute house dust mite (HDM)‐driven allergic airway inflammation mouse model. The number of eosinophils, T lymphocytes, neutrophils, and macrophages in bronchoalveolar lavage fluid was significantly decreased in HDM‐treated PDE3A−/− mice and PDE3B−/− mice as compared with wild‐type mice. Moreover, the proportion of IL‐5‐ and IL‐13‐positive CD4+ T cells in bronchoalveolar lavage fluid was significantly decreased in HDM‐treated PDE3A−/− and PDE3B−/− mice compared with wild‐type mice, suggesting that PDE3 may act as a novel anti‐inflammatory target in allergic airway inflammation (Beute et al., 2018). Regarding bronchodilation, a substantial body of evidence shows that PDE3 inhibitors (siguazodan, SK&F94120, and org9935) are potent relaxants in airway smooth muscle (Bernareggi, Belvisi, Patel, Barnes, & Giembycz, 1999; Nicholson et al., 1995; Torphy et al., 1993), while contrasting findings are reported regarding PDE4 inhibition in various animal models. Such differences may be explained by differential expression patterns of PDE4 in airway smooth muscle cells from various species (Zuo et al., 2018). Oral administration of roflumilast, a PDE4 inhibitor, has been approved for the treatment of severe COPD patients associated with bronchitis and with a history of frequent exacerbations. However, side effects still limit the extensive usage of PDE4 inhibitors (Giembycz & Maurice, 2014). A strategy to overcome the side effects is to deliver the drugs by inhalation; however, none of the very potent inhaled PDE4 inhibitors (GSK256066 and CHF6001) have shown any convincing evidence of efficacy in the treatment of respiratory diseases so far, thereby suggesting that the clinical benefits of PDE4 inhalation may arise from systemic effects.
The field of compartmentalized cAMP signalling may also offer answers to yet unresolved questions underlying the distinct stages of the EMT process that is linked to a diverse subset of lung responses. Certainly, EMT plays a vital role during organ fibrosis, including pulmonary fibrosis (Jolly et al., 2018; Rout‐Pitt et al., 2018). In addition, accumulating evidence indicates that an active EMT process is operational in experimental models of COPD, asthma, and IPF. Recent evidence also indicates that cAMP scaffolds maintained by a diverse subset of receptors, PDEs, PKA, Epac, and members of the AKAP superfamily have the potential to target distinct aspects of the EMT process, with the EMT process being closely related to factors such as TGF‐β1, TNF‐α, and/or IL‐13. Intriguingly, Jia et al. (2018) reported recently that the expression of the AKAP family member ezrin is closely related to the severity of asthma. The loss of ezrin correlated with the IL‐13‐induced damage of bronchial epithelial cells in both patients and experimental models of asthma, suggesting that ezrin may serve as a potential biomarker to control asthma (Jia et al., 2018). Similarly, expression of ezrin was subject to alterations in both airway smooth muscle exposed to cigarette smoke and lung tissue from COPD patients (Poppinga et al., 2015), implying that ezrin plays a crucial role in obstructive lung diseases. In addition, ezrin directly interacted with Epac1 and promoted the nuclear translocation of β‐catenin (Jansen et al., 2016). These findings indicated that cAMP scaffolds encompassing ezrin and Epac1 have the potential to initiate the canonical β‐catenin‐dependent signalling of Wnt receptors, with Wnt signalling being known to play an important role in promoting epithelial repair (Skronska‐Wasek, Gosens, Königshoff, & Baarsma, 2018). Ezrin acts as a regulator for the Rho signalling pathway, a pathway that regulates cell migration, phenotypical alterations, and metastatic cellular potential (Jansen et al., 2016) and therefore may be of central importance at different stages of the EMT process. Another study linked the AKAP family member AKAP13 to IPF in a process involving Rho and the prostanoid receptors (Allen et al., 2017). To date, only a very limited number of drugs are available to target lung fibrosis. Therefore, further studies with a special focus on the distinct role of the AKAP family members such as ezrin and AKAP13 will identify new therapeutic targets and hence novel drugs in the treatment of lung fibrosis (Gourdie, Dimmeler, & Kohl, 2016; Kalluri, 2016; Mora, Rojas, Pardo, & Selman, 2017).
7. CONCLUSIONS
Potential links between diverse lung disorders such as asthma, COPD and IPF might be represented by oxidative stress and air pollution. Oxidative stress, either induced by inflammatory cells or by inhaled noxious compounds, is an important player in the pathophysiology of these obstructive lung disorders (Anathy et al., 2018; Bernardo et al., 2015; Comhair & Erzurum, 2010; Domej, Oettl, & Renner, 2014; Nadeem, Masood, & Siddiqui, 2008). Air pollution is a major environmental threat not only in Europe but worldwide as it represents a global threat as far as human health and the social economic burden in the long term. The World Health Organization (WHO) report indicates that each year about 7 million people die as a result of exposure to air pollution. Despite the decrease in air pollutants over the past decades, air pollutant concentrations in urbanized areas still exceed reference values. Long‐term and peak exposure to ground level ozone, nitrogen dioxide and particulate matter (PM) pose serious health risks, with PM 2.5 in air being estimated to reduce life expectancy by at least eight months. Such devastating effects are related to the heterogeneous nature of PM in size and composition. For instance, fine PM from diesel exhaust represent a considerable percentage of urban PM which contains polycyclic aromatic hydrocarbons. Certain groups of people are identified as more susceptible to health effects due to air pollution, and among these and of particular concern are elderly people, children, and people with pre‐existing lung disease such as asthma and COPD, specifically the groups suffering from exacerbations (Annesi‐Maesano, 2017). Interestingly, it has been shown that ultrafine particulate matter initiates the process of EMT in BEAS‐2B cells (Thevenot et al., 2013)—a process accompanied by a loss of E‐cadherin and a gain in α‐SMA. Recent studies reported on alterations of the expression of the AKAP member: AKAP5, AKAP12, ezrin and AKAP9 in experimental models of COPD (Oldenburger, Poppinga, et al., 2014; Poppinga et al., 2015), next to a change in the expression of Epacs and PDEs (Oldenburger, Timens, et al., 2014; Zuo et al., 2018), clearly indicating that oxidative stress alters the subcellular composition of cAMP scaffolds. Therefore, future studies should aim to target cAMP scaffolds either by stabilizing their composition or modifying their composition. A better understanding of compartmentalized cellular cAMP signalling might further increase our knowledge about the distinct stages of EMT phenotypes thereby bridging potential disconnections between in vitro and in vivo findings.
