

Abstract
Myocardial fibrosis is a key histopathological component that drives the progression of heart disease leading to heart failure and constitutes a therapeutic target. Recent preclinical and clinical studies have implicated galectin‐3 (Gal‐3) as a pro‐fibrotic molecule and a biomarker of heart disease and fibrosis. However, our knowledge is poor on the mechanism(s) that determine the blood level or regulate cardiac expression of Gal‐3. Recent studies have demonstrated that enhanced β‐adrenoceptor activity is a determinant of both circulating concentration and cardiac expression of Gal‐3. Pharmacological or transgenic activation of β‐adrenoceptors leads to increased blood levels of Gal‐3 and up‐regulated cardiac Gal‐3 expression, effect that can be reversed with the use of β‐adrenoceptor antagonists. Conversely, Gal‐3 gene deletion confers protection against isoprenaline‐induced cardiotoxicity and fibrogenesis. At the transcription level, β‐adrenoceptor stimulation activates cardiac mammalian sterile‐20‐like kinase 1, a pivotal kinase of the Hippo signalling pathway, which is associated with Gal‐3 up‐regulation. Recent studies have suggested a role for the β‐adrenoceptor‐Hippo signalling pathway in the regulation of cardiac Gal‐3 expression thereby contributing to the onset and progression of heart disease. This implies a therapeutic potential of the suppression of Gal‐3 expression. In this review, we discuss the effects of β‐adrenoceptor activity on Gal‐3 as a biomarker and causative mediator in the setting of heart disease and point out pivotal knowledge gaps.
Linked Articles
This article is part of a themed section on Adrenoceptors—New Roles for Old Players. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.14/issuetoc
Abbreviations
- AF
atrial fibrillation
- BIM
Bcl‐2 interacting mediator of cell death
- CRD
carbohydrate recognition and binding domain
- CTGF
connective tissue growth factor
- DCM
dilated cardiomyopathy
- Gal‐3
galectin‐3
- HF
heart failure
- IR
ischaemia–reperfusion
- KO
knockout
- Lats
large tumour suppressor homolog
- MACE
major adverse cardiovascular events
- MCP
modified citrus pectin
- MI
myocardial infarction
- Mst1
mammalian sterile‐20 like kinase‐1
- TAZ
transcriptional co‐activator with PDZ‐binding motif
- TG
transgenic
- YAP
Yes‐associated protein
1. INTRODUCTION
Galectin‐3 (Gal‐3) is a β‐galactoside‐binding lectin of the galectin family that consists of 15 members in mammals. The common molecular structure of all galectins is the C‐terminal carbohydrate recognition/binding domain (CRD; Figure 1), through which galectins bind to and regulate activity of glycoproteins (Cooper, 2002; Rabinovich & Toscano, 2009). With its unique structure different from other members of the galectin family, Gal‐3 is able to form pentamers, when the concentration of Gal‐3 monomers is high, with enhanced capacity and stability of ligand binding (Figure 1; Cooper, 2002; Rabinovich & Toscano, 2009; Suthahar et al., 2018). Gal‐3 binds to glycoproteins as its endogenous ligands and alters their functionality. Glycoproteins are known to account for 50% to 70% of total proteins and are widely distributed in the nucleus, cytoplasm, cellular membrane, and extracellular matrix. This allows Gal‐3 to exert pleiotropic actions under pathological conditions (Figure 1).
Figure 1.
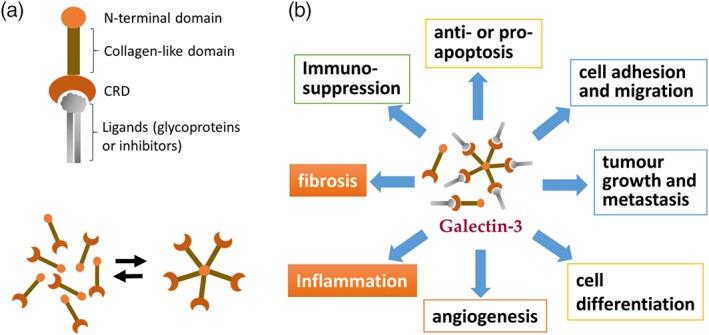
Structure of Gal‐3 and its biological actions through binding to glycoproteins. (a) Gal‐3 is one of 15 members of the β‐galactoside‐binding lectin family. A common structure of galectins is C‐terminal carbohydrate recognition‐binding domain (CRD) by which galectins bind with glycoproteins. When concentration of Gal‐3 is higher, Gal‐3 monomers form pentamers with increased binding capability to glycoproteins via CRD and stability of the complex. (b) The pleotropic bioactivities of Gal‐3 are achieved through interactions with numerous glycoproteins localized within cells, on cellular membrane, or extracellular matrix
With its expression significantly and rapidly induced under diseased conditions, Gal‐3 has received great interest over other galectins for its role in a variety of pathological conditions including cancer, diabetes, inflammation, and heart disease (Figure 1). In almost all animal models of heart disease studied so far, cardiac Gal‐3 expression is up‐regulated (Calvier et al., 2015; Frunza et al., 2016; Gonzalez et al., 2014, 2016; Nguyen, Su, Vizi, et al., 2018; Vergaro et al., 2015; Yu et al., 2013). Current development of anti‐Gal‐3 drugs is to target the CRD to neutralize Gal‐3 from binding with glycoproteins (Table 1). There is good evidence that Gal‐3 promotes inflammation and fibrosis leading to adverse cardiac remodelling and decompensation (Rabinovich & Toscano, 2009). The pro‐fibrotic role of Gal‐3 has been indicated by an increasing number of studies in vitro and in vivo. By inhibiting Gal‐3 with genetic or pharmacological means, preclinical studies have shown cardiac benefits (Arrieta et al., 2017; Calvier et al., 2013; Jaquenod De Giusti, Ure, Rivadeneyra, Schattner, & Gomez, 2015; Yu et al., 2013). Clinical studies have reported high circulating levels of Gal‐3 in patients with heart failure (HF), myocardial infarction (MI), or atrial fibrillation (AF; de Boer et al., 2011; Gurses et al., 2015; Kornej et al., 2015; Wu, Su, et al., 2015). High Gal‐3 levels have been shown to predict the long‐term risk of HF, AF, or total mortality in patients with heart disease or in cohorts of general population (de Boer et al., 2011, 2018; Ho et al., 2012, 2013, 2014). Based on these findings, Gal‐3 has been regarded as a biomarker and a causative mediator of heart disease. However, more recent studies have also revealed inconsistency regarding Gal‐3 as a biomarker in predicting disease severity or adverse prognosis with reasons currently unclear (vide infra).
Table 1.
List of Gal‐3 inhibitors recently tested in preclinical studies. All inhibitors target the carbohydrate recognition and binding domain (CRD) of Gal‐3
| Drugs | Features | Reference |
|---|---|---|
| TDG | Thiodigalactoside and derivatives | Bum‐Erdene et al. (2013) |
| Modified citrus pectins (MCP) | Shorter, non‐branched, galactose‐rich carbohydrate chains from plant citrus pectins via pH and temperature modifications; MW >1,000 kDa | Vergaro et al. (2016) |
| GM‐CT‐01, GM‐CT‐02 | Galactomannan, MW ~50 kDa | Takemoto et al. (2016) |
| 33DFTG | 3,3′‐dideoxy‐3,3′‐bis‐[4‐(3‐fluorophenyl)‐1H‐1,2,3‐triazol‐1‐yl]‐1,1′‐sulfanediyl‐di‐β‐D‐galactopyranoside; K D = 14 nM | Chen, Cao, Leffler, Nilsson, and Panjwani (2017) |
| TD139 | Thiodigalactoside analogue, rapidly absorbed and inhaled, t 1/2 7 hr, MW 648, K D = 14 nM | Mackinnon et al. (2012) |
| GCS‐100 | A modified citrus pectin carbohydrate | Streetly et al. (2010) |
| Lactulosyl‐L‐leucine (Lac‐L‐Leu) | Synthetic low‐molecular weight carbohydrate‐based compound | |
| N‐acetyllactosamine (N‐Lac) | Semi‐synthetic carbohydrate‐based compound that simulates the component of many glycoproteins, MW 383 | Yu et al. (2013) |
| Ac‐SDKP | N‐acetyl‐seryl‐aspartyl‐lysyl‐proline | Liu et al. (2009) |
| bis‐(3‐deoxy‐3‐(3‐methoxybenzamido)‐β‐D‐galactopyranosyl) sulfane | Thiodigalactoside bis‐benzamido derivative, K D = 60 nM | MacKinnon et al. (2008) |
In the setting of heart disease and HF, the sympathetic nervous system is activated leading to enhanced and sustained stimulation of cardiac β‐adrenoceptors (Cohn et al., 1984 ; Kaye et al., 1994 ; Lymperopoulos, Rengo, & Koch, 2013 ; Triposkiadis et al., 2009), which forms the basis for therapy with β‐adrenoceptor antagonists (β‐blockers). There is emerging evidence that activation of β‐adrenoceptors affects the role of Gal‐3 in heart disease, either as a biomarker or as a disease mediator. Importantly, recent studies have revealed that activation of β‐adrenoceptors increases circulating levels of Gal‐3 and directly regulates expression of Gal‐3 in the heart.
2. GAL‐3 AS A BIOMARKER OF HEART DISEASE: INFLUENCE OF β‐ADRENOCEPTOR ACTIVATION
2.1. Clinical findings of Gal‐3 as a biomarker of heart disease
Clinical studies have suggested a role of increased circulating levels of Gal‐3 in predicting the severity of heart disease or a high risk of major adverse cardiovascular events (MACE) including HF (Ho et al., 2013; van der Velde et al., 2016), poor survival (Bayes‐Genis et al., 2014; Lok et al., 2013), post‐MI cardiac events (Di Tano et al., 2017; Grandin et al., 2012; Maiolino et al., 2015), as well as diastolic dysfunction (Wu, Li, et al., 2015) and severity of atrial fibrosis (Hernandez‐Romero et al., 2017; Takemoto et al., 2016; Wu, Su, et al., 2015). French et al. reported in HF patients (n = 1,385) that elevated Gal‐3 level was associated with a greater risk of MACE (hazard ratio = 1.96 for each doubling in Gal‐3) and that such association was most pronounced among patients diagnosed as HF with preserved ejection fraction (hazard ratio = 3.30; French et al., 2016). In chronic HF patients, high Gal‐3 level predicts increased risk of MACE, with the predictive power increased when Gal‐3 was persistently high (Motiwala et al., 2013). Studies have consistently shown that in patients with AF who had undergone ablation therapy, high Gal‐3 level is an independent predictor of AF recurrence (Clementy et al., 2016; Wu, Su, et al., 2015). In patients with ischaemic heart disease or in healthy population cohorts, high Gal‐3 level predicts the risk for newly onset of HF or AF (Grandin et al., 2012; Ho et al., 2012, 2013, 2014; van der Velde, Meijers, et al., 2016).
2.2. Mechanisms responsible for increase in circulating Gal‐3 level
Although the usefulness of Gal‐3 as a biomarker of heart disease has gained support from the findings of many clinical studies, more recent studies have revealed inconsistency, questioning the usefulness of Gal‐3 in predicting MACE, even though higher blood levels of Gal‐3 are consistent findings in almost all reports (Abou Ezzeddine et al., 2016; Arangalage et al., 2016; Begg et al., 2018; Besler et al., 2017; de Boer et al., 2018; Huang et al., 2018; Jansen et al., 2016; Lopez et al., 2015; Stoltze Gaborit et al., 2016). For instance, in studies on community cohorts or HF patients, multiple biomarkers were employed and compared for individual prognostic power and Gal‐3 was found to have insignificant clinical value (Bayes‐Genis et al., 2014; de Boer et al., 2018). In patients with non‐ischaemic cardiomyopathy, presence of myocardial fibrosis, detected by late‐gadolinium enhancement of cardiac magnetic resonance, but not Gal‐3 level, have prognostic value for MACE (Hu et al., 2016). The reason for the controversies regarding Gal‐3 as a biomarker remains unclear but is related, at least in part, to our poor understanding on the mechanisms that determine the elevated blood level of Gal‐3.
Animal studies have shown up‐regulation of Gal‐3 in diseased hearts (de Boer et al., 2014; Frunza et al., 2016; Martinez‐Martinez, Calvier, et al., 2015; Nguyen, Su, Vizi, et al., 2018; Yu et al., 2013). A few clinical studies, in which cardiac biopsies were obtained from patients with HF or AF, consistently showed increased cardiac Gal‐3 expression (Besler et al., 2017; Hernandez‐Romero et al., 2017; Lopez‐Andres et al., 2012; Takemoto et al., 2016). These findings led to the assumption that increased blood levels of Gal‐3 are derived from the diseased heart. However, in patients with AF or severe HF due to cardiomyopathy, cardiac release of Gal‐3 is not evident, indicated by the absence of a trans‐cardiac Gal‐3 gradient (Kornej et al., 2015; Nguyen, Su, Vizi, et al., 2018; Truong et al., 2014). In HF patients, a positive correlation between blood and myocardial levels of Gal‐3 was reported in one study (Lopez et al., 2015), but not in another (Besler et al., 2017). Clinical studies have consistently reported a strong and inverse correlation between circulating Gal‐3 levels and renal dysfunction (estimated GFR), indicating that the impaired renal clearance of Gal‐3 leads to elevation of blood Gal‐3 (Figure 2; Abou Ezzeddine et al., 2015; Gopal et al., 2012; Meijers et al., 2014; Motiwala et al., 2013; Zamora et al., 2014). Indeed, Gal‐3 levels are markedly increased in patients who are under dialysis therapy due to end‐stage renal failure (Drechsler et al., 2015).
Figure 2.
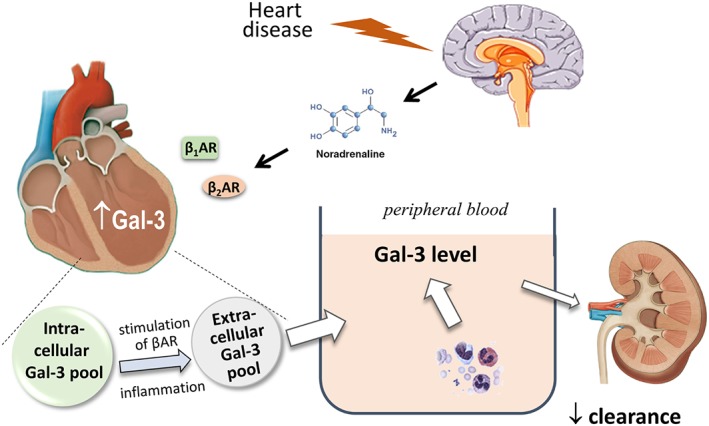
Schematic diagram depicting mechanisms responsible for elevated circulating levels of Gal‐3 in heart disease. Haemodynamic stress associated with heart disease mediates activation of the sympathetic nervous system. Diseases such as cardiomyopathy or myocardial infarction are associated with increased cardiac content of Gal‐3. Cardiac Gal‐3 presents as a non‐releasable intracellular pool or a releasable extracellular pool. Cardiac release of Gal‐3 into the circulation may not necessarily occur when the extracellular Gal‐3 pool is limited despite a sizable intracellular Gal‐3 pool. Cardiac release of Gal‐3 is evident in the presence of myocardial inflammation or β‐adrenoceptor (β1AR, β2AR) stimulation. Systemic inflammation with activation of circulating inflammatory cells contributes to elevation of circulating Gal‐3. Furthermore, reduced renal clearance causes elevated circulating Gal‐3 levels
2.3. β‐Adrenoceptor activity influences the circulating level of Gal‐3
A characteristic of heart disease is activation of the sympatho‐β‐adrenergic system (Cohn et al., 1984; Hartupee & Mann, 2017; Kaye et al., 1994) but there are no studies on the interactions between β‐blocking therapy and Gal‐3 levels. Sanders‐van Wijk et al. analysed data from two HF trials, TIME‐CHF and GISSI‐HF, both consisting of newly diagnosed HF patients, and explored the possible association between a single Gal‐3 concentration measured at the time of diagnosis and therapeutic effects by medications including β‐blockers. By dividing patients into Gal‐3 low‐ and high‐subgroups, use of β‐blockers was associated with improvement of the 5‐year survival in Gal‐3 low‐ but not in Gal‐3 high‐subgroup patients (Sanders‐van Wijk et al., 2016). However, this study has several limitations including a single measure of Gal‐3 and lack of data on renal function (Sanders‐van Wijk et al., 2016). In this context, treatment with the β‐blocker carvedilol in patients with end‐stage renal failure had no effect on circulating Gal‐3 levels measured 12 months following treatment (Roberts et al., 2017).
A recent study has investigated patients with different types of cardiomyopathy as well as four animal models to explore the mechanisms responsible for elevation of circulating Gal‐3 level. In patients with hypertrophic cardiomyopathy, ischaemic cardiomyopathy, or dilated cardiomyopathy (DCM), elevation of circulating Gal‐3 level is entirely dependent on the presence of renal dysfunction (by estimated GFR) without evidence for cardiac release, measured as the trans‐cardiac Gal‐3 gradient (Nguyen, Su, Vizi, et al., 2018). Experimentally, elevated cardiac content of Gal‐3 was observed in all four heart disease models (Nguyen, Su, Vizi, et al., 2018): isoprenaline cardiotoxicity, transgenic (TG) cardiomyopathy mice with cardiac overexpression of human β2‐adrenoceptors (β2‐TG; Nguyen et al., 2015; Xu et al., 2011), TG mice with DCM due to cardiac overexpression of mammalian sterile‐20 like kinase‐1 (Mst1‐TG; Yamamoto et al., 2003), and ischaemia–reperfusion (IR). Circulating Gal‐3 level is elevated in β2‐TG, IR, and isoprenaline models, and the cardiac source of circulating Gal‐3 was confirmed in these mouse models by demonstrating a trans‐cardiac gradient of Gal‐3 (Figure 2; Nguyen, Su, Vizi, et al., 2018). In the Mst1‐TG model, however, while the cardiac content of Gal‐3 is the highest (approximately 50‐fold) relative to other models (sevenfold to 18‐fold), circulating Gal‐3 concentration was unchanged (Nguyen, Su, Vizi, et al., 2018). These findings suggest the presence of a non‐releasable “intracellular Gal‐3 pool” and a releasable “extracellular Gal‐3 pool” (Figure 2). The proposed existence of two different Ga‐3 pools may be helpful to explain the findings from the DCM model of Mst1‐TG mice, in which cardiac Gal‐3 content is markedly elevated without change in circulation levels of Gal‐3, relative to control mice (Nguyen, Su, Vizi, et al., 2018), suggesting the absence of the “extracellular pool.” In contrast, in other disease models studied, cardiac content of Gal‐3 is in proportion to the increment of circulating Gal‐3 levels, indicating the presence of both Gal‐3 pools leading to cardiac Gal‐3 spillover into the circulation (Nguyen, Su, Vizi, et al., 2018). Administration of isoprenaline to wild‐type mice induced parallel increases in cardiac and circulating content of Gal‐3, which can be effectively inhibited by β‐adrenoceptor antagonists (Nguyen et al., 2019). Interestingly, in the Mst1‐TG mice without increase in the circulating Gal‐3 level at basal conditions, isoprenaline treatment for 2 days markedly increased circulating levels of Gal‐3, implying a “permissive” role of β‐adrenoceptor activation in mediating expansion of the “extracellular Gal‐3 pool” and cardiac release of Gal‐3 in this DCM model (Figure 2; Nguyen, Su, Vizi, et al., 2018).
Gal‐3 is expressed in a variety of cell types, most notably macrophages (MacKinnon et al., 2008; Rabinovich & Toscano, 2009). The Mst1‐TG model showed a lack of of inflammatory cell infiltration into the myocardium, which again differs from the other three animal models (vide supra), including isoprenaline treatment, which exhibited high densities of inflammatory cells in the myocardium and circulating levels of Gal‐3 (Nguyen, Su, Vizi, et al., 2018; Nguyen et al., 2019). This implies an association between the elevated blood level of Gal‐3 and cardiac inflammatory status. It has been increasingly appreciated that myocardial inflammation contributes to cardiac fibrosis and HF (Dick & Epelman, 2016). In patients with either DCM or inflammatory cardiomyopathy, cardiac expression of Gal‐3 is correlated with the density of inflammatory cells in the myocardium (Besler et al., 2017). An extreme situation is MI or IR that triggers regional and systemic inflammation (Fang, Du, Gao, & Dart, 2010; Liu et al., 2011; Nguyen, Su, Vizi, et al., 2018), and there are studies on patients with acute MI showing increased blood levels of inflammatory biomarkers including MMP‐9, TNF‐α, and Gal‐3 (Fang et al., 2010; Meijers, van der Velde, Pascual‐Figal, & de Boer, 2015; van der Velde et al., 2016).
Many different factors increase circulating levels of Gal‐3 in heart disease. As illustrated in Figure 2, renal dysfunction is associated with high Gal‐3 levels because of a reduced renal clearance of Gal‐3, while activation of circulating inflammatory cells leads to Gal‐3 secretion directly into the circulation. Although the cardiac content of Gal‐3 is increased in almost all types of heart disease, the cardiac release of Gal‐3 into the circulation is disease‐specific, depending on presence of the releasable “extracellular Gal‐3 pool” (Figure 2). Isoprenaline treatment increased total Gal‐3 content together with increased “extracellular pool” leading to cardiac Gal‐3 spillover into the circulation (Nguyen, Su, Vizi, et al., 2018). Thus, changes in circulating Gal‐3 is aetiology‐specific and may not necessarily reflect the cardiac Gal‐3 content (Nguyen, Su, Vizi, et al., 2018). Importantly, β‐adrenoceptor‐mediated increase in both circulating and cardiac Gal‐3 levels can be effectively reversed by the use of the corresponding receptor antagonists (Nguyen, Su, Vizi, et al., 2018; Zhao et al., 2019). These findings call for clinical research to explore the influence of β‐blocking therapy on dynamic changes in the level of Gal‐3, in relation to the overall therapeutic efficacy of β‐blockers.
Under conditions of cardiac pressure overload or MI, histological examination showed that increased Gal‐3 expression is mainly due to regional inflammatory cells and fibroblasts (Frunza et al., 2016; Yu et al., 2013). Cardiomyocytes express ample amounts of β‐adrenoceptors and activation of these receptors by pharmacological and genetic means is known to promote cardiomyocyte death, myocardial inflammation, and fibrosis (Engelhardt et al., 2001; Hartupee & Mann, 2017; Nguyen, Su, Vizi, et al., 2018; Xiao et al., 2018; Xu et al., 2011). In this regard, inflammatory cells express β‐adrenoceptors (β2‐adrenoceptors are the predominant subtype) and there is evidence for activation of inflammatory cells mediated by β2‐adrenoceptors (Padro & Sanders, 2014). Following a short‐term stimulation with isoprenaline or in β2‐TG mice, up‐regulation of cardiac Gal‐3 expression is derived from cardiomyocytes, in addition to inflammatory cells and fibroblasts (Nguyen, Su, Kiriazis, et al., 2018; Nguyen, Su, Vizi, et al., 2018; Nguyen et al., 2019). This is particularly the case for the Mst1‐TG model of DCM with very high cardiac Gal‐3 content and interstitial fibrosis without inflammatory cell infiltration (Nguyen, Su, Vizi, et al., 2018; Nguyen et al., 2019). In this model, immunofluorescence staining of the LV reveals that cardiomyocytes, but not other cell types, are strongly stained for Gal‐3 (Nguyen et al., 2019). Thus, cardiomyocytes express Gal‐3 at a high level. Cardiac Gal‐3 content is increased in β2‐TG mice at a young age and then it progressively increases with ageing in parallel to the progression of cardiomyopathy, myocardial fibrosis, and inflammation (Nguyen, Su, Kiriazis, et al., 2018; Xu et al., 2011). Thus, the progressive increase in cardiac Gal‐3 content in this model is likely to be attributable to the age‐dependent increase in the cardiac densities of inflammatory cells and fibroblasts (Nguyen et al., 2015; Nguyen, Su, Vizi, et al., 2018).
3. ROLE OF Gal‐3 IN HEART DISEASE AS A CAUSATIVE MEDIATOR
3.1. Preclinical findings on the role of Gal‐3 in heart disease
A consistent finding from preclinical and clinical studies is the elevated Gal‐3 content in the diseased heart due to a variety of aetiologies (de Boer et al., 2014; Frunza et al., 2016; Martinez‐Martinez, Calvier, et al., 2015; Nguyen, Su, Vizi, et al., 2018; Yu et al., 2013). In mouse models of cardiomyopathy, cardiac expression of other members of the galectin family, such as galectin‐1, ‐8, and ‐9, was either unchanged or only moderately increased (Nguyen, Su, Kiriazis, et al., 2018; Nguyen et al., 2019), leading to the speculation that Gal‐3 was the causative mediator. The role of Gal‐3 in heart disease has been explored preclinically by testing effects of Gal‐3 inhibitors or use of Gal‐3 gene knockout (Gal‐3 KO) mice (Table 2).
Table 2.
Summary of cardiovascular benefits of Gal‐3 gene deletion in comparison with that of Gal‐3 inhibitor in similar disease models
| Disease model (species) | Effect (relative to control) | Reference |
|---|---|---|
| Atherosclerosis by ApoE KO (aged mouse) |
 inflammation↓, atherosclerotic lesions↓ inflammation↓, atherosclerotic lesions↓ |
Nachtigal et al. (2008) |
| Atherosclerosis by ApoE KO + high fat diet (mouse) |
 atherosclerotic lesions↓, aortic plaque volume↓, plaque collagen↓, plaque size↓ atherosclerotic lesions↓, aortic plaque volume↓, plaque collagen↓, plaque size↓ |
MacKinnon et al. (2013) |
 atherosclerotic lesions ↓ atherosclerotic lesions ↓ | ||
| Hypertension by aldosterone + high salt (mouse) |
 inflammation↓, fibrosis ↓ (by 50%), vascular hypertrophy↓ inflammation↓, fibrosis ↓ (by 50%), vascular hypertrophy↓ |
Calvier et al. (2013) |
 inflammation ↓, fibrosis ↓ (by 30%), vascular hypertrophy ↓ inflammation ↓, fibrosis ↓ (by 30%), vascular hypertrophy ↓ | ||
| TAC (mouse) |
 fibrosis ↓, hypertrophy ↔, LV function↑ fibrosis ↓, hypertrophy ↔, LV function↑ |
Yu et al. (2013) |
| Chronic Ang II infusion (mouse) |
 fibrosis↓, LV function↑ fibrosis↓, LV function↑ |
Yu et al. (2013) |
| Myocardial infarction (mouse) |
 inflammation↓, fibrosis↓, LV remodelling↑, LV function↓ inflammation↓, fibrosis↓, LV remodelling↑, LV function↓ |
Gonzalez et al. (2014) |
| Hypertension by aldosterone + high salt (mouse) |
 inflammation↓, fibrosis↓, cardiac and renal hypertrophy ↓ inflammation↓, fibrosis↓, cardiac and renal hypertrophy ↓ |
Calvier et al. (2015) |
 inflammation↓, fibrosis↓, cardiac and renal hypertrophy ↓ inflammation↓, fibrosis↓, cardiac and renal hypertrophy ↓ | ||
| Hypertension by aldosterone + high salt (mouse) |
 inflammation↓ inflammation↓ |
Martinez‐Martinez, Calvier, et al. (2015) |
| CVB3‐induced acute myocarditis (mouse) |
 inflammation ↓, fibrosis ↓ (by 51%) inflammation ↓, fibrosis ↓ (by 51%) |
Jaquenod De Giusti et al. (2015) |
 inflammation ↓, fibrosis ↓ (by 32%) inflammation ↓, fibrosis ↓ (by 32%) | ||
| Autoimmune myocarditis (mouse) |
 inflammation ↓ inflammation ↓ |
Kovacevic et al. (2018) |
| Chronic Ang II infusion (mouse) |
 inflammation ↓, fibrosis ↓, hypertrophy ↔ inflammation ↓, fibrosis ↓, hypertrophy ↔ |
Gonzalez et al. (2016) |
| TAC (mouse) |
 fibrosis ↔, cardiomyocyte hypertrophy ↑ (early phase), fibrosis ↔, cardiomyocyte hypertrophy ↑ (early phase),LV remodelling and dysfunction ↔ |
Frunza et al. (2016) |
| Trypanosoma cruzi infection (mouse) |
 inflammation ↓, fibrosis ↓ inflammation ↓, fibrosis ↓ |
Pineda et al. (2015) |
| Acute myocardial infarction (mouse) |
 inflammation ↓ inflammation ↓ |
Mosleh et al. (2018) |
| Isoprenaline cardiotoxicity (mouse) |
 inflammation ↓, fibrosis ↓, hypertrophy ↔, LV function ↑ inflammation ↓, fibrosis ↓, hypertrophy ↔, LV function ↑ |
Zhao et al. (2019) |
| Cardiomyopathy due to Mst1‐TG (mouse) |
 chamber dilatation ↓, organ congestion ↓, fibrosis ↓, LV function ↑ chamber dilatation ↓, organ congestion ↓, fibrosis ↓, LV function ↑ |
Nguyen et al. (2019) |
 chamber dilatation ↔, organ congestion ↔, fibrosis ↔, cardiac contractile function ↔ chamber dilatation ↔, organ congestion ↔, fibrosis ↔, cardiac contractile function ↔ | ||
| Cardiomyopathy due to β2AR‐TG (mouse) |
 inflammation ↔, fibrosis ↔, chamber dilatation ↔, inflammation ↔, fibrosis ↔, chamber dilatation ↔,organ congestion ↔, LV function ↔ |
Nguyen, Su, Kiriazis, et al. (2018) |
 inflammation ↔, fibrosis ↔, chamber dilatation ↔, organ congestion ↔, LV function ↔ inflammation ↔, fibrosis ↔, chamber dilatation ↔, organ congestion ↔, LV function ↔ |
Note:  : Gal‐3 gene deletion;
: Gal‐3 gene deletion;  : treatment with Gal‐3 inhibitors; ISO, isoprenaline; ↔ no change; ↓ reduced; ↑ improved; Ang II, angiotensin II; KO, gene knockout; LV, left ventricle; TAC, transverse aortic constriction; TG, cardiomyocyte‐restricted transgenic overexpression.
: treatment with Gal‐3 inhibitors; ISO, isoprenaline; ↔ no change; ↓ reduced; ↑ improved; Ang II, angiotensin II; KO, gene knockout; LV, left ventricle; TAC, transverse aortic constriction; TG, cardiomyocyte‐restricted transgenic overexpression.
Experimental studies in vitro and in vivo have yielded several lines of evidence for Gal‐3 as a mediator of heart disease. First, when tested ex vivo, Gal‐3 promoted fibroblast proliferation and expression of fibrogenic genes (Frunza et al., 2016; Ibarrola et al., 2018; Mackinnon et al., 2012; Takemoto et al., 2016; Vergaro et al., 2016; Yu et al., 2013). Gal‐3 also promoted the innate immune response by stimulating monocyte migration and expression of inflammatory cytokines and chemokines (Jaquenod De Giusti et al., 2015; Nachtigal et al., 2008; Sharma et al., 2004; Vasta, 2012). Moreover, in a rat study, chronic delivery of Gal‐3 via implanted osmotic minipump with a cannula inserted into the pericardium caused cardiac histopathology including fibrosis (Sharma et al., 2004). Second, studies have shown positive correlation between the cardiac Gal‐3 level or Gal‐3 positive cell density and the extent of LV remodelling and dysfunction (Frunza et al., 2016; Nguyen, Su, Kiriazis, et al., 2018). Third, in models of pressure overload (e.g., hypertension or transverse aortic constriction), myocarditis or drug‐induced cardiotoxicity, treatment with Gal‐3 inhibitors such as N‐acetyl‐lactosamine, modified citrus pectin (MCP), or galactomannan polysaccharide attenuated cardiovascular inflammation and fibrosis (Arrieta et al., 2017; Calvier et al., 2015; Jaquenod De Giusti et al., 2015; Lopez et al., 2015; Martinez‐Martinez, Lopez‐Andres, et al., 2015; Martinez‐Martinez, Calvier, et al., 2015; Sharma et al., 2004, 2008; Yu et al., 2013). In a sheep model of rapid pacing‐induced AF, atrial levels of both TGF‐β1 and Gal‐3 were significantly elevated, which was reversed by treatment with the Gal‐3 inhibitor GM‐CT‐01 (Takemoto et al., 2016). Fourth, in animal models of heart disease due to increased LV afterload (e.g., hypertension and aortic constriction), Gal‐3 KO mice exhibited less severe cardiac fibrosis and inflammation and a better preservation of cardiac function relative to their wild‐type controls (Calvier et al., 2013, 2015; Martinez‐Martinez, Lopez‐Andres, et al., 2015; Yu et al., 2013; Table 2). Based on these findings, Gal‐3 is likely to be a disease mediator forming a therapeutic target. However, there are studies reporting contradictory findings questioning the efficacy of Gal‐3 inhibitors or even the Gal‐3 KO model (vide infra).
3.2. Effect of Gal‐3 inhibitors or Gal‐3 gene deletion on heart disease with enhanced β‐adrenoceptor activity
Using TG mice with hyperaldosteronism (due to overexpression of aldosterone synthase gene), Vergaro et al. studied effects of the Gal‐3 inhibitor MCP on cardiac damage induced by isoprenaline (300 mg·kg−1, s.c., twice daily for 2 days; Vergaro et al., 2016). MCP treatment, commenced 2 days after administration of isoprenaline, significantly reduced cardiac levels of Gal‐3, inflammatory markers, and fibrosis by approximately 50–60% (Vergaro et al., 2016). In a recent study, Gal‐3 KO and wild‐type control mice were subjected to continuous isoprenaline administration (30 mg·kg−1·day−1 for 7 days via osmotic minipump). Minipumps were removed at Day 7 to washout isoprenaline overnight prior to echocardiography. In WT mice, isoprenaline treatment increased cardiac Gal‐3 content by eightfold, induced LV hypertrophy by 20%, and suppressed LV fractional shortening by 35%. Myocardial collagen content was increased by 50% together with up‐regulation of a number of fibrotic and inflammatory genes (Zhao et al., 2019; Table 2). All these phenotypes at global and molecular levels were not present in Gal‐3 KO mice, except for a similar degree of LV hypertrophy (Zhao et al., 2019). Studies have shown sustained elevation of cardiac Gal‐3 by a 3‐week period of administration with isoprenaline at 30 mg·kg−1·day−1 (Wang et al., 2016).
There are also controversial reports regarding the effectiveness of currently available Gal‐3 inhibitors (Frunza et al., 2016; Kirk & Frangogiannis, 2018; Nguyen et al., 2019). One of the models that did not respond to Gal‐3 inhibitor therapy is the β2‐TG model in mice. Cardiac overexpression of β2‐adrenoceptors leads to an age‐dependent progression of fibrosis, inflammation, hypertrophy, LV dilatation, spontaneous ventricular arrhythmias, and premature death due either to malignant arrhythmias or HF (Du et al., 2000; Gao et al., 2003; Liggett et al., 2000; Nguyen et al., 2015; Xu et al., 2011). At the molecular level, the β2‐TG mouse strains show enhancement of oxidative stress, p38‐MAPK activity, fibrotic and inflammatory signalling, and cardiomyocyte apoptosis (Gao et al., 2003; Lee et al., 2013; Peter et al., 2007; Xu et al., 2011). In the failing heart, β1‐adrenoceptors are known to undergo down‐regulation and desensitization while the density of β2‐adrenoceptors is maintained (Bristow, 2000) and undergoes redistribution across the cellular membrane of cardiomyocytes thereby mediating a broad intracellular signalling similar to that of β1‐adrenoceptors (Gorelik, Wright, Lyon, & Harding, 2013; Nikolaev et al., 2010). Thus, the β2‐TG mice represent a useful model for study on the role of β‐adrenoceptor signalling in heart disease. Using β2‐TG mice, a recent study has tested the effect of Gal‐3 inhibitor N‐acetyl‐lactosamine (5 mg·kg−1 i.p., three injections per week, 3 weeks) or MCP (200–500 mg−1·kg−1·day−1 for 3 months; Nguyen, Su, Kiriazis, et al., 2018) and observed that both drugs conferred no benefits as measured by Gal‐3 expression, myocardial fibrosis, or cardiac dysfunction (Nguyen, Su, Kiriazis, et al., 2018). The ineffectiveness of Gal‐3 inhibitors in this model is unexpected. One possible reason is the high cardiac Gal‐3 content in the β2‐TG heart (up to 18‐fold) relative to other animal models of heart disease that responded to MCP treatment (by twofold to fivefold; Calvier et al., 2015; Frunza et al., 2016; Gonzalez et al., 2016; Vergaro et al., 2015; Yu et al., 2013). Gal‐3 monomers undergo self‐association to form pentamers under conditions of high Gal‐3 content and the presence of glycoprotein ligands (Figure 1; Ahmad et al., 2004; Suthahar et al., 2018). It is likely that in a diseased myocardium with a high content of Gal‐3, the proportion of Gal‐3 in the form of pentamers is higher. It remains unknown whether the current Gal‐3 neutralizing inhibitors are equipotent or less potent in inhibiting pentamerized Gal‐3 relative to Gal‐3 monomers. A similar ineffectiveness of MCP treatment (up to 500 mg−1·kg−1·day−1 for 4 months) was also observed in the DCM model of Mst1‐TG mice in which cardiac content of Gal‐3 is very high (Nguyen et al., 2019). Another uncertainty of the current CRD‐targeted Gal‐3 inhibitors (Table 1) is their limited pharmacological profile including capability of penetrating cellular membranes required for neutralizing intracellular and nuclear Gal‐3, considering their lipophobic nature and/or large molecular sizes (Table 1). These questions call for further investigation.
4. ACTIVATION OF β‐ADRENOCEPTORS UPREGULATES GAL‐3 EXPRESSION IN CARDIOVASCULAR DISEASE: THERAPEUTIC POTENTIAL
4.1. Experimental evidence for cardiovascular benefits by suppression of Gal‐3 gene expression
Elevated Gal‐3 content in the diseased heart has been well documented. By using Gal‐3 KO mice subjected to a variety of diseases, a number of studies have reported cardiovascular benefits by Gal‐3 gene deletion (Table 2). In some disease models, cardiovascular benefits, measured as reduced severity in cardiac fibrosis, inflammation, or dysfunction, appear to be greater after Gal‐3 gene deletion than that after treatment with Gal‐3 inhibitors (Calvier et al., 2013; Jaquenod De Giusti et al., 2015; Nguyen et al., 2019; Table 2). The only exception is MI, in which Gal‐3 KO mice suffered from more severe ventricular remodelling and a higher mortality due to insufficient inflammation and fibrotic healing (Gonzalez et al., 2014; Table 2). Collectively, these studies indicate suppression of Gal‐3 expression as a potential therapeutic approach complementing the current development of CRD‐targeted Gal‐3 inhibitors. To achieve this, understanding the signal mechanism(s) regulating Gal‐3 expression is critical to identify the therapeutic target(s). However, our knowledge is very limited on the mechanism responsible for Gal‐3 up‐regulation (Suthahar et al., 2018).
4.2. The β‐adrenoceptor‐Hippo pathway regulates Gal‐3 expression
Very recent studies have demonstrated a pivotal role of the β‐adrenoceptor ‐Hippo signalling pathway in regulating Gal‐3 expression in the heart. The Hippo signalling pathway is the essential regulator of organ size (Yu, Zhao, & Guan, 2015). This pathway was discovered initially in Drosophila and recently in mammals with Mst1/2 as the mammalian orthologue of Drosophila Hippo kinase (Figure 3; Yu et al., 2015). The core of this pathway is Mst/large tumour suppressor homologue (Lats) forming a kinase cascade that regulates activity of the down‐stream transcription co‐regulator yes‐activated protein (YAP) and its paralogue transcriptional co‐activator with PDZ‐binding motif (TAZ). YAP/TAZ forms the main signal output of the Hippo pathway through binding to numerous transcription factors and epigenetic factors to regulate expression of a range of target genes thereby promoting cell proliferation and survival (Ikeda & Sadoshima, 2016; Plouffe, Hong, & Guan, 2015; Yu et al., 2015).
Figure 3.
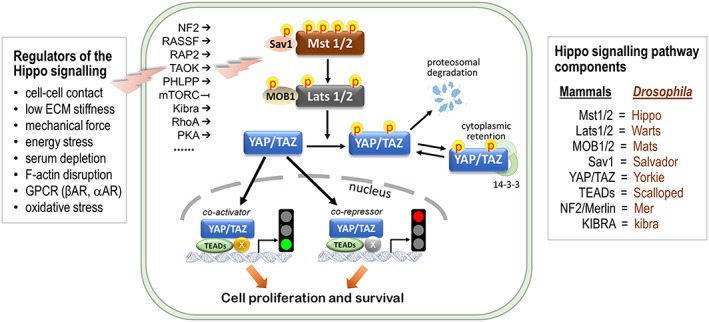
The Hippo signalling pathway: key components and regulatory mechanisms. The Hippo pathway is highly conserved in Drosophila and in mammals, for example, mammalian sterile‐20‐like kinase (Mst) is the orthologue of Hippo, as listed in the diagram for comparison (right). The effector of the Hippo pathway is yes‐activated protein (YAP) and its paralogue transcriptional coactivator with PDZ‐binding motif (TAZ). As transcription co‐activator or co‐repressor, nuclear YAP/TAZ interacts with numerous transcriptional and epigenetic factors (indicated as “×”) controlling expression of target genes to promote cell proliferation and survival. A number of factors have been shown to regulate Mst1/Lats (large tumour suppressor homologue) kinase cascade through complex signalling mechanisms currently only partially understood (left, ➜, activation; ⊣ inhibition). Mst1/2 and Lats1/2 mediates YAP/TAZ phosphorylation/inactivation. This leads to cytoplasm retention of YAP/TAZ after binding with 14‐3‐3 or proteosomic degradation. ECM: extracellular matrix; MOB1/2: Mps one binder kinase activator like 1/2; mTORC: mammalian target of rapamycin complex; NF2: neurofibromin 2 (also known as merlin); PCMT1: protein‐L‐isoaspartate(D‐aspartate) O‐methyltransferase; PHLPP: PH domain and leucine rich repeat protein phosphatase; RAP2: Ras‐related GTPase; RASSF: Ras association domain‐containing proteins; RhoA: a small GTPase; Sav1: salvador homologue 1; TAOK: Tao kinase; TEADs: tea domain family members; β‐ or α‐AR: β‐ or α‐adrenoceptor
Factors such as cell–cell contact, mechanical force, soft extracellular matrix stiffness, or energy stress lead to activation of the Mst1/2–Lats1/2 kinase cascade via a number of signalling molecules that regulate activity of the Hippo pathway partly through mediating phosphorylation of Mst1 at different sites (Figure 3; Del Re et al., 2010; Matsuda et al., 2016; Meng et al., 2018; Plouffe et al., 2016; Yu et al., 2015). Mst1/2 interacts with the scaffold protein Salvador homologue (Sav) to form an active enzyme complex that phosphorylates and activates Lats1/2. Lats1/2 then binds to the kinase Mob1 homologue (MOB1) to induce inhibitory phosphorylation of YAP/TAZ. The nuclear localization of YAP/TAZ is essential for its transcription activity. YAP phosphorylation at Ser127, localized within the binding domain of 14‐3‐3, promotes YAP binding to 14‐3‐3 with increased cytoplasmic retention, while Ser397 phosphorylation might initiate ubiquitinated degradation (Figure 3).
Recent studies have shown regulation of the Hippo pathway by hormones and membrane receptors including GPCRs (Ikeda & Sadoshima, 2016; Yu et al., 2012, 2015). Coupling of the β‐adrenoceptors with the Hippo pathway was first proposed by Yu et al. (Yu et al., 2012) who discovered in mammalian cell lines that overexpression of glucagon receptor, β2‐adrenoceptors or a constitutively active form of Gαs increased YAP phosphorylation. In cell lines, such as mouse embryonic fibroblasts, Kim et al. also showed that activation of PKA using the AC activator forskolin induced YAP phosphorylation indicating YAP inactivation by cAMP/PKA signalling (Kim et al., 2013). Dethlefsen et al. recently demonstrated in MCF‐7 cells (human adenocarcinoma cell line) that stimulation with catecholamines induced YAP phosphorylation, which was inhibited by propranolol (Dethlefsen et al., 2017). Yu et al. observed that activation of a variety of other GPCRs that couple to G12/13, Gq, Gi, or Go, including α1B‐adrenoceptors and angiotensin AT1 receptors, mediate YAP de‐phosphorylation including the Ser127 site with increased nuclear localization of YAP (Yu et al., 2012).
Activation of the sympatho‐β‐adrenergic system is a characteristic feature of heart disease (Cohn et al., 1984; Kaye et al., 1994), and an increasing body of research has implicated a role of the Hippo pathway in heart disease (Chen et al., 2014; Del Re et al., 2013; Ikeda & Sadoshima, 2016; Leach et al., 2017; Plouffe et al., 2016; Wang, Liu, Heallen, & Martin, 2018). Thus, it is likely that activation of the β‐adrenoceptor‐Hippo signalling with resultant suppression of YAP activity might contribute to the pathogenesis of heart disease. This possibility has not been addressed in the in vitro studies already mentioned (Dethlefsen et al., 2017; Kim et al., 2013; Yu et al., 2012). For instance, whereas Lats1/2 and YAP activity can be directly regulated bypassing Mst1 (Plouffe et al., 2016; Wang et al., 2018; Yu et al., 2015), it is unclear whether activation of the Hippo pathway by β‐adrenoceptor stimulation involves Mst1 in the heart. It is also unknown what are the downstream target genes that are regulated by the β‐adrenoceptor‐Hippo signalling pathway and contribute to the development and worsening of heart disease. Recent studies have provided several lines of evidence for regulation of Gal‐3 expression by the cardiac β‐adrenoceptor‐Hippo signalling pathway. First, pharmacological or TG activation of β‐adrenoceptors (isoprenaline treatment or β2‐TG) induces cardiac up‐regulation of Mst1 and Gal‐3 as well as YAP phosphorylation, which were all dependent on isoprenaline dosage and treatment duration (Zhao et al., 2019). These in vivo findings were supported by a recent study in cultured rat cardiomyocytes showing that isoprenaline stimulation induced rapid phosphorylation of both Mst1/2 and YAP (Dalal, Connelly, Singh, & Singh, 2018). In addition, cell experiments showed that the isoprenaline‐mediated effects were abolished by PKA inhibitors (H89 and PKI 14‐22) but mimicked with the use of forskolin (Zhao et al., 2019), implying PKA‐dependent signalling. These effects of isoprenaline were partly suppressed by selective or non‐selective β‐adrenoceptor antagonists including propranolol, carvedilol, atenolol, or ICI‐118551 (Nguyen, Su, Vizi, et al., 2018; Zhao et al., 2019). Second, in mice with TG cardiac expression of dominant‐negative kinase‐dead Mst1 gene (dnMst1‐TG) that inhibits native Mst1 activity of cardiomyocytes (Yamamoto et al., 2003), isoprenaline‐induced up‐regulation of Gal‐3 in dnMst1‐TG hearts was significantly inhibited (Zhao et al., 2019). In contrast, cardiac Gal‐3 expression is increased by up to 50‐fold in Mst1‐TG hearts, together with inhibitory YAP hyper‐phosphorylation (Nguyen et al., 2019; Zhao et al., 2019). In the Mst1‐TG model with activated cardiac Hippo signalling, isoprenaline‐induced Gal‐3 expression was 10 times greater than that in wild‐type mouse hearts (Zhao et al., 2019). Third, in cardiomyoblast H9c2 cells, isoprenaline induces YAP phosphorylation and Gal‐3 up‐regulation, and siRNA‐mediated knockdown of YAP is associated with an augmented expression of Gal‐3 (Zhao et al., 2019). This finding indicates that YAP is a transcription co‐repressor for Gal‐3 expression. Hence removal of such inhibitory “STOP” signal following activation of the β‐adrenoceptor‐Hippo pathway results in Gal‐3 up‐regulation (Figure 4). Recent studies have also shown that YAP/TAZ acts as transcriptional co‐repressor and hence its inhibition leads to up‐regulation of certain target genes (Figure 3; Beyer et al., 2013; Kim, Kim, Johnson, & Lim, 2015; Valencia‐Sama et al., 2015; Zhao et al., 2019). Further support of this view comes from a recent study on the Mst1‐TG model, known to have YAP‐Ser127 hyper‐phosphorylation and inactivation (Zhao et al., 2019), showing that RNA sequencing of the Mst1‐TG heart revealed that 68% of YAP‐target genes are up‐regulated (Zhao et al., 2019).
Figure 4.
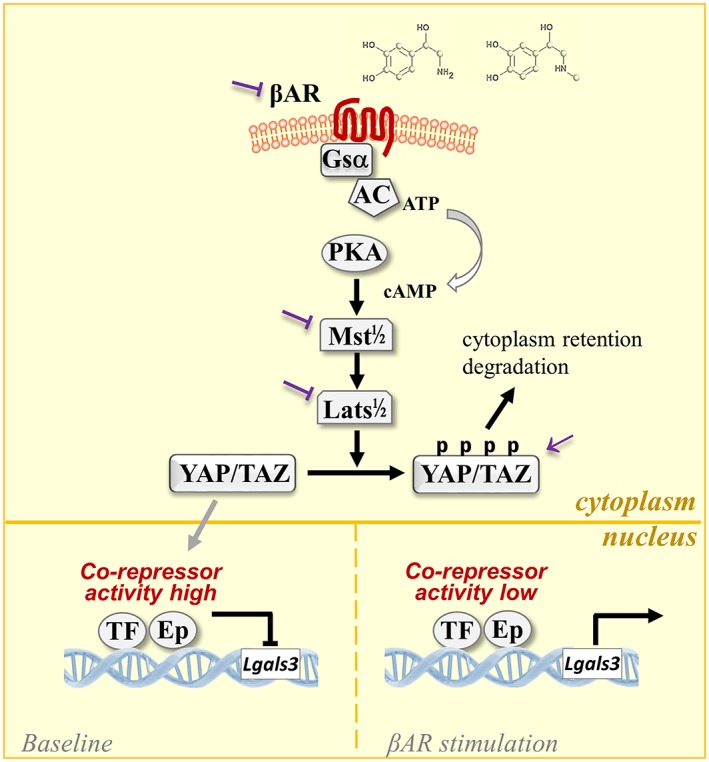
Schematic representation showing stimulation of β‐adrenoceptors (βAR) leading to activation of the Hippo signalling pathway through phosphorylation and inactivation of the transcription co‐regulator of YAP with resultant disinhibition of expression of Gal‐3 gene (Lgals3). Ep: epigenetic factors; Lats1/2: large tumour suppressor homolog1/2; Mst1/2: mammalian sterile‐20 like kinase1/2; P: phosphorylation; TAZ: transcriptional coactivator with PDZ‐binding motif; TF: transcription factors; YAP: yes‐activated protein. Possible therapeutic targets for drug intervention are indicated as inhibition (⊣) or activation (→)
4.3. Targeting the β‐adrenoceptor‐Hippo‐Gal‐3 signalling in heart disease
Cardiac Mst1 is up‐regulated in experimental models of MI (Odashima et al., 2007), pathological hypertrophy (Odashima et al., 2007), arrhythmic cardiomyopathy (Chen et al., 2014), diabetic cardiomyopathy (Hu et al., 2018), or cardiomyopathy due to cardiac overexpression of β1‐ or β2‐adrenoceptors (Chen et al., 2014; Lee, Yan, Vatner, & Vatner, 2015; Peter et al., 2007; Zhao et al., 2019). In cardiac biopsy samples from patients with DCM or arrhythmogenic cardiomyopathy, and HF due to ischaemic cardiomyopathy or DCM, YAP phosphorylation at Ser127 is increased implying activation of the Hippo signalling pathway (Chen et al., 2014; Hou et al., 2017; Leach et al., 2017).
Lee et al. studied the effect of cardiomyocyte‐specific inactivation of Mst1 in the β1‐TG cardiomyopathy model by crossing the β1‐TG mice with dnMst1‐TG mice to generate dual TG mice and mice with control genotypes (Lee et al., 2015). They studied cardiac phenotype in animals at 18–20 months of age and revealed inhibition by approximately 60% of cardiomyocyte apoptosis and myocardial fibrosis together with improved LV function and survival (Lee et al., 2015). In Mst1 gene deleted mice, similar protection against cardiotoxicity by isoprenaline treatment (60 mg−1·kg−1·day−1 for 24 hr via minipump) was observed (Lee et al., 2015). These findings underscore a pivotal role of Mst1 signalling in this β1‐TG model of cardiomyopathy, albeit potential changes in Gal‐3 were not studied.
Nguyen et al. studied the effect of Gal‐3 gene deletion on cardiomyopathy phenotype of the β2‐TG mice with up‐regulation of Gal‐3. Gal‐3 KO mice were crossed with the β2‐TG mice to generate TG/KO mice and mice with control genotypes (Nguyen, Su, Kiriazis, et al., 2018). Animals of different genotypes were studied at 3 and 9 months of age, representing early and advanced phase of cardiomyopathy respectively. Despite complete elimination of Gal‐3, no detectable improvement in cardiomyopathy phenotype in TG/KO versus β2‐TG mice including myocardial fibrosis, expression of fibrotic or inflammatory genes, and cardiac dysfunction by echocardiography (Nguyen, Su, Kiriazis, et al., 2018; Table 2). These findings are in contrast to several reports showing beneficial effects of Gal‐3 gene deletion in the setting of heart disease (Table 2; Calvier et al., 2015; Martinez‐Martinez, Calvier, et al., 2015; Martinez‐Martinez, Lopez‐Andres, et al., 2015; Pineda et al., 2015; Vergaro et al., 2015; Yu et al., 2013). The reason for different outcomes following Gal‐3 gene deletion is unclear but suggest that the causative role of Gal‐3 in heart disease is aetiology‐specific and that in the absence of Gal‐3, redundant pathways of inflammatory and fibrotic signalling are able to drive cardiac fibrogenesis in the β2‐TG model.
Another relevant model is the Mst1‐TG with overexpression of Mst1 restricted to cardiomyocytes (Yamamoto et al., 2003). This model exhibits cardiac chamber dilatation, profound contractile and diastolic dysfunction, absence of compensatory hypertrophy, and severe interstitial fibrosis (Nguyen et al., 2019; Pretorius et al., 2009; Yamamoto et al., 2003). In this model of DCM, cardiac expression of Gal‐3 increased up to 50‐fold at mRNA and protein levels (Nguyen et al., 2019; Zhao et al., 2019). RNA sequencing of Mst1‐TG hearts revealed dysregulated transcriptsome including activated apoptotic and fibrotic signalling, metabolic disturbance, and up‐regulation of components of the Hippo pathway including Lats1/2, Mob1, Sav1, YAP/TAZ as well as 14‐3‐3 (Nguyen et al., 2019). The effect of Gal‐3 gene deletion was recently studied by crossing Mst1‐TG mice with Gal‐3 KO mice (Nguyen et al., 2019). Although having limited effect on the dysregulated cardiac transcriptome, Gal‐3 gene deletion in Mst1‐TG mice significantly reduced myocardial collagen content by 25% and 40%, respectively, when studied at 3 and 8 months of age (Nguyen et al., 2019; Table 2). This was associated with reversal of numerous up‐regulated fibrotic genes by RNA sequencing and validated by RT‐PCR including α‐smooth muscle actin, connective tissue growth factor (CTGF), TGF‐β1, lysyl oxidase, fibronectin, and procollagen (Nguyen et al., 2019).
The therapeutic potential of the β‐adrenoceptor‐Hippo ‐Gal‐3 signalling pathway is indicated by the findings that isoprenaline‐induced cardiac dysfunction and fibrosis, but not myocardial hypertrophy, are prevented in mice with Gal‐3 gene deletion (Zhao et al., 2019). CTGF is an important pro‐fibrotic growth factor with its expression up‐regulated by 7‐ 10‐fold by isoprenaline treatment or in the Mst1‐TG heart (Nguyen, Su, Kiriazis, et al., 2018; Nguyen, Su, Vizi, et al., 2018; Nguyen et al., 2019; Zhao et al., 2019). Interestingly, Gal‐3 gene deletion largely or totally reversed CTGF up‐regulation induced either by isoprenaline treatment (Zhao et al., 2019) or by TG overexpression of Mst1 (Nguyen et al., 2019), which implies that up‐regulation of CTGF by isoprenaline treatment or TG activation of Mst1 is indirectly regulated through Gal‐3. Bcl‐2 interacting mediator of cell death (BIM) is a BH3‐only pro‐apoptotic protein of the Bcl‐2 family (Shamas‐Din, Brahmbhatt, Leber, & Andrews, 2011). As an upstream apoptosis initiator, BIM concomitantly inhibits anti‐apoptotic Bcl‐2 proteins (Bcl‐2 and MCL‐1) while activates proapoptotic Bcl‐2 proteins (Bax and Bak; Bouillet & O'Reilly, 2009; Czabotar, Colman, & Huang, 2009). A recent study showed that cardiac expression of the pro‐apoptotic protein BIM is similarly up‐regulated by the β‐adrenoceptor‐Hippo pathway (Zhao et al., 2019). An earlier study demonstrated that isoprenaline‐induced cardiomyocygte apoptosis was abolished in BIM KO mouse hearts (Lee et al., 2013). Considering that apoptotic cardiomyocyte loss is a key mechanism in the progression of cardiomyopathy and HF (Konstantinidis, Whelan, & Kitsis, 2012; Zhu & Sun, 2018), future study is warranted to investigate the regulation of BIM expression by cardiac β‐adrenoceptor‐Hippo signalling pathway and its role in the pathogenesis of cardiomyopathy. It is important to identify other disease‐related genes that are up‐regulated by the β‐adrenoceptor‐Hippo signalling pathway.
A number of studies have provided strong evidence for an anti‐fibrotic and anti‐apoptotic property of YAP in the heart. Del Re et al. reported that cardiomyocyte‐restricted KO of YAP gene resulted in severe fibrosis, myocyte apoptosis, cardiac dilatation, poor systolic function, and premature death due to HF (Del Re et al., 2013). Several studies have shown that genetic activation of YAP suppressed post‐MI fibrosis with improved cardiac regeneration (Del Re et al., 2013; Heallen et al., 2013; Leach et al., 2017; Lin et al., 2014; Odashima et al., 2007; Xin et al., 2013). dnMst1‐TG mice subjected to MI exhibited attenuation of cardiac fibrosis, myocyte apoptosis, and LV dysfunction (Odashima et al., 2007). Furthermore, genetic activation of cardiac Mst1 led to DCM with severe interstitial fibrosis (Nguyen et al., 2019; Pretorius et al., 2009; Yamamoto et al., 2003). In addition, endothelial‐targeted expression of Mst1 exacerbates diabetic cardiomyopathy with more pronounced LV systolic and diastolic dysfunction (Hu et al., 2018). As previously indicated, genetic inhibition of cardiac Mst1‐activity significantly reduced myocardial fibrosis in the β1‐TG model (Lee et al., 2015). Thus, targeting the cardiac Hippo signalling pathway might have potential therapeutic implications for patients with heart disease.
5. CONCLUDING REMARKS
Activation of the sympathetic nervous system is a hallmark of heart disease. The finding that the sympatho‐β‐adrenergic activity is a determinant of the circulating Gal‐3 level adds to our current interpretation of Gal‐3 as a clinical biomarker in heart disease. In addition, this raises question on the potential influence of treatments with β‐blockers on Gal‐3 levels and the reciprocal effects on the efficacy of β‐adrenoceptor antagonists in patients with heart disease. This calls for preclinical and clinical studies with serial measurements of Gal‐3 during the study period in relation to the use of β‐blockers. The significance of the β‐adrenoceptor‐Hippo signalling pathway is twofold. First, enhanced expression of Gal‐3 and BIM driven by this signalling pathway enriches our understanding on the mechanisms responsible for the adverse biological action of β‐adrenoceptor activation, particularly pathogenesis of myocardial fibrosis and cardiomyocyte loss. Second, it is feasible to target the β‐adrenoceptor‐Hippo signalling pathway to suppress cardiac de novo expression of disease‐driven genes, like those for Gal‐3, BIM, and others yet to be identified. This could be achieved by inhibition of Mst/Lats activity, restoration of cardiac YAP activity, or by targeting the down‐stream transcriptional machinery once it is further clarified (Figure 4). Existence of the β‐adrenoceptor‐Hippo‐Gal‐3 signalling pathway provides a potential means for therapeutic inhibition of Gal‐3 expression without affecting the compensatory cardiac β‐adrenoceptor signalling.
5.1. Nomenclature of drugs and molecular targets
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017; Alexander et al., 2017; Alexander, Christopoulos et al., 2017).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
This work is funded by the National Health and Medical Research Council (NHMRC) of Australia (ID 1081710), the NHMRC fellowship to X.J.D. (ID 1043026), and the Nature Science Fund of China (81870300). W.B.Z. and Q.L. are funded by Chinese Overseas Scholarship Council (File numbers: 201603170211 and 201706285111).
Du X‐J, Zhao W‐B, Nguyen M‐N, Lu Q, Kiriazis H. β‐Adrenoceptor activation affects galectin‐3 as a biomarker and therapeutic target in heart disease. Br J Pharmacol. 2019;176:2449–2464. 10.1111/bph.14620
REFERENCES
- Abou Ezzeddine, O. F. , Haines, P. , Stevens, S. , Nativi‐Nicolau, J. , Felker, G. M. , Borlaug, B. A. , … Redfield, M. M. (2015). Galectin‐3 in heart failure with preserved ejection fraction. A RELAX Trial Substudy. JACC Heart Failure, 3, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abou Ezzeddine, O. F. , McKie, P. M. , Scott, C. G. , Rodeheffer, R. J. , Chen, H. H. , Michael Felker, G. , … Redfield, M. M. (2016). Biomarker‐based risk prediction in the community. European Journal of Heart Failure, 18, 1342–1350. 10.1002/ejhf.663 [DOI] [PubMed] [Google Scholar]
- Ahmad, N. , Gabius, H. J. , Andre, S. , Kaltner, H. , Sabesan, S. , Roy, R. , … Brewer, C. F. (2004). Galectin‐3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross‐linked complexes. The Journal of Biological Chemistry, 279, 10841–10847. [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174(Suppl 1), S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174(Suppl 1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. British Journal of Pharmacology, 174(Suppl 1), S1–S16. 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arangalage, D. , Nguyen, V. , Robert, T. , Melissopoulou, M. , Mathieu, T. , Estellat, C. , … Messika‐Zeitoun, D. (2016). Determinants and prognostic value of Galectin‐3 in patients with aortic valve stenosis. Heart, 102, 862–868. 10.1136/heartjnl-2015-308873 [DOI] [PubMed] [Google Scholar]
- Arrieta, V. , Martinez‐Martinez, E. , Ibarrola, J. , Alvarez, V. , Sadaba, R. , Garcia‐Pena, A. , … López‐Andrés, N. (2017). A role for galectin‐3 in the development of early molecular alterations in short‐term aortic stenosis. Clinical Science, 131, 935–949. [DOI] [PubMed] [Google Scholar]
- Bayes‐Genis, A. , de Antonio, M. , Vila, J. , Penafiel, J. , Galan, A. , Barallat, J. , … Lupón, J. (2014). Head‐to‐head comparison of 2 myocardial fibrosis biomarkers for long‐term heart failure risk stratification: ST2 versus galectin‐3. Journal of the American College of Cardiology, 63, 158–166. 10.1016/j.jacc.2013.07.087 [DOI] [PubMed] [Google Scholar]
- Begg, G. A. , Karim, R. , Oesterlein, T. , Graham, L. N. , Hogarth, A. J. , Page, S. P. , … Tayebjee, M. H. (2018). Left atrial voltage, circulating biomarkers of fibrosis, and atrial fibrillation ablation. A prospective cohort study. PLoS ONE, 13, e0189936 10.1371/journal.pone.0189936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besler, C. , Lang, D. , Urban, D. , Rommel, K. P. , von Roeder, M. , Fengler, K. , … Lurz, P. (2017). Plasma and cardiac Galectin‐3 in patients with heart failure reflects both inflammation and fibrosis: Implications for Its Use as a Biomarker. Circulation: Heart Failure, 10: Pii, e003804. [DOI] [PubMed] [Google Scholar]
- Beyer, T. A. , Weiss, A. , Khomchuk, Y. , Huang, K. , Ogunjimi, A. A. , Varelas, X. , & Wrana, J. L. (2013). Switch enhancers interpret TGF‐β and Hippo signaling to control cell fate in human embryonic stem cells. Cell Reports, 5, 1611–1624. [DOI] [PubMed] [Google Scholar]
- Bouillet, P. , & O'Reilly, L. A. (2009). CD95, BIM and T cell homeostasis. Nature Reviews. Immunology, 9, 514–519. [DOI] [PubMed] [Google Scholar]
- Bristow, M. R. (2000). β‐Adrenergic receptor blockade in chronic heart failure. Circulation, 101, 558–569. 10.1161/01.CIR.101.5.558 [DOI] [PubMed] [Google Scholar]
- Bum‐Erdene, K. , Gagarinov, I. A. , Collins, P. M. , Winger, M. , Pearson, A. G. , Wilson, J. C. , … Blanchard, H. (2013). Investigation into the feasibility of thioditaloside as a novel scaffold for galectin‐3‐specific inhibitors. Chembiochem, 14, 1331–1342. 10.1002/cbic.201300245 [DOI] [PubMed] [Google Scholar]
- Calvier, L. , Martinez‐Martinez, E. , Miana, M. , Cachofeiro, V. , Rousseau, E. , Sadaba, J. R. , … López‐Andrés, N. (2015). The impact of galectin‐3 inhibition on aldosterone‐induced cardiac and renal injuries. JACC Heart Failure, 3, 59–67. [DOI] [PubMed] [Google Scholar]
- Calvier, L. , Miana, M. , Reboul, P. , Cachofeiro, V. , Martinez‐Martinez, E. , de Boer, R. A. , … López‐Andrés, N. (2013). Galectin‐3 mediates aldosterone‐induced vascular fibrosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 33, 67–75. 10.1161/ATVBAHA.112.300569 [DOI] [PubMed] [Google Scholar]
- Chen, S. N. , Gurha, P. , Lombardi, R. , Ruggiero, A. , Willerson, J. T. , & Marian, A. J. (2014). The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circulation Research, 114, 454–468. 10.1161/CIRCRESAHA.114.302810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. S. , Cao, Z. , Leffler, H. , Nilsson, U. J. , & Panjwani, N. (2017). Galectin‐3 inhibition by a small‐molecule inhibitor reduces both pathological corneal neovascularization and fibrosis. Investigative Ophthalmology & Visual Science, 58, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementy, N. , Benhenda, N. , Piver, E. , Pierre, B. , Bernard, A. , Fauchier, L. , … Babuty, D. (2016). Serum Galectin‐3 levels predict recurrences after ablation of atrial fibrillation. Science Reports, 6, 34357 10.1038/srep34357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn, J. N. , Levine, T. B. , Olivari, M. T. , Garberg, V. , Lura, D. , Francis, G. S. , … Rector, T. (1984). Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. The New England Journal of Medicine, 311, 819–823. [DOI] [PubMed] [Google Scholar]
- Cooper, D. N. (2002). Galectinomics: Finding themes in complexity. Biochimica et Biophysica Acta, 1572, 209–231. [DOI] [PubMed] [Google Scholar]
- Czabotar, P. E. , Colman, P. M. , & Huang, D. C. (2009). Bax activation by Bim? Cell Death and Differentiation, 16, 1187–1191. [DOI] [PubMed] [Google Scholar]
- Dalal, S. , Connelly, B. , Singh, M. , & Singh, K. (2018). NF2 signaling pathway plays a pro‐apoptotic role in β‐adrenergic receptor stimulated cardiac myocyte apoptosis. PLoS ONE, 13, e0196626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer, R. A. , Lok, D. J. , Jaarsma, T. , van der Meer, P. , Voors, A. A. , Hillege, H. L. , & van Veldhuisen, D. J. (2011). Predictive value of plasma galectin‐3 levels in heart failure with reduced and preserved ejection fraction. Annals of Medicine, 43, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer, R. A. , Nayor, M. , deFilippi, C. R. , Enserro, D. , Bhambhani, V. , Kizer, J. R. , … Ho, J. E. (2018). Association of cardiovascular biomarkers with incident heart failure with preserved and reduced ejection fraction. JAMA Cardiology, 3, 215–224. 10.1001/jamacardio.2017.4987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer, R. A. , van der Velde, A. R. , Mueller, C. , van Veldhuisen, D. J. , Anker, S. D. , Peacock, W. F. , … Maisel, A. (2014). Galectin‐3: A modifiable risk factor in heart failure. Cardiovascular Drugs and Therapy, 28, 237–246. 10.1007/s10557-014-6520-2 [DOI] [PubMed] [Google Scholar]
- Del Re, D. P. , Matsuda, T. , Zhai, P. , Gao, S. , Clark, G. J. , Van Der Weyden, L. , & Sadoshima, J. (2010). Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. The Journal of Clinical Investigation, 120, 3555–3567. 10.1172/JCI43569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Re, D. P. , Yang, Y. , Nakano, N. , Cho, J. , Zhai, P. , Yamamoto, T. , … Sadoshima, J. (2013). Yes‐associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. The Journal of Biological Chemistry, 288, 3977–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen, C. , Hansen, L. S. , Lillelund, C. , Andersen, C. , Gehl, J. , Christensen, J. F. , … Hojman, P. (2017). Exercise‐induced catecholamines activate the hippo tumor suppressor pathway to reduce risks of breast cancer development. Cancer Research, 77, 4894–4904. 10.1158/0008-5472.CAN-16-3125 [DOI] [PubMed] [Google Scholar]
- Di Tano, G. , Caretta, G. , De Maria, R. , Parolini, M. , Bassi, L. , Testa, S. , & Pirelli, S. (2017). Galectin‐3 predicts left ventricular remodelling after anterior‐wall myocardial infarction treated by primary percutaneous coronary intervention. Heart, 103, 71–77. 10.1136/heartjnl-2016-309673 [DOI] [PubMed] [Google Scholar]
- Dick, S. A. , & Epelman, S. (2016). Chronic heart failure and inflammation: What do we really know? Circulation Research, 119, 159–176. [DOI] [PubMed] [Google Scholar]
- Drechsler, C. , Delgado, G. , Wanner, C. , Blouin, K. , Pilz, S. , Tomaschitz, A. , … de Boer, R. A. (2015). Galectin‐3, renal function, and clinical outcomes: Results from the LURIC and 4D studies. Journal of the American Society of Nephrology, 26, 2213–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, X. J. , Gao, X. M. , Wang, B. , Jennings, G. L. , Woodcock, E. A. , & Dart, A. M. (2000). Age‐dependent cardiomyopathy and heart failure phenotype in mice overexpressing β2‐adrenergic receptors in the heart. Cardiovascular Research, 48, 448–454. [DOI] [PubMed] [Google Scholar]
- Engelhardt, S. , Boknik, P. , Keller, U. , Neumann, J. , Lohse, M. J. , & Hein, L. (2001). Early impairment of calcium handling and altered expression of junctin in hearts of mice overexpressing the β1‐adrenergic receptor. The FASEB Journal, 15, 2718–2720. 10.1096/fj.01-0107fje [DOI] [PubMed] [Google Scholar]
- Fang, L. , Du, X. J. , Gao, X. M. , & Dart, A. M. (2010). Activation of peripheral blood mononuclear cells and extracellular matrix and inflammatory gene profile in acute myocardial infarction. Clinical Science, 119, 175–183. 10.1042/CS20100011 [DOI] [PubMed] [Google Scholar]
- French, B. , Wang, L. , Ky, B. , Brandimarto, J. , Basuray, A. , Fang, J. C. , … Cappola, T. P. (2016). Prognostic value of Galectin‐3 for adverse outcomes in chronic heart failure. Journal of Cardiac Failure, 22, 256–262. 10.1016/j.cardfail.2015.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frunza, O. , Russo, I. , Saxena, A. , Shinde, A. V. , Humeres, C. , Hanif, W. , … Frangogiannis, N. G. (2016). Myocardial Galectin‐3 expression is associated with remodeling of the pressure‐overloaded heart and may delay the hypertrophic response without affecting survival, dysfunction, and cardiac fibrosis. The American Journal of Pathology, 186, 1114–1127. 10.1016/j.ajpath.2015.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, X. M. , Agrotis, A. , Autelitano, D. J. , Percy, E. , Woodcock, E. A. , Jennings, G. L. , … du, X. J. (2003). Sex hormones and cardiomyopathic phenotype induced by cardiac β2‐adrenergic receptor overexpression. Endocrinology, 144, 4097–4105. 10.1210/en.2002-0214 [DOI] [PubMed] [Google Scholar]
- Gonzalez, G. E. , Cassaglia, P. , Noli Truant, S. , Fernandez, M. M. , Wilensky, L. , Volberg, V. , … Gelpi, R. J. (2014). Galectin‐3 is essential for early wound healing and ventricular remodeling after myocardial infarction in mice. International Journal of Cardiology, 176, 1423–1425. [DOI] [PubMed] [Google Scholar]
- Gonzalez, G. E. , Rhaleb, N. E. , D'Ambrosio, M. A. , Nakagawa, P. , Liao, T. D. , Peterson, E. L. , … Carretero, O. A. (2016). Cardiac‐deleterious role of galectin‐3 in chronic angiotensin II‐induced hypertension. American Journal of Physiology. Heart and Circulatory Physiology, 311, H1287–H1296. 10.1152/ajpheart.00096.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal, D. M. , Kommineni, M. , Ayalon, N. , Koelbl, C. , Ayalon, R. , Biolo, A. , … Colucci, W. S. (2012). Relationship of plasma galectin‐3 to renal function in patients with heart failure: Effects of clinical status, pathophysiology of heart failure, and presence or absence of heart failure. Journal of the American Heart Association, 1, e000760 10.1161/JAHA.112.000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik, J. , Wright, P. T. , Lyon, A. R. , & Harding, S. E. (2013). Spatial control of the βAR system in heart failure: The transverse tubule and beyond. Cardiovascular Research, 98, 216–224. 10.1093/cvr/cvt005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandin, E. W. , Jarolim, P. , Murphy, S. A. , Ritterova, L. , Cannon, C. P. , Braunwald, E. , & Morrow, D. A. (2012). Galectin‐3 and the development of heart failure after acute coronary syndrome: pilot experience from PROVE IT‐TIMI 22. Clinical Chemistry, 58, 267–273. [DOI] [PubMed] [Google Scholar]
- Gurses, K. M. , Yalcin, M. U. , Kocyigit, D. , Canpinar, H. , Evranos, B. , Yorgun, H. , … Aytemir, K. (2015). Effects of persistent atrial fibrillation on serum galectin‐3 levels. The American Journal of Cardiology, 115, 647–651. [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartupee, J. , & Mann, D. L. (2017). Neurohormonal activation in heart failure with reduced ejection fraction. Nature Reviews. Cardiology, 14, 30–38. 10.1038/nrcardio.2016.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heallen, T. , Morikawa, Y. , Leach, J. , Tao, G. , Willerson, J. T. , Johnson, R. L. , & Martin, J. F. (2013). Hippo signaling impedes adult heart regeneration. Development, 140, 4683–4690. 10.1242/dev.102798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Romero, D. , Vilchez, J. A. , Lahoz, A. , Romero‐Aniorte, A. I. , Jover, E. , Garcia‐Alberola, A. , … Marín, F. (2017). Galectin‐3 as a marker of interstitial atrial remodelling involved in atrial fibrillation. Science Reports, 7, 40378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, J. E. , Liu, C. , Lyass, A. , Courchesne, P. , Pencina, M. J. , Vasan, R. S. , … Levy, D. (2012). Galectin‐3, a marker of cardiac fibrosis, predicts incident heart failure in the community. Journal of the American College of Cardiology, 60, 1249–1256. 10.1016/j.jacc.2012.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, J. E. , Lyass, A. , Lee, D. S. , Vasan, R. S. , Kannel, W. B. , Larson, M. G. , & Levy, D. (2013). Predictors of new‐onset heart failure: Differences in preserved versus reduced ejection fraction. Circulation. Heart Failure, 6, 279–286. 10.1161/CIRCHEARTFAILURE.112.972828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, J. E. , Yin, X. , Levy, D. , Vasan, R. S. , Magnani, J. W. , Ellinor, P. T. , … Benjamin, E. J. (2014). Galectin 3 and incident atrial fibrillation in the community. American Heart Journal, 167, 729–734.e721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, N. , Wen, Y. , Yuan, X. , Xu, H. , Wang, X. , Li, F. , & Ye, B. (2017). Activation of Yap1/Taz signaling in ischemic heart disease and dilated cardiomyopathy. Experimental and Molecular Pathology, 103, 267–275. 10.1016/j.yexmp.2017.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, D. J. , Xu, J. , Du, W. , Zhang, J. X. , Zhong, M. , & Zhou, Y. N. (2016). Cardiac magnetic resonance and galectin‐3 level as predictors of prognostic outcomes for non‐ischemic cardiomyopathy patients. The International Journal of Cardiovascular Imaging, 32, 1725–1733. [DOI] [PubMed] [Google Scholar]
- Hu, J. , Wang, S. , Xiong, Z. , Cheng, Z. , Yang, Z. , Lin, J. , … Sun, D. (2018). Exosomal Mst1 transfer from cardiac microvascular endothelial cells to cardiomyocytes deteriorates diabetic cardiomyopathy. Biochimica et Biophysica Acta ‐ Molecular Basis of Disease, 1864, 3639–3649. 10.1016/j.bbadis.2018.08.026 [DOI] [PubMed] [Google Scholar]
- Huang, Z. , Zhong, J. , Ling, Y. , Zhang, Y. , Lin, W. , Tang, L. , … Li, S. (2018). Diagnostic value of novel biomarkers for heart failure: A meta‐analysis. Herz.. 10.1007/s00059-018-4702-6 [DOI] [PubMed] [Google Scholar]
- Ibarrola, J. , Arrieta, V. , Sadaba, R. , Martinez‐Martinez, E. , Garcia‐Pena, A. , Alvarez, V. , … López‐Andrés, N. (2018). Galectin‐3 down‐regulates antioxidant peroxiredoxin‐4 in human cardiac fibroblasts: A new pathway to induce cardiac damage. Clinical Science, 132, 1471–1485. 10.1042/CS20171389 [DOI] [PubMed] [Google Scholar]
- Ikeda, S. , & Sadoshima, J. (2016). Regulation of myocardial cell growth and death by the Hippo pathway. Circulation Journal, 80, 1511–1519. 10.1253/circj.CJ-16-0476 [DOI] [PubMed] [Google Scholar]
- Jansen, H. , Koenig, W. , Jaensch, A. , Mons, U. , Breitling, L. P. , Scharnagl, H. , … Rothenbacher, D. (2016). Prognostic utility of Galectin‐3 for recurrent cardiovascular events during long‐term follow‐up in patients with stable coronary heart disease: Results of the KAROLA Study. Clinical Chemistry, 62, 1372–1379. [DOI] [PubMed] [Google Scholar]
- Jaquenod De Giusti, C. , Ure, A. E. , Rivadeneyra, L. , Schattner, M. , & Gomez, R. M. (2015). Macrophages and galectin 3 play critical roles in CVB3‐induced murine acute myocarditis and chronic fibrosis. Journal of Molecular and Cellular Cardiology, 85, 58–70. 10.1016/j.yjmcc.2015.05.010 [DOI] [PubMed] [Google Scholar]
- Kaye, D. M. , Lambert, G. W. , Lefkovits, J. , Morris, M. , Jennings, G. , & Esler, M. D. (1994). Neurochemical evidence of cardiac sympathetic activation and increased central nervous system norepinephrine turnover in severe congestive heart failure. Journal of the American College of Cardiology, 23, 570–578. 10.1016/0735-1097(94)90738-2 [DOI] [PubMed] [Google Scholar]
- Kim, M. , Kim, M. , Lee, S. , Kuninaka, S. , Saya, H. , Lee, H. , … Lim, D. S. (2013). cAMP/PKA signalling reinforces the LATS‐YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. The EMBO Journal, 32, 1543–1555. 10.1038/emboj.2013.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M. , Kim, T. , Johnson, R. L. , & Lim, D. S. (2015). Transcriptional co‐repressor function of the hippo pathway transducers YAP and TAZ. Cell Reports, 11, 270–282. 10.1016/j.celrep.2015.03.015 [DOI] [PubMed] [Google Scholar]
- Kirk, J. A. , & Frangogiannis, N. G. (2018). Galectin‐3 in the pathogenesis of heart failure: A causative mediator or simply a biomarker? American Journal of Physiology. Heart and Circulatory Physiology, 314, H1256–H1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis, K. , Whelan, R. S. , & Kitsis, R. N. (2012). Mechanisms of cell death in heart disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 32, 1552–1562. 10.1161/ATVBAHA.111.224915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornej, J. , Schmidl, J. , Ueberham, L. , John, S. , Daneschnejad, S. , Dinov, B. , … Bollmann, A. (2015). Galectin‐3 in patients with atrial fibrillation undergoing radiofrequency catheter ablation. PLoS ONE, 10, e0123574 10.1371/journal.pone.0123574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacevic, M. M. , Pejnovic, N. , Mitrovic, S. , Jovicic, N. , Petrovic, I. , Arsenijevic, N. , … Ljujic, B. (2018). Galectin‐3 deficiency enhances type 2 immune cell‐mediated myocarditis in mice. Immunologic Research, 66, 491–502. [DOI] [PubMed] [Google Scholar]
- Leach, J. P. , Heallen, T. , Zhang, M. , Rahmani, M. , Morikawa, Y. , Hill, M. C. , … Martin, J. F. (2017). Hippo pathway deficiency reverses systolic heart failure after infarction. Nature, 550, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, G. J. , Yan, L. , Vatner, D. E. , & Vatner, S. F. (2015). Mst1 inhibition rescues β1‐adrenergic cardiomyopathy by reducing myocyte necrosis and non‐myocyte apoptosis rather than myocyte apoptosis. Basic Research in Cardiology, 110(2), 7 10.1007/s00395-015-0461-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. Y. , Moujalled, D. , Doerflinger, M. , Gangoda, L. , Weston, R. , Rahimi, A. , … Puthalakath, H. (2013). CREB‐binding protein (CBP) regulates β‐adrenoceptor (β‐AR)‐mediated apoptosis. Cell Death and Differentiation, 20, 941–952. 10.1038/cdd.2013.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggett, S. B. , Tepe, N. M. , Lorenz, J. N. , Canning, A. M. , Jantz, T. D. , Mitarai, S. , … Dorn, G. W. 2nd (2000). Early and delayed consequences of β2‐adrenergic receptor overexpression in mouse hearts: Critical role for expression level. Circulation, 101, 1707–1714. [DOI] [PubMed] [Google Scholar]
- Lin, Z. , von Gise, A. , Zhou, P. , Gu, F. , Ma, Q. , Jiang, J. , … Pu, W. T. (2014). Cardiac‐specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circulation Research, 115, 354–363. 10.1161/CIRCRESAHA.115.303632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Gao, X. M. , Fang, L. , Jennings, N. L. , Su, Y. , Q, X. , … du, X. J. (2011). Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arteriosc Thromb Vasc Biol, 31, 834–841. 10.1161/ATVBAHA.110.220467 [DOI] [PubMed] [Google Scholar]
- Liu, Y. H. , D'Ambrosio, M. , Liao, T. D. , Peng, H. , Rhaleb, N. E. , Sharma, U. , … Carretero, O. A. (2009). N‐acetyl‐seryl‐aspartyl‐lysyl‐proline prevents cardiac remodeling and dysfunction induced by galectin‐3, a mammalian adhesion/growth‐regulatory lectin. American Journal of Physiology. Heart and Circulatory Physiology, 296, H404–H412. 10.1152/ajpheart.00747.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok, D. J. , Lok, S. I. , Bruggink‐Andre de la Porte, P. W. , Badings, E. , Lipsic, E. , van Wijngaarden, J. , … van der Meer, P. (2013). Galectin‐3 is an independent marker for ventricular remodeling and mortality in patients with chronic heart failure. Clinical Research in Cardiology, 102, 103–110. 10.1007/s00392-012-0500-y [DOI] [PubMed] [Google Scholar]
- Lopez, B. , Gonzalez, A. , Querejeta, R. , Zubillaga, E. , Larman, M. , & Diez, J. (2015). Galectin‐3 and histological, molecular and biochemical aspects of myocardial fibrosis in heart failure of hypertensive origin. European Journal of Heart Failure, 17, 385–392. [DOI] [PubMed] [Google Scholar]
- Lopez‐Andres, N. , Rossignol, P. , Iraqi, W. , Fay, R. , Nuee, J. , Ghio, S. , … Lacolley, P. (2012). Association of galectin‐3 and fibrosis markers with long‐term cardiovascular outcomes in patients with heart failure, left ventricular dysfunction, and dyssynchrony: insights from the CARE‐HF (Cardiac Resynchronization in Heart Failure) trial. European Journal of Heart Failure, 14, 74–81. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos, A. , Rengo, G. , & Koch, W. J. (2013). Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circulation Research, 113, 739–753. 10.1161/CIRCRESAHA.113.300308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon, A. C. , Farnworth, S. L. , Hodkinson, P. S. , Henderson, N. C. , Atkinson, K. M. , Leffler, H. , … Sethi, T. (2008). Regulation of alternative macrophage activation by galectin‐3. Journal of Immunology, 180, 2650–2658. 10.4049/jimmunol.180.4.2650 [DOI] [PubMed] [Google Scholar]
- Mackinnon, A. C. , Gibbons, M. A. , Farnworth, S. L. , Leffler, H. , Nilsson, U. J. , Delaine, T. , … Sethi, T. (2012). Regulation of transforming growth factor‐β1‐driven lung fibrosis by galectin‐3. Am J Resp Crit Care Med, 185, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon, A. C. , Liu, X. , Hadoke, P. W. , Miller, M. R. , Newby, D. E. , & Sethi, T. (2013). Inhibition of galectin‐3 reduces atherosclerosis in apolipoprotein E‐deficient mice. Glycobiology, 23, 654–663. 10.1093/glycob/cwt006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiolino, G. , Rossitto, G. , Pedon, L. , Cesari, M. , Frigo, A. C. , Azzolini, M. , … Rossi, G. P. (2015). Galectin‐3 predicts long‐term cardiovascular death in high‐risk patients with coronary artery disease. Arteriosc Thromb Vasc Biol, 35, 725–732. [DOI] [PubMed] [Google Scholar]
- Martinez‐Martinez, E. , Calvier, L. , Fernandez‐Celis, A. , Rousseau, E. , Jurado‐Lopez, R. , Rossoni, L. V. , … López‐Andrés, N. (2015). Galectin‐3 blockade inhibits cardiac inflammation and fibrosis in experimental hyperaldosteronism and hypertension. Hypertension, 66, 767–775. 10.1161/HYPERTENSIONAHA.115.05876 [DOI] [PubMed] [Google Scholar]
- Martinez‐Martinez, E. , Lopez‐Andres, N. , Jurado‐Lopez, R. , Rousseau, E. , Bartolome, M. V. , Fernandez‐Celis, A. , … Cachofeiro, V. (2015). Galectin‐3 participates in cardiovascular remodeling associated with obesity. Hypertension, 66, 961–969. [DOI] [PubMed] [Google Scholar]
- Matsuda, T. , Zhai, P. , Sciarretta, S. , Zhang, Y. , Jeong, J. I. , Ikeda, S. , … Del Re, D. P. (2016). NF2 activates hippo signaling and promotes ischemia/reperfusion injury in the heart. Circulation Research, 119, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijers, W. C. , Januzzi, J. L. , deFilippi, C. , Adourian, A. S. , Shah, S. J. , van Veldhuisen, D. J. , & de Boer, R. A. (2014). Elevated plasma galectin‐3 is associated with near‐term rehospitalization in heart failure: A pooled analysis of 3 clinical trials. Am Heart J, 167, 853–860.e854. [DOI] [PubMed] [Google Scholar]
- Meijers, W. C. , van der Velde, A. R. , Pascual‐Figal, D. A. , & de Boer, R. A. (2015). Galectin‐3 and post‐myocardial infarction cardiac remodeling. European Journal of Pharmacology, 763, 115–121. 10.1016/j.ejphar.2015.06.025 [DOI] [PubMed] [Google Scholar]
- Meng, Z. , Qiu, Y. , Lin, K. C. , Kumar, A. , Placone, J. K. , Fang, C. , … Guan, K. L. (2018). RAP2 mediates mechanoresponses of the Hippo pathway. Nature, 560, 655–660. 10.1038/s41586-018-0444-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosleh, W. , Chaudhari, M. R. , Sonkawade, S. , Mahajan, S. , Khalil, C. , Frodey, K. , … Sharma, U. C. (2018). The therapeutic potential of blocking Galectin‐3 expression in acute myocardial infarction and mitigating inflammation of infarct region: A clinical outcome‐based translational study. Biomarker Insights, 13 10.1177/1177271918771969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motiwala, S. R. , Szymonifka, J. , Belcher, A. , Weiner, R. B. , Baggish, A. L. , Sluss, P. , … Januzzi, J. L. (2013). Serial measurement of galectin‐3 in patients with chronic heart failure: Results from the ProBNP Outpatient Tailored Chronic Heart Failure Therapy (PROTECT) study. European Journal of Heart Failure, 15, 1157–1163. 10.1093/eurjhf/hft075 [DOI] [PubMed] [Google Scholar]
- Nachtigal, M. , Ghaffar, A. , & Mayer, E. P. (2008). Galectin‐3 gene inactivation reduces atherosclerotic lesions and adventitial inflammation in ApoE‐deficient mice. The American Journal of Pathology, 172, 247–255. 10.2353/ajpath.2008.070348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, M. N. , Kiriazis, H. , Ruggiero, D. , Gao, X. M. , Su, Y. , Jian, A. , … du, X. J. (2015). Spontaneous ventricular tachyarrhythmias in β2‐adrenoceptor transgenic mice in relation to cardiac interstitial fibrosis. American Journal of Physiology. Heart and Circulatory Physiology, 309, H946–H957. 10.1152/ajpheart.00405.2015 [DOI] [PubMed] [Google Scholar]
- Nguyen, M. N. , Su, Y. , Kiriazis, H. , Yang, Y. , Gao, X. M. , McMullen, J. R. , … Du, X. J. (2018). Upregulated galectin‐3 is not a critical disease mediator of cardiomyopathy induced by β2‐adrenoceptor overexpression. American Journal of Physiology. Heart and Circulatory Physiology, 314(6), H1169–H1178. 10.1152/ajpheart.00337.2017 [DOI] [PubMed] [Google Scholar]
- Nguyen, M. N. , Su, Y. , Vizi, D. , Fang, L. , Ellims, A. H. , Zhao, W. B. , … Du, X.‐J. (2018). Mechanisms responsible for increased circulating levels of galectin‐3 in cardiomyopathy and heart failure. Sci Reports, 8(1), 8213 10.1038/s41598-018-26115-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, M. N. , Ziemann, M. , Kiriazis, H. , Su, Y. , Donner, D. G. , Zhao, W. B. , … Du, X. J. (2019). Galectin‐3 deficiency ameliorates fibrosis and remodeling in dilated cardiomyopathy mice with enhanced Mst1 signaling. Am J Physiol Heart Circ Physiology, 316, H45–H60. 10.1152/ajpheart.00609.2018 [DOI] [PubMed] [Google Scholar]
- Nikolaev, V. O. , Moshkov, A. , Lyon, A. R. , Miragoli, M. , Novak, P. , Paur, H. , … Gorelik, J. (2010). β2‐Adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science, 327, 1653–1657. 10.1126/science.1185988 [DOI] [PubMed] [Google Scholar]
- Odashima, M. , Usui, S. , Takagi, H. , Hong, C. , Liu, J. , Yokota, M. , & Sadoshima, J. (2007). Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circulation Research, 100, 1344–1352. 10.1161/01.RES.0000265846.23485.7a [DOI] [PubMed] [Google Scholar]
- Padro, C. J. , & Sanders, V. M. (2014). Neuroendocrine regulation of inflammation. Seminars in Immunology, 26, 357–368. 10.1016/j.smim.2014.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter, P. S. , Brady, J. E. , Yan, L. , Chen, W. , Engelhardt, S. , Wang, Y. , … Vatner, D. E. (2007). Inhibition of p38α MAPK rescues cardiomyopathy induced by overexpressed β2‐adrenergic receptor, but not β1‐adrenergic receptor. The Journal of Clinical Investigation, 117, 1335–1343. 10.1172/JCI29576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda, M. A. , Cuervo, H. , Fresno, M. , Soto, M. , & Bonay, P. (2015). Lack of Galectin‐3 prevents cardiac fibrosis and effective immune responses in a murine model of Trypanosoma cruzi infection. J Infectious Dis, 212, 1160–1171. [DOI] [PubMed] [Google Scholar]
- Plouffe, S. W. , Hong, A. W. , & Guan, K. L. (2015). Disease implications of the Hippo/YAP pathway. Trends in Molecular Medicine, 21, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe, S. W. , Meng, Z. , Lin, K. C. , Lin, B. , Hong, A. W. , Chun, J. V. , & Guan, K. L. (2016). Characterization of Hippo pathway components by gene inactivation. Molecular Cell, 64, 993–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretorius, L. , Du, X. J. , Woodcock, E. A. , Kiriazis, H. , Lin, R. C. , Marasco, S. , … McMullen, J. R. (2009). Reduced phosphoinositide 3‐kinase (p110α) activation increases the susceptibility to atrial fibrillation. The American Journal of Pathology, 175, 998–1009. 10.2353/ajpath.2009.090126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich, G. A. , & Toscano, M. A. (2009). Turning ‘sweet’ on immunity: Galectin‐glycan interactions in immune tolerance and inflammation. Nature Reviews. Immunology, 9, 338–352. [DOI] [PubMed] [Google Scholar]
- Roberts, M. A. , Darssan, D. , Badve, S. V. , Carroll, R. P. , Fahim, M. A. , Haluska, B. A. , … Ierino, F. L. (2017). Carvedilol and cardiac biomarkers in dialysis patients: Secondary analysis of a randomized controlled trial. Kidney & Blood Pressure Research, 42, 1033–1044. 10.1159/000485589 [DOI] [PubMed] [Google Scholar]
- Sanders‐van Wijk, S. , Masson, S. , Milani, V. , Rickenbacher, P. , Gorini, M. , Tavazzi, L. T. , … TIME‐CHF Investigators and the GISSI‐HF Investigators . (2016). Interaction of Galectin‐3 concentrations with the treatment effects of β‐blockers and RAS blockade in patients with systolic heart failure: A derivation‐validation study from TIME‐CHF and GISSI‐HF. Clinical Chemistry, 62, 605–616. 10.1373/clinchem.2015.246850 [DOI] [PubMed] [Google Scholar]
- Shamas‐Din, A. , Brahmbhatt, H. , Leber, B. , & Andrews, D. W. (2011). BH3‐only proteins: Orchestrators of apoptosis. Biochimica et Biophysica Acta, 1813, 508–520. [DOI] [PubMed] [Google Scholar]
- Sharma, U. , Rhaleb, N. E. , Pokharel, S. , Harding, P. , Rasoul, S. , Peng, H. , & Carretero, O. A. (2008). Novel anti‐inflammatory mechanisms of N‐Acetyl‐Ser‐Asp‐Lys‐Pro in hypertension‐induced target organ damage. American Journal of Physiology. Heart and Circulatory Physiology, 294, H1226–H1232. 10.1152/ajpheart.00305.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, U. C. , Pokharel, S. , van Brakel, T. J. , van Berlo, J. H. , Cleutjens, J. P. , Schroen, B. , … Pinto, Y. M. (2004). Galectin‐3 marks activated macrophages in failure‐prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation, 110, 3121–3128. 10.1161/01.CIR.0000147181.65298.4D [DOI] [PubMed] [Google Scholar]
- Stoltze Gaborit, F. , Bosselmann, H. , Kistorp, C. , Iversen, K. , Kumler, T. , Gustafsson, F. , … Schou, M. (2016). Galectin 3: Association to neurohumoral activity, echocardiographic parameters and renal function in outpatients with heart failure. BMC Cardiovasc Disorders, 16, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streetly, M. J. , Maharaj, L. , Joel, S. , Schey, S. A. , Gribben, J. G. , & Cotter, F. E. (2010). GCS‐100, a novel galectin‐3 antagonist, modulates MCL‐1, NOXA, and cell cycle to induce myeloma cell death. Blood, 115, 3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthahar, N. , Meijers, W. C. , Sillje, H. H. W. , Ho, J. E. , Liu, F. T. , & de Boer, R. A. (2018). Galectin‐3 activation and inhibition in heart failure and cardiovascular disease: An update. Theranostics, 8, 593–609. 10.7150/thno.22196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto, Y. , Ramirez, R. J. , Yokokawa, M. , Kaur, K. , Ponce‐Balbuena, D. , Sinno, M. C. , … Jalife, J. (2016). Galectin‐3 regulates atrial fibrillation remodeling and predicts catheter ablation outcomes. JACC Basic Translational Science, 1, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triposkiadis, F. , Karayannis, G. , Giamouzis, G. , Skoularigis, J. , Louridas, G. , & Butler, J. (2009). The sympathetic nervous system in heart failure. Physiology, Pathophysiology, and Clinical Implications. J Am Coll Cardiol, 54, 1747–1762. [DOI] [PubMed] [Google Scholar]
- Truong, Q. A. , Januzzi, J. L. , Szymonifka, J. , Thai, W. E. , Wai, B. , Lavender, Z. , … Singh, J. P. (2014). Coronary sinus biomarker sampling compared to peripheral venous blood for predicting outcomes in patients with severe heart failure undergoing cardiac resynchronization therapy: the BIOCRT study. Heart Rhythm, 11, 2167–2175. 10.1016/j.hrthm.2014.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia‐Sama, I. , Zhao, Y. , Lai, D. , Janse van Rensburg, H. J. , Hao, Y. , & Yang, X. (2015). Hippo component TAZ functions as a co‐repressor and negatively regulates δNp63 transcription through TEA domain (TEAD) transcription factor. The Journal of Biological Chemistry, 290, 16906–16917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velde, A. R. , Lexis, C. P. , Meijers, W. C. , van der Horst, I. C. , Lipsic, E. , Dokter, M. M. , … de Boer, R. A. (2016). Galectin‐3 and sST2 in prediction of left ventricular ejection fraction after myocardial infarction. Clin Chimica Acta, 452, 50–57. [DOI] [PubMed] [Google Scholar]
- van der Velde, A. R. , Meijers, W. C. , Ho, J. E. , Brouwers, F. P. , Rienstra, M. , Bakker, S. J. L. , … de Boer, R. A. (2016). Serial galectin‐3 and future cardiovascular disease in the general population. Heart, 102, 1134–1141. 10.1136/heartjnl-2015-308975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasta, G. R. (2012). Galectins as pattern recognition receptors: Structure, function and evolution. Adv Exp Med Biol, 946, 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergaro, G. , Del Franco, A. , Giannoni, A. , Prontera, C. , Ripoli, A. , Barison, A. , … Emdin, M. (2015). Galectin‐3 and myocardial fibrosis in nonischemic dilated cardiomyopathy. Internat J Cardiol, 184, 96–100. 10.1016/j.ijcard.2015.02.008 [DOI] [PubMed] [Google Scholar]
- Vergaro, G. , Prud'homme, M. , Fazal, L. , Merval, R. , Passino, C. , Emdin, M. , … Delcayre, C. (2016). Inhibition of Galectin‐3 pathway prevents isoproterenol‐induced left ventricular dysfunction and fibrosis in mice. Hypertension, 67, 606–612. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Liu, S. , Heallen, T. , & Martin, J. F. (2018). The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nature Reviews. Cardiology, 15, 672–684. [DOI] [PubMed] [Google Scholar]
- Wang, J. J. , Rau, C. , Avetisyan, R. , Ren, S. , Romay, M. C. , Stolin, G. , … Lusis, A. J. (2016). Genetic dissection of cardiac remodeling in an isoproterenol‐induced heart failure mouse model. PLoS Genetics, 12, e1006038 10.1371/journal.pgen.1006038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. K. , Su, M. Y. , Lee, J. K. , Chiang, F. T. , Hwang, J. J. , Lin, J. L. , … Tsai, C.‐T. (2015). Galectin‐3 level and the severity of cardiac diastolic dysfunction using cellular and animal models and clinical indices. Sci Reports, 5, 17007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X. Y. , Li, S. N. , Wen, S. N. , Nie, J. G. , Deng, W. N. , Bai, R. , … Ma, C. S. (2015). Plasma galectin‐3 predicts clinical outcomes after catheter ablation in persistent atrial fibrillation patients without structural heart disease. Europace, 7, 1541–1547. [DOI] [PubMed] [Google Scholar]
- Xiao, H. , Li, H. , Wang, J. J. , Zhang, J. S. , Shen, J. , An, X. B. , … Zhang, Y. Y. (2018). IL‐18 cleavage triggers cardiac inflammation and fibrosis upon β‐adrenergic insult. European Heart Journal, 39, 60–69. 10.1093/eurheartj/ehx261 [DOI] [PubMed] [Google Scholar]
- Xin, M. , Kim, Y. , Sutherland, L. B. , Murakami, M. , Qi, X. , McAnally, J. , … Olson, E. N. (2013). Hippo pathway effector Yap promotes cardiac regeneration. Proceedings of the National Academy of Sciences of the United States of America, 110, 13839–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Q. , Dalic, A. , Fang, L. , Kiriazis, H. , Ritchie, R. H. , Sim, K. , … Du, X.‐J. (2011). Myocardial oxidative stress contributes to transgenic β2‐adrenoceptor activation‐induced cardiomyopathy and heart failure. British Journal of Pharmacology, 162, 1012–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, S. , Yang, G. , Zablocki, D. , Liu, J. , Hong, C. , Kim, S. J. , … Sadoshima, J. (2003). Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. The Journal of Clinical Investigation, 111, 1463–1474. 10.1172/JCI17459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, F.‐X. , Zhao, B. , & Guan, K.‐L. (2015). Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell, 163, 811–828. 10.1016/j.cell.2015.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, F. X. , Zhao, B. , Panupinthu, N. , Jewell, J. L. , Lian, I. , Wang, L. H. , … Guan, K. L. (2012). Regulation of the Hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell, 150, 780–791. 10.1016/j.cell.2012.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L. , Ruifrok, W. P. , Meissner, M. , Bos, E. M. , van Goor, H. , Sanjabi, B. , … de Boer, R. A. (2013). Genetic and pharmacological inhibition of galectin‐3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circulation. Heart Failure, 6, 107–117. [DOI] [PubMed] [Google Scholar]
- Zamora, E. , Lupon, J. , de Antonio, M. , Galan, A. , Domingo, M. , Urrutia, A. , … Bayes‐Genis, A. (2014). Renal function largely influences Galectin‐3 prognostic value in heart failure. Internat J Cardiol, 177, 171–177. 10.1016/j.ijcard.2014.09.011 [DOI] [PubMed] [Google Scholar]
- Zhao, W. B. , Lu, Q. , Nguyen, M. N. , Su, Y. , Ziemann, M. , Wang, L. N. , … Du, X. J. (2019). β‐Adrenergic receptor stimulation upregulates cardiac expression of galectin‐3 and BIM through the Hippo signaling pathway. Br J Pharmacol. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, H. , & Sun, A. (2018). Programmed necrosis in heart disease: Molecular mechanisms and clinical implications. Journal of Molecular and Cellular Cardiology, 116, 125–134. [DOI] [PubMed] [Google Scholar]