

Abstract
Background and Purpose
Both innate immunity and the renin‐angiotensin system (RAS) play important roles in the pathogenesis of diabetic nephropathy (DN). Myeloid differentiation factor 2 (MD2) is a co‐receptor of toll‐like receptor 4 (TLR4) in innate immunity. While TLR4 is involved in the development of DN, the role of MD2 in DN has not been characterized. It also remains unclear whether the MD2/TLR4 signalling pathway is associated with RAS activation in diabetes.
Experimental Approach
MD2 was blocked using siRNA or the low MW inhibitor, L6H9, in renal proximal tubular cells (NRK‐52E cells) exposed to high concentrations of glucose (HG). In vivo, C57BL/6 and MD2−/− mice were injected with streptozotocin to induce Type 1 diabetes and nephropathy.
Key Results
Inhibition of MD2 by genetic knockdown or the inhibitor L6H9 suppressed HG‐induced expression of ACE and angiotensin receptors and production of angiotensin II in NRK‐52E cells, along with decreased fibrosis markers (TGF‐β and collagen IV). Inhibition of the MD2/TLR4‐MAPKs pathway did not affect HG‐induced renin overproduction. In vivo, using the streptozotocin‐induced diabetic mice, MD2 was overexpressed in diabetic kidney. MD2 gene knockout or L6H9 attenuated renal fibrosis and dysfunction by suppressing local RAS activation and inflammation.
Conclusions and Implications
Hyperglycaemia activated the MD2/TLR4‐MAPKs signalling cascade to induce renal RAS activation, leading to renal fibrosis and dysfunction. Pharmacological inhibition of MD2 may be considered as a therapeutic approach to mitigate DN and the low MW inhibitor L6H9 could be a candidate for such therapy.
Abbreviations
- Ang II
angiotensin II
- DM
diabetes mellitus
- DN
diabetic nephropathy
- HG
high glucose
- KO
knockout
- MD2
myeloid differentiation factor 2
- RAS
renin‐angiotensin system
- STZ
streptozotocin
What is already known
Both innate immunity and renin‐angiotensin system play important roles in the pathogenesis of diabetic nephropathy.
TLR4 has been reported to mediate inflammatory diabetic nephropathy.
What this study adds
Blockade of MD2 attenuates diabetic nephropathy by suppressing local RAS activation and inflammation.
Hyperglycaemia induces renal ACE/AT1 expression and Ang II production via activating MD2/TLR4‐MyD88‐MAPKs signalling cascade.
What is the clinical significance
Pharmacological inhibition of MD2 is a potential therapeutic approach for diabetic nephropathy.
1. INTRODUCTION
There is evidence for an important role for the renin‐angiotensin system (RAS) in the pathogenesis of diabetic complications. Activation of local RAS is one of the major pathogenetic factors in diabetic nephropathy (DN). Renal RAS is activated in DN along with an elevated production of local angiotensin II (Ang II; Roscioni, Heerspink, & de Zeeuw, 2014; Ruster & Wolf, 2006; Zain & Awan, 2014). Ang II, which binds its classic membrane receptor, the AT1 receptor, was originally identified as a vasoactive peptide that constricts the afferent and the efferent arterioles as well as the interlobular arteries, causing reduced renal blood flow, increased intraglomerular pressure, and increased GFR (Denton, Fennessy, Alcorn, & Anderson, 1992). Local Ang II also functions as a growth factor activating interstitial and tubular fibrosis (Singh, Alavi, Singh, & Leehey, 1999) and triggers inflammation through promoting production of inflammatory cytokine and adhesion molecules (Vaziri et al., 2007). Furthermore, RAS blockade through either inhibition of renin and ACE or AT1 receptor antagonists exhibits beneficial effects against DN (Barnett et al., 2004). Despite the critical role of activated local RAS in DN, the local regulation of each component by hyperglycaemia has not been fully clarified. Current anti‐RAS therapies for the treatment of DN focus on the inhibition/antagonism of ACE or AT1 receptors, but few studies have been performed to understand the upstream mechanism and targets that mediate the up‐regulation of renal ACE/AT1 receptor expression under the condition of hyperglycaemia. It remains poorly understood how hyperglycaemia activates local RAS and increases local Ang II level in kidney.
Using some specific low MW inhibitors, we previously demonstrated that the MAPKs signalling pathway could regulate ACE expression in renal proximal tubular NRK‐52E cells under high glucose (HG) stimulation, which contributed to the increase in renal Ang II level and the development of DN (Pan et al., 2014). In innate immunity, activation of MAPKs was associated with the toll‐like receptor 4 (TLR4) signalling pathway. TLR4, together with its co‐receptor myeloid differentiation factor 2 (MD2), is essential for the response of the innate immunity system to LPS. Activated MD2/TLR4 recruits its downstream factor MyD88 to activate MAPKs signalling pathways, leading to production of inflammatory cytokines (Kawai & Akira, 2006). In addition, recent studies have demonstrated that TLR4 plays an important role in the pathogenesis of DN (Verzola et al., 2014; Wada & Makino, 2016). TLR4 activation promotes renal inflammation, podocyte and tubular epithelial cell injury, and interstitial fibrosis, and TLR4−/− mice were protected against the development of DN (Ma et al., 2014). However, the role of MD2 in DN has not been characterized, and it remains unclear whether diabetes‐induced RAS activation including ACE/ AT1 receptor expression and Ang II production is associated with the TLR4 or the MD2/TLR4 signalling pathways.
Based on this analysis, we hypothesized that activation of MD2/TLR4 by hyperglycaemia may induce local RAS activation and Ang II production, via an MAPK‐dependent manner. In this study, using pharmacological and genetic interventions both in vitro and in vivo, we characterized the role of MD2/TLR4‐MAPKs in hyperglycaemia‐mediated local RAS activation and renal fibrosis in a model of Type 1 diabetes and found that blockade of MD2 significantly attenuated DN via reducing renal RAS activation.
2. METHODS
2.1. Cell culture
NRK‐52E cells were obtained from the Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China) and cultured in DMEM medium (Gibco, Eggenstein, Germany) containing 10% FBS, 100 U·ml−1 of penicillin, and 100 mg·ml−1 of streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37°C. In the normal and control cell culture, the NRK‐52E cells were incubated with 5.5‐mM glucose. When cells were exposed to HG conditions, the cells were incubated with 33‐mM glucose.
2.2. Real‐time quantitative PCR
Cells or kidney tissues (80–100 mg) were homogenized in TRIZOL (Invitrogen, Carlsbad, CA, USA) for extraction of RNA according to each manufacturer's protocol. Both reverse transcription and quantitative PCR were carried out using a two‐step M‐MLV Platinum SYBR Green qPCR SuperMix‐UDG kit (Invitrogen). Eppendorf Mastercycler realplex detection system (Eppendorf, Hamburg, Germany) was used for qPCR analysis. The primers of genes used here were obtained from Invitrogen (Shanghai, China) and the primer sequences are listed in Table S1.
For in vitro samples, each treatment condition was run in triplicate (technical replicates). The same experiment was independently repeated for five times. Gene expression was normalized to β‐actin mRNA levels in the same sample and then normalized to the average value of the control group in each experiment according to the 2−∆∆Ct algorithm. Data represented five independent experiments and expressed as means ± SEM. Since we calculated the ∆CT value in each experiment independently, the means ± SEM of the relative ∆Ct values of the control group is 1.0 ± 0.0 (n = 5). However, when we performed the statistical analysis, we put the original data in five‐time qPCR experiments together, and the gene expression was normalized to β‐actin mRNA levels in the same sample and was then normalized to the ∆Ct value of the control group in the first‐time repeat.
For in vivo samples, the qPCR analysis of the kidney tissues from seven mice were performed together (in one plate). The gene expression was normalized to β‐actin mRNA levels in the same sample and was then normalized to one randomly selected mouse in control group according to the 2−∆∆Ct algorithm.
2.3. Western