7.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017a, Alexander, Fabbro et al., 2017b; Alexander, Kelly et al., 2017; Alexander, Striessnig, et al., 2017).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
This work was supported by the Ubbo Emmius Programme (grant to H.Z.), a sandwich PhD scholarship from the Brazilian Federal Agency for Support and Evaluation of Graduate Education (CAPES [055/14], grant to I.C.‐C and S.S.V.), an FSE Fellowship (grant to N.M.), and the Deutsche Forschungsgemeinschaft (grant to M.S.).
Zuo H, Cattani‐Cavalieri I, Valença SS, Musheshe N, Schmidt M. Function of cAMP scaffolds in obstructive lung disease: Focus on epithelial‐to‐mesenchymal transition and oxidative stress. Br J Pharmacol. 2019;176:2402–2415. 10.1111/bph.14605
REFERENCES
- Acloque, H. , Adams, M. S. , Fishwick, K. , Bronner‐Fraser, M. , & Nieto, M. A. (2009). Epithelial–mesenchymal transitions: The importance of changing cell state in development and disease. The Journal of Clinical Investigation, 119, 1438–1449. 10.1172/JCI38019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal, S. , Loder, S. , Cholok, D. , Peterson, J. , Li, J. , Fireman, D. , … Levi, B. (2016). Local and circulating endothelial cells undergo endothelial to mesenchymal transition (EndMT) in response to musculoskeletal injury. Scientific Reports, 6, 32514 10.1038/srep32514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … Collaborators, C. G. T. P. (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … Collaborators, C. G. T. P. (2017b). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … Collaborators, C. G. T. P. (2017). The Concise Guide to PHARMACOLOGY 2017/18: Other ion channels. British Journal of Pharmacology, 174, S195–S207. 10.1111/bph.13881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Striessnig, J. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … Collaborators, C. G. T. P. (2017). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. British Journal of Pharmacology, 174, S160–S194. 10.1111/bph.13884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, R. J. , Porte, J. , Braybrooke, R. , Flores, C. , Fingerlin, T. E. , Oldham, J. M. , … Jenkins, R. G. (2017). Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome‐wide association study. The Lancet Respiratory Medicine, 5, 869–880. 10.1016/S2213-2600(17)30387-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anathy, V. , Lahue, K. G. , Chapman, D. G. , Chia, S. B. , Casey, D. T. , Aboushousha, R. , … Janssen‐Heininger, Y. M. W. (2018). Reducing protein oxidation reverses lung fibrosis. Nature Medicine, 24, 1128–1135. 10.1038/s41591-018-0090-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annesi‐Maesano, I. (2017). The air of Europe: Where are we going? European Respiratory Review, 26, 170024 10.1183/16000617.0024-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antus, B. , & Kardos, Z. (2015). Oxidative stress in COPD: Molecular background and clinical monitoring. Current Medicinal Chemistry, 22, 627–650. 10.2174/092986732205150112104411 [DOI] [PubMed] [Google Scholar]
- Baarsma, H. A. , Spanjer, A. I. R. , Haitsma, G. , Engelbertink, L. H. J. M. , Meurs, H. , Jonker, M. R. , … Gosens, R. (2011). Activation of WNT/β‐catenin signaling in pulmonary fibroblasts by TGF‐β₁ is increased in chronic obstructive pulmonary disease. PLoS ONE, 6, e25450 10.1371/journal.pone.0025450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber, R. , Baillie, G. S. , Bergmann, R. , Shepherd, M. C. , Sepper, R. , Houslay, M. D. , & Heeke, G. V. (2004). Differential expression of PDE4 cAMP phosphodiesterase isoforms in inflammatory cells of smokers with COPD, smokers without COPD, and nonsmokers. Am J Physiol—Lung Cell Mol Physiol, 287, L332–L343. 10.1152/ajplung.00384.2003 [DOI] [PubMed] [Google Scholar]
- Barnes, P. J. , Burney, P. G. J. , Silverman, E. K. , Celli, B. R. , Vestbo, J. , Wedzicha, J. A. , & Wouters, E. F. (2015). Chronic obstructive pulmonary disease. Nature Reviews Disease Primers, 1, 15076. [DOI] [PubMed] [Google Scholar]
- Bartis, D. , Mise, N. , Mahida, R. Y. , Eickelberg, O. , & Thickett, D. R. (2014). Epithelial–mesenchymal transition in lung development and disease: Does it exist and is it important? Thorax, 69, 760–765. 10.1136/thoraxjnl-2013-204608 [DOI] [PubMed] [Google Scholar]
- Beavo, J. A. , & Brunton, L. L. (2002). Cyclic nucleotide research—Still expanding after half a century. Nature Reviews. Molecular Cell Biology, 3, 710–718. 10.1038/nrm911 [DOI] [PubMed] [Google Scholar]
- Beene, D. L. , & Scott, J. D. (2007). A‐kinase anchoring proteins take shape. Current Opinion in Cell Biology, 19, 192–198. 10.1016/j.ceb.2007.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo, I. , Bozinovski, S. , & Vlahos, R. (2015). Targeting oxidant‐dependent mechanisms for the treatment of COPD and its comorbidities. Pharmacology & Therapeutics, 155, 60–79. 10.1016/j.pharmthera.2015.08.005 [DOI] [PubMed] [Google Scholar]
- Bernareggi, M. M. , Belvisi, M. G. , Patel, H. , Barnes, P. J. , & Giembycz, M. A. (1999). Anti‐spasmogenic activity of isoenzyme‐selective phosphodiesterase inhibitors in guinea‐pig trachealis. British Journal of Pharmacology, 128, 327–336. 10.1038/sj.bjp.0702779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet, J. , Rall, T. W. , & Sutherland, E. W. (1957). The relationship of epinephrine and glucagon to liver phosphorylase. IV. Effect of epinephrine and glucagon on the reactivation of phosphorylase in liver homogenates. J Biol Chem, 224, 463–475. [PubMed] [Google Scholar]
- Beute, J. , Lukkes, M. , Koekoek, E. P. , Nastiti, H. , Ganesh, K. , de Bruijn, M. J. , … KleinJan, A. (2018). A pathophysiological role of PDE3 in allergic airway inflammation. JCI Insight, 3, pii: 94888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel, M. , & Michalakis, S. (2009). Cyclic nucleotide‐gated channels. Handbook of Experimental Pharmacology, 191, 111–136. 10.1007/978-3-540-68964-5_7. [DOI] [PubMed] [Google Scholar]
- Billington, C. K. , Penn, R. B. , & Hall, I. P. (2017). β2 agonists. Handbook of Experimental Pharmacology, 237, 23–40. 10.1007/164_2016_64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosmann, M. , Grailer, J. J. , Zhu, K. , Matthay, M. A. , Sarma, J. V. , Zetoune, F. S. , & Ward, P. A. (2012). Anti‐inflammatory effects of β2 adrenergic receptor agonists in experimental acute lung injury. The FASEB Journal, 26, 2137–2144. 10.1096/fj.11-201640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, H. A. (2011). Epithelial–mesenchymal interactions in pulmonary fibrosis. Annual Review of Physiology, 73, 413–435. 10.1146/annurev-physiol-012110-142225 [DOI] [PubMed] [Google Scholar]
- Charlton, S. J. (2009). Agonist efficacy and receptor desensitization: From partial truths to a fuller picture. British Journal of Pharmacology, 158, 165–168. 10.1111/j.1476-5381.2009.00352.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.‐J. , Gao, X.‐J. , Xu, L.‐N. , Liu, T.‐F. , Liu, X.‐H. , & Liu, L.‐X. (2014). Ezrin is required for epithelial–mesenchymal transition induced by TGF‐β1 in A549 cells. International Journal of Oncology, 45, 1515–1522. 10.3892/ijo.2014.2554 [DOI] [PubMed] [Google Scholar]
- Chung, K. Y. , Rasmussen, S. G. F. , Liu, T. , Li, S. , DeVree, B. T. , Chae, P. S. , … Sunahara, R. K. (2011). Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature, 477, 611–615. 10.1038/nature10488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comhair, S. A. A. , & Erzurum, S. C. (2010). Redox control of asthma: Molecular mechanisms and therapeutic opportunities. Antioxidants & Redox Signaling, 12, 93–124. 10.1089/ars.2008.2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrotto, P. , Yakymovych, I. , Yakymovych, M. , & Souchelnytskyi, S. (2007). Interactome of transforming growth factor‐β type I receptor (TβRI): Inhibition of TGFβ signaling by Epac1. Journal of Proteome Research, 6, 287–297. 10.1021/pr060427q [DOI] [PubMed] [Google Scholar]
- Conti, M. , Richter, W. , Mehats, C. , Livera, G. , Park, J.‐Y. , & Jin, C. (2003). Cyclic AMP‐specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. The Journal of Biological Chemistry, 278, 5493–5496. 10.1074/jbc.R200029200 [DOI] [PubMed] [Google Scholar]
- Dekkers, B. G. J. , Racké, K. , & Schmidt, M. (2013). Distinct PKA and Epac compartmentalization in airway function and plasticity. Pharmacology & Therapeutics, 137, 248–265. 10.1016/j.pharmthera.2012.10.006 [DOI] [PubMed] [Google Scholar]
- Diviani, D. , Soderling, J. , & Scott, J. D. (2001). AKAP‐Lbc anchors protein kinase A and nucleates Gα12‐selective Rho‐mediated stress fiber formation. The Journal of Biological Chemistry, 276, 44247–44257. 10.1074/jbc.M106629200 [DOI] [PubMed] [Google Scholar]
- Dohadwala, M. , Yang, S.‐C. , Luo, J. , Sharma, S. , Batra, R. K. , Huang, M. , … Dubinett, S. M. (2006). Cyclooxygenase‐2‐dependent regulation of E‐cadherin: Prostaglandin E2 induces transcriptional repressors ZEB1 and snail in non‐small cell lung cancer. Cancer Research, 66, 5338–5345. 10.1158/0008-5472.CAN-05-3635 [DOI] [PubMed] [Google Scholar]
- Domej, W. , Oettl, K. , & Renner, W. (2014). Oxidative stress and free radicals in COPD—Implications and relevance for treatment. International Journal of Chronic Obstructive Pulmonary Disease, 9, 1207–1224. 10.2147/COPD.S51226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, B. E. , Meens, J. A. , SenGupta, S. K. , Louvard, D. , & Arpin, M. (2005). The membrane cytoskeletal crosslinker ezrin is required for metastasis of breast carcinoma cells. Breast Cancer Research BCR, 7, R365–R373. 10.1186/bcr1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels, P. , Fichtel, K. , & Lübbert, H. (1994). Expression and regulation of human and rat phosphodiesterase type IV isogenes. FEBS Letters, 350, 291–295. 10.1016/0014-5793(94)00788-8 [DOI] [PubMed] [Google Scholar]
- Eurlings, I. M. J. , Reynaert, N. L. , van den Beucken, T. , Gosker, H. R. , de Theije, C. C. , Verhamme, F. M. , … Dentener, M. A. (2014). Cigarette smoke extract induces a phenotypic shift in epithelial cells; involvement of HIF1α in mesenchymal transition. PLoS ONE, 9, e107757 10.1371/journal.pone.0107757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, B. , Wiestler, M. , Kropp, S. , Hemminki, K. , Spurdle, A. B. , Sutter, C. , … Burwinkel, B. (2008). Association of a common AKAP9 variant with breast cancer risk: A collaborative analysis. Journal of the National Cancer Institute, 100, 437–442. 10.1093/jnci/djn037 [DOI] [PubMed] [Google Scholar]
- Fuhrmann, M. , Jahn, H.‐U. , Seybold, J. , Neurohr, C. , Barnes, P. J. , Hippenstiel, S. , … Suttorp, N. (1999). Identification and function of cyclic nucleotide phosphodiesterase isoenzymes in airway epithelial cells. American Journal of Respiratory Cell and Molecular Biology, 20, 292–302. 10.1165/ajrcmb.20.2.3140 [DOI] [PubMed] [Google Scholar]
- Giembycz, M. A. , Corrigan, C. J. , Seybold, J. , Newton, R. , & Barnes, P. J. (1996). Identification of cyclic AMP phosphodiesterases 3, 4 and 7 in human CD4+ and CD8+ T‐lymphocytes: Role in regulating proliferation and the biosynthesis of interleukin‐2. British Journal of Pharmacology, 118, 1945–1958. 10.1111/j.1476-5381.1996.tb15629.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giembycz, M. A. , & Maurice, D. H. (2014). Cyclic nucleotide‐based therapeutics for chronic obstructive pulmonary disease. Current Opinion in Pharmacology, 16, 89–107. 10.1016/j.coph.2014.04.001 [DOI] [PubMed] [Google Scholar]
- Giembycz, M. A. , & Newton, R. (2006). Beyond the dogma: Novel β2‐adrenoceptor signalling in the airways. The European Respiratory Journal, 27, 1286–1306. 10.1183/09031936.06.00112605 [DOI] [PubMed] [Google Scholar]
- Gonzalez, D. M. , & Medici, D. (2014). Signaling mechanisms of the epithelial–mesenchymal transition. Science Signaling, 7, re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourdie, R. G. , Dimmeler, S. , & Kohl, P. (2016). Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nature Reviews. Drug Discovery, 15, 620–638. 10.1038/nrd.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandoch, M. , Roscioni, S. S. , & Schmidt, M. (2010). The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. British Journal of Pharmacology, 159, 265–284. 10.1111/j.1476-5381.2009.00458.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad, J. J. , Land, S. C. , Tarnow‐Mordi, W. O. , Zembala, M. , Kowalczyk, D. , & Lauterbach, R. (2002). Immunopharmacological potential of selective phosphodiesterase inhibition. I. Differential regulation of lipopolysaccharide‐mediated proinflammatory cytokine (interleukin‐6 and tumor necrosis factor‐α) biosynthesis in alveolar epithelial cells. The Journal of Pharmacology and Experimental Therapeutics, 300, 559–566. 10.1124/jpet.300.2.559 [DOI] [PubMed] [Google Scholar]
- Halls, M. L. , & Cooper, D. M. F. (2017). Adenylyl cyclase signalling complexes—Pharmacological challenges and opportunities. Pharmacology & Therapeutics, 172, 171–180. 10.1016/j.pharmthera.2017.01.001 [DOI] [PubMed] [Google Scholar]
- Hallsworth, M. P. , Twort, C. H. , Lee, T. H. , & Hirst, S. J. (2001). β2‐Adrenoceptor agonists inhibit release of eosinophil‐activating cytokines from human airway smooth muscle cells. British Journal of Pharmacology, 132, 729–741. 10.1038/sj.bjp.0703866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes, J. , Srivastava, J. , Madson, N. , Wittmann, T. , & Barber, D. L. (2011). Dynamic actin remodeling during epithelial–mesenchymal transition depends on increased moesin expression. Molecular Biology of the Cell, 22, 4750–4764. 10.1091/mbc.e11-02-0119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Z.‐Y. , Liu, Y.‐P. , Xie, L.‐Y. , Wang, X.‐Y. , Yang, F. , Chen, S.‐Y. , & Li, Z. G. (2016). AKAP‐9 promotes colorectal cancer development by regulating Cdc42 interacting protein 4. Biochimica et Biophysica Acta, 1862, 1172–1181. 10.1016/j.bbadis.2016.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, H.‐Y. , Li, C.‐F. , Fang, F.‐M. , Tsai, J.‐W. , Li, S.‐H. , Lee, Y.‐T. , & Wei, H. M. (2010). Prognostic implication of ezrin overexpression in myxofibrosarcomas. Annals of Surgical Oncology, 17, 3212–3219. 10.1245/s10434-010-1185-y [DOI] [PubMed] [Google Scholar]
- Insel, P. A. , Murray, F. , Yokoyama, U. , Romano, S. , Yun, H. , Brown, L. , … Aroonsakool, N. (2012). cAMP and Epac in the regulation of tissue fibrosis. British Journal of Pharmacology, 166, 447–456. 10.1111/j.1476-5381.2012.01847.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, S. , Gosens, R. , Wieland, T. , & Schmidt, M. (2018). Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacology & Therapeutics, 183, 1–21. 10.1016/j.pharmthera.2017.09.002 [DOI] [PubMed] [Google Scholar]
- Jansen, S. R. , Poppinga, W. J. , de Jager, W. , Lezoualc'h, F. , Cheng, X. , Wieland, T. , … Schmidt, M. (2016). Epac1 links prostaglandin E2 to β‐catenin‐dependent transcription during epithelial‐to‐mesenchymal transition. Oncotarget, 7, 46354–46370. 10.18632/oncotarget.10128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins, R. G. , Su, X. , Su, G. , Scotton, C. J. , Camerer, E. , Laurent, G. J. , … Sheppard, D. (2006). Ligation of protease‐activated receptor 1 enhances αvβ6 integrin‐dependent TGF‐β activation and promotes acute lung injury. The Journal of Clinical Investigation, 116, 1606–1614. 10.1172/JCI27183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, J.‐H. , Jang, H. J. , Kwak, S. , Sung, G.‐J. , Park, S.‐H. , Song, J.‐H. , … Choi, K. C. (2019). Novel TGF‐β1 inhibitor antagonizes TGF‐β1‐induced epithelial–mesenchymal transition in human A549 lung cancer cells. Journal of Cellular Biochemistry, 120, 977–987. 10.1002/jcb.27460 [DOI] [PubMed] [Google Scholar]
- Jia, M. , Yan, X. , Jiang, X. , Wu, Y. , Xu, J. , Meng, Y. , … Yao X. (2018). Ezrin, a membrane cytoskeleton cross linker protein, as a marker of epithelial damage in asthma. American Journal of Respiratory and Critical Care Medicine, 199(4), 496–507. 10.1164/rccm.201802-0373OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly, M. K. , Ward, C. , Eapen, M. S. , Myers, S. , Hallgren, O. , Levine, H. , & Sohal, S. S. (2018). Epithelial–mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease. Developmental Dynamics, 247, 346–358. 10.1002/dvdy.24541 [DOI] [PubMed] [Google Scholar]
- Jolly, M. K. , Ware, K. E. , Gilja, S. , Somarelli, J. A. , & Levine, H. (2017). EMT and MET: Necessary or permissive for metastasis? Molecular Oncology, 11, 755–769. 10.1002/1878-0261.12083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabbarah, O. , Nogueira, C. , Feng, B. , Nazarian, R. M. , Bosenberg, M. , Wu, M. , … Chin, L. (2010). Integrative genome comparison of primary and metastatic melanomas. PLoS ONE, 5, e10770 10.1371/journal.pone.0010770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri, R. (2016). The biology and function of fibroblasts in cancer. Nature Reviews. Cancer, 16, 582–598. 10.1038/nrc.2016.73 [DOI] [PubMed] [Google Scholar]
- Kalluri, R. , & Neilson, E. G. (2003). Epithelial–mesenchymal transition and its implications for fibrosis. The Journal of Clinical Investigation, 112, 1776–1784. 10.1172/JCI200320530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri, R. , & Weinberg, R. A. (2009). The basics of epithelial–mesenchymal transition. The Journal of Clinical Investigation, 119, 1420–1428. 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani, S. , Yamauchi, Y. , Kawasaki, S. , Takami, K. , Takizawa, H. , Nagase, T. , & Kohyama, T. (2011). Simultaneous stimulation with TGF‐β1 and TNF‐α induces epithelial mesenchymal transition in bronchial epithelial cells. International Archives of Allergy and Immunology, 155, 119–128. 10.1159/000318854 [DOI] [PubMed] [Google Scholar]
- Karvonen, H. M. , Lehtonen, S. T. , Harju, T. , Sormunen, R. T. , Lappi‐Blanco, E. , Mäkinen, J. M. , … Kaarteenaho, R. L. (2013). Myofibroblast expression in airways and alveoli is affected by smoking and COPD. Respiratory Research, 14, 84 10.1186/1465-9921-14-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp, U. B. , & Seifert, R. (2002). Cyclic nucleotide‐gated ion channels. Physiological Reviews, 82, 769–824. 10.1152/physrev.00008.2002 [DOI] [PubMed] [Google Scholar]
- Kim, D. H. , Xing, T. , Yang, Z. , Dudek, R. , Lu, Q. , & Chen, Y.‐H. (2017). Epithelial mesenchymal transition in embryonic development, tissue repair and cancer: A comprehensive overview. Journal of Clinical Medicine, 7 10.3390/jcm7010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K. K. , Kugler, M. C. , Wolters, P. J. , Robillard, L. , Galvez, M. G. , Brumwell, A. N. , … Chapman, H. A. (2006). Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proceedings of the National Academy of Sciences, 10, 13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkham, P. A. , & Barnes, P. J. (2013). Oxidative stress in COPD. Chest, 144, 266–273. 10.1378/chest.12-2664 [DOI] [PubMed] [Google Scholar]
- Kolosionek, E. , Savai, R. , Ghofrani, H. A. , Weissmann, N. , Guenther, A. , Grimminger, F. , … Pullamsetti, S. S. (2009). Expression and activity of phosphodiesterase isoforms during epithelial mesenchymal transition: The role of phosphodiesterase 4. Molecular Biology of the Cell, 20, 4751–4765. 10.1091/mbc.e09-01-0019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambers, C. , Qi, Y. , Eleni, P. , Costa, L. , Zhong, J. , Tamm, M. , … Roth, M. (2014). Extracellular matrix composition is modified by β₂‐agonists through cAMP in COPD. Biochemical Pharmacology, 91, 400–408. 10.1016/j.bcp.2014.07.026 [DOI] [PubMed] [Google Scholar]
- Lamouille, S. , Xu, J. , & Derynck, R. (2014). Molecular mechanisms of epithelial–mesenchymal transition. Nature Reviews. Molecular Cell Biology, 15, 178–196. 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudette, M. , Zuo, H. , Lezoualc'h, F. , & Schmidt, M. (2018). Epac function and cAMP scaffolds in the heart and lung. Journal of Cardiovascular Development and Disease, 5, pii: E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz, R. J. , Roth, J. , & Pastan, I. (1970). Radioreceptor assay of adrenocorticotropic hormone: New approach to assay of polypeptide hormones in plasma. Science, 170, 633–635. 10.1126/science.170.3958.633 [DOI] [PubMed] [Google Scholar]
- Lefkowitz, R. J. , Roth, J. , Pricer, W. , & Pastan, I. (1970). ACTH receptors in the adrenal: Specific binding of ACTH‐125I and its relation to adenyl cyclase. Proceedings of the National Academy of Sciences of the United States of America, 65, 745–752. 10.1073/pnas.65.3.745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Gao, H. , Xu, H. , Wang, X. , Pan, Y. , Hao, F. , … Wang, E. (2012). Expression of ezrin correlates with malignant phenotype of lung cancer, and in vitro knockdown of ezrin reverses the aggressive biological behavior of lung cancer cells. Tumor Biology, 33, 1493–1504. 10.1007/s13277-012-0400-9 [DOI] [PubMed] [Google Scholar]
- López‐Novoa, J. M. , & Nieto, M. A. (2009). Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Molecular Medicine, 1, 303–314. 10.1002/emmm.200900043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood, M. Q. , Sohal, S. S. , Shukla, S. D. , Ward, C. , Hardikar, A. , Noor, W. D. , … Walters, E. H. (2015). Epithelial mesenchymal transition in smokers: Large versus small airways and relation to airflow obstruction. International Journal of Chronic Obstructive Pulmonary Disease, 10, 1515–1524. 10.2147/COPD.S81032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji, K. J. , Dikshit, A. K. , Arora, M. , & Deshpande, A. (2018). Estimating premature mortality attributable to PM2.5 exposure and benefit of air pollution control policies in China for 2020. Sci Total Environ, 612, 683–693. 10.1016/j.scitotenv.2017.08.254 [DOI] [PubMed] [Google Scholar]
- Majumdar, M. , Seasholtz, T. M. , Buckmaster, C. , Toksoz, D. , & Brown, J. H. (1999). A Rho exchange factor mediates thrombin and Gα12‐induced cytoskeletal responses. The Journal of Biological Chemistry, 274, 26815–26821. 10.1074/jbc.274.38.26815 [DOI] [PubMed] [Google Scholar]
- Manganiello, V. (2002). Short‐term regulation of PDE4 activity. British Journal of Pharmacology, 136, 339–340. 10.1038/sj.bjp.0704741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martorana, P. A. , Beume, R. , Lucattelli, M. , Wollin, L. , & Lungarella, G. (2005). Roflumilast fully prevents emphysema in mice chronically exposed to cigarette smoke. American Journal of Respiratory and Critical Care Medicine, 172, 848–853. 10.1164/rccm.200411-1549OC [DOI] [PubMed] [Google Scholar]
- Milara, J. , Peiró, T. , Serrano, A. , Artigues, E. , Aparicio, J. , Tenor, H. , … Cortijo, J. (2015). Simvastatin increases the ability of roflumilast N‐oxide to inhibit cigarette smoke‐induced epithelial to mesenchymal transition in well‐differentiated human bronchial epithelial cells in vitro. COPD, 12, 320–331. 10.3109/15412555.2014.948995 [DOI] [PubMed] [Google Scholar]
- Milara, J. , Peiró, T. , Serrano, A. , & Cortijo, J. (2013). Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax, 68, 410–420. 10.1136/thoraxjnl-2012-201761 [DOI] [PubMed] [Google Scholar]
- Milara, J. , Peiró, T. , Serrano, A. , Guijarro, R. , Zaragozá, C. , Tenor, H. , & Cortijo, J. (2014). Roflumilast N‐oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulmonary Pharmacology & Therapeutics, 28, 138–148. 10.1016/j.pupt.2014.02.001 [DOI] [PubMed] [Google Scholar]
- Molloy, E. L. , Adams, A. , Moore, J. B. , Masterson, J. C. , Madrigal‐Estebas, L. , Mahon, B. P. , & O'Dea, S. (2008). BMP4 induces an epithelial–mesenchymal transition‐like response in adult airway epithelial cells. Growth Factors, 26, 12–22. 10.1080/08977190801987166 [DOI] [PubMed] [Google Scholar]
- Mora, A. L. , Rojas, M. , Pardo, A. , & Selman, M. (2017). Emerging therapies for idiopathic pulmonary fibrosis, a progressive age‐related disease. Nature Reviews. Drug Discovery, 16, 755–772. 10.1038/nrd.2017.170 [DOI] [PubMed] [Google Scholar]
- Musheshe, N. , Schmidt, M. , & Zaccolo, M. (2018). cAMP: From long‐range second messenger to nanodomain signalling. Trends in Pharmacological Sciences, 39, 209–222. 10.1016/j.tips.2017.11.006 [DOI] [PubMed] [Google Scholar]
- Nadeem, A. , Masood, A. , & Siddiqui, N. (2008). Oxidant–antioxidant imbalance in asthma: Scientific evidence, epidemiological data and possible therapeutic options. Therapeutic Advances in Respiratory Disease, 2, 215–235. 10.1177/1753465808094971 [DOI] [PubMed] [Google Scholar]
- Neisch, A. L. , & Fehon, R. G. (2011). Ezrin, radixin and moesin: Key regulators of membrane–cortex interactions and signaling. Current Opinion in Cell Biology, 23, 377–382. 10.1016/j.ceb.2011.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L. P. , Al‐Sawalha, N. A. , Parra, S. , Pokkunuri, I. , Omoluabi, O. , Okulate, A. A. , … Bond, R. A. (2017). β2‐Adrenoceptor signaling in airway epithelial cells promotes eosinophilic inflammation, mucous metaplasia, and airway contractility. Proceedings of the National Academy of Sciences of the United States of America, 114, E9163–E9171. 10.1073/pnas.1710196114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, C. D. , Shahid, M. , Bruin, J. , Barron, E. , Spiers, I. , de Boer, J. , … Dent, G. (1995). Characterization of ORG 20241, a combined phosphodiesterase IV/III cyclic nucleotide phosphodiesterase inhibitor for asthma. The Journal of Pharmacology and Experimental Therapeutics, 274, 678–687. [PubMed] [Google Scholar]
- Nieto, M. A. (2011). The ins and outs of the epithelial to mesenchymal transition in health and disease. Annual Review of Cell and Developmental Biology, 27, 347–376. 10.1146/annurev-cellbio-092910-154036 [DOI] [PubMed] [Google Scholar]
- Nishioka, M. , Venkatesan, N. , Dessalle, K. , Mogas, A. , Kyoh, S. , Lin, T.‐Y. , … Hamid, Q. (2015). Fibroblast–epithelial cell interactions drive epithelial–mesenchymal transition differently in cells from normal and COPD patients. Respiratory Research, 16, 72 10.1186/s12931-015-0232-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani, K. , Sakamoto, H. , Rutherford, T. , Chen, Z. , Satoh, K. , & Naftolin, F. (1999). Ezrin, a membrane–cytoskeletal linking protein, is involved in the process of invasion of endometrial cancer cells. Cancer Letters, 147, 31–38. 10.1016/S0304-3835(99)00272-4 [DOI] [PubMed] [Google Scholar]
- Oldenburger, A. , Poppinga, W. J. , Kos, F. , de Bruin, H. G. , Rijks, W. F. , Heijink, I. H. , … Schmidt, M. (2014). A‐kinase anchoring proteins contribute to loss of E‐cadherin and bronchial epithelial barrier by cigarette smoke. American Journal of Physiology. Cell Physiology, 306, C585–C597. 10.1152/ajpcell.00183.2013 [DOI] [PubMed] [Google Scholar]
- Oldenburger, A. , Roscioni, S. S. , Jansen, E. , Menzen, M. H. , Halayko, A. J. , Timens, W. , … Schmidt, M. (2012). Anti‐inflammatory role of the cAMP effectors Epac and PKA: Implications in chronic obstructive pulmonary disease. PLoS ONE, 7, e31574 10.1371/journal.pone.0031574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburger, A. , Timens, W. , Bos, S. , Smit, M. , Smrcka, A. V. , Laurent, A.‐C. , … Schmidt, M. (2014). Epac1 and Epac2 are differentially involved in inflammatory and remodeling processes induced by cigarette smoke. The FASEB Journal, 28, 4617–4628. 10.1096/fj.13-248930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburger, A. , van Basten, B. , Kooistra, W. , Meurs, H. , Maarsingh, H. , Krenning, G. , … Schmidt, M. (2014). Interaction between Epac1 and miRNA‐7 in airway smooth muscle cells. Naunyn‐Schmiedeberg's Archives of Pharmacology, 387, 795–797. 10.1007/s00210-014-1015-z [DOI] [PubMed] [Google Scholar]
- Omori, K. , & Kotera, J. (2007). Overview of PDEs and their regulation. Circulation Research, 100, 309–327. 10.1161/01.RES.0000256354.95791.f1 [DOI] [PubMed] [Google Scholar]
- Page, C. P. , & Spina, D. (2012). Selective PDE inhibitors as novel treatments for respiratory diseases. Current Opinion in Pharmacology, 12, 275–286. 10.1016/j.coph.2012.02.016 [DOI] [PubMed] [Google Scholar]
- Parnell, E. , Palmer, T. M. , & Yarwood, S. J. (2015). The future of EPAC‐targeted therapies: Agonism versus antagonism. Trends in Pharmacological Sciences, 36, 203–214. 10.1016/j.tips.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppinga, W. J. , Heijink, I. H. , Holtzer, L. J. , Skroblin, P. , Klussmann, E. , Halayko, A. J. , … Schmidt, M. (2015). A‐kinase‐anchoring proteins coordinate inflammatory responses to cigarette smoke in airway smooth muscle. American Journal of Physiology. Lung Cellular and Molecular Physiology, 308, L766–L775. 10.1152/ajplung.00301.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppinga, W. J. , Muñoz‐Llancao, P. , González‐Billault, C. , & Schmidt, M. (2014). A‐kinase anchoring proteins: cAMP compartmentalization in neurodegenerative and obstructive pulmonary diseases. British Journal of Pharmacology, 171, 5603–5623. 10.1111/bph.12882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen, S. G. F. , Choi, H.‐J. , Fung, J. J. , Pardon, E. , Casarosa, P. , Chae, P. S. , … Kobilka, B. K. (2011). Structure of a nanobody‐stabilized active state of the β2 adrenoceptor. Nature, 469, 175–180. 10.1038/nature09648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen, S. G. F. , DeVree, B. T. , Zou, Y. , Kruse, A. C. , Chung, K. Y. , Kobilka, T. S. , … Kobilka, B. K. (2011). Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature, 477, 549–555. 10.1038/nature10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux, W. G. , & Cheng, X. (2018). Intracellular cAMP sensor EPAC: Physiology, pathophysiology, and therapeutics development. Physiological Reviews, 98, 919–1053. 10.1152/physrev.00025.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout‐Pitt, N. , Farrow, N. , Parsons, D. , & Donnelley, M. (2018). Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respiratory Research, 19, 136 10.1186/s12931-018-0834-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler, R. F. R. , & Brand, T. (2016). The popeye domain containing protein family—A novel class of cAMP effectors with important functions in multiple tissues. Progress in Biophysics and Molecular Biology, 120, 28–36. 10.1016/j.pbiomolbio.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, M. , Dekker, F. J. , & Maarsingh, H. (2013). Exchange protein directly activated by cAMP (epac): A multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacological Reviews, 65, 670–709. 10.1124/pr.110.003707 [DOI] [PubMed] [Google Scholar]
- Scotton, C. J. , & Chambers, R. C. (2007). Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest, 132, 1311–1321. 10.1378/chest.06-2568 [DOI] [PubMed] [Google Scholar]
- Shen, H. , Sun, Y. , Zhang, S. , Jiang, J. , Dong, X. , Jia, Y. , … Yan, X. F. (2014). Cigarette smoke‐induced alveolar epithelial–mesenchymal transition is mediated by Rac1 activation. Biochimica et Biophysica Acta, 1840, 1838–1849. 10.1016/j.bbagen.2014.01.033 [DOI] [PubMed] [Google Scholar]
- Singh, T. , & Katiyar, S. K. (2013). Honokiol inhibits non‐small cell lung cancer cell migration by targeting PGE₂‐mediated activation of β‐catenin signaling. PLoS ONE, 8, e60749 10.1371/journal.pone.0060749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skroblin, P. , Grossmann, S. , Schäfer, G. , Rosenthal, W. , & Klussmann, E. (2010). Mechanisms of protein kinase A anchoring. International Review of Cell and Molecular Biology, 283, 235–330. 10.1016/S1937-6448(10)83005-9 [DOI] [PubMed] [Google Scholar]
- Skronska‐Wasek, W. , Gosens, R. , Königshoff, M. , & Baarsma, H. A. (2018). WNT receptor signalling in lung physiology and pathology. Pharmacology & Therapeutics, 187, 150–166. 10.1016/j.pharmthera.2018.02.009 [DOI] [PubMed] [Google Scholar]
- Sohal, S. S. , Reid, D. , Soltani, A. , Ward, C. , Weston, S. , Muller, H. K. , … Walters, E. H. (2010). Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology, 15, 930–938. 10.1111/j.1440-1843.2010.01808.x [DOI] [PubMed] [Google Scholar]
- Soltani, A. , Reid, D. W. , Sohal, S. S. , Wood‐Baker, R. , Weston, S. , Muller, H. K. , & Walters, E. H. (2010). Basement membrane and vascular remodelling in smokers and chronic obstructive pulmonary disease: A cross‐sectional study. Respiratory Research, 11, 105 10.1186/1465-9921-11-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, E.‐J. , Olsson, A.‐K. , & Moustakas, A. (2015). Reprogramming during epithelial to mesenchymal transition under the control of TGFβ. Cell Adhesion & Migration, 9, 233–246. 10.4161/19336918.2014.983794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, S. S. , Knighton, D. R. , Zheng, J. , Ten Eyck, L. F. , & Sowadski, J. M. (1992). Structural framework for the protein kinase family. Annual Review of Cell Biology, 8, 429–462. 10.1146/annurev.cb.08.110192.002241 [DOI] [PubMed] [Google Scholar]
- Thevenot, P. T. , Saravia, J. , Jin, N. , Giaimo, J. D. , Chustz, R. E. , Mahne, S. , … Cormier, S. A. (2013). Radical‐containing ultrafine particulate matter initiates epithelial‐to‐mesenchymal transitions in airway epithelial cells. American Journal of Respiratory Cell and Molecular Biology, 48, 188–197. 10.1165/rcmb.2012-0052OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery, J. P. , Acloque, H. , Huang, R. Y. J. , & Nieto, M. A. (2009). Epithelial–mesenchymal transitions in development and disease. Cell, 139, 871–890. 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- Torphy, T. J. , Undem, B. J. , Cieslinski, L. B. , Luttmann, M. A. , Reeves, M. L. , & Hay, D. W. (1993). Identification, characterization and functional role of phosphodiesterase isozymes in human airway smooth muscle. The Journal of Pharmacology and Experimental Therapeutics, 265, 1213–1223. [PubMed] [Google Scholar]
- Tröger, J. , Moutty, M. C. , Skroblin, P. , & Klussmann, E. (2012). A‐kinase anchoring proteins as potential drug targets. British Journal of Pharmacology, 166, 420–433. 10.1111/j.1476-5381.2011.01796.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong, T. , Sauter, W. , McKay, J. D. , Hosgood, H. D. , Gallagher, C. , Amos, C. I. , … Hung, R. J. (2010). International Lung Cancer Consortium: Coordinated association study of 10 potential lung cancer susceptibility variants. Carcinogenesis, 31, 625–633. 10.1093/carcin/bgq001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita, S. , & Yonemura, S. (1999). Cortical actin organization: Lessons from ERM (ezrin/radixin/moesin) proteins. The Journal of Biological Chemistry, 274, 34507–34510. 10.1074/jbc.274.49.34507 [DOI] [PubMed] [Google Scholar]
- Vogelmeier, C. F. , Criner, G. J. , Martinez, F. J. , Anzueto, A. , Barnes, P. J. , Bourbeau, J. , … Agustí, A. (2017). Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. American Journal of Respiratory and Critical Care Medicine, 195, 557–582. 10.1164/rccm.201701-0218PP [DOI] [PubMed] [Google Scholar]
- Walker, J. K. L. , Penn, R. B. , Hanania, N. A. , Dickey, B. F. , & Bond, R. A. (2011). New perspectives regarding β2‐adrenoceptor ligands in the treatment of asthma. British Journal of Pharmacology, 163, 18–28. 10.1111/j.1476-5381.2010.01178.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Xu, J. , Yang, L. , Xu, Y. , Zhang, X. , Bai, C. , … He, J. (2018). Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): A national cross‐sectional study. Lancet, 391, 1706–1717. 10.1016/S0140-6736(18)30841-9 [DOI] [PubMed] [Google Scholar]
- Wang, Q. , Wang, Y. , Zhang, Y. , Zhang, Y. , & Xiao, W. (2013). The role of uPAR in epithelial–mesenchymal transition in small airway epithelium of patients with chronic obstructive pulmonary disease. Respiratory Research, 14, 67 10.1186/1465-9921-14-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, B. C. , Liebler, J. M. , Luby‐Phelps, K. , Nicholson, A. G. , Crandall, E. D. , du Bois, R. M. , & Borok, Z. (2005). Induction of epithelial–mesenchymal transition in alveolar epithelial cells by transforming growth factor‐β1: Potential role in idiopathic pulmonary fibrosis. The American Journal of Pathology, 166, 1321–1332. 10.1016/S0002-9440(10)62351-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, M. Y. , Porte, J. , Knox, A. J. , Weinreb, P. H. , Maher, T. M. , Violette, S. M. , … Jenkins, G. (2009). Lysophosphatidic acid induces αvβ6 integrin‐mediated TGF‐β activation via the LPA2 receptor and the small G protein Gαq . The American Journal of Pathology, 174, 1264–1279. 10.2353/ajpath.2009.080160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg, M. , & Neilson, E. G. (2009). Biomarkers for epithelial–mesenchymal transitions. The Journal of Clinical Investigation, 119, 1429–1437. 10.1172/JCI36183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, A. , Dong, Z. , & Yang, T. (2006). Prostaglandin D2 inhibits TGF‐β1‐induced epithelial‐to‐mesenchymal transition in MDCK cells. American Journal of Physiology. Renal Physiology, 291, F1332–F1342. 10.1152/ajprenal.00131.2006 [DOI] [PubMed] [Google Scholar]
- Zhang, S. , Da, L. , Yang, X. , Feng, D. , Yin, R. , Li, M. , … Xu, L. (2014). Celecoxib potentially inhibits metastasis of lung cancer promoted by surgery in mice, via suppression of the PGE2‐modulated β‐catenin pathway. Toxicology Letters, 225, 201–207. 10.1016/j.toxlet.2013.12.014 [DOI] [PubMed] [Google Scholar]
- Zhou, W. , Mo, X. , Cui, W. , Zhang, Z. , Li, D. , Li, L. , … Gao, J. (2016). Nrf2 inhibits epithelial–mesenchymal transition by suppressing snail expression during pulmonary fibrosis. Scientific Reports, 6, 38646 10.1038/srep38646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo, H. , Han, B. , Poppinga, W. J. , Ringnalda, L. , Kistemaker, L. E. M. , Halayko, A. J. , … Schmidt, M. (2018). Cigarette smoke up‐regulates PDE3 and PDE4 to decrease cAMP in airway cells. British Journal of Pharmacology, 175, 2988–3006. 10.1111/bph.14347 [DOI] [PMC free article] [PubMed] [Google Scholar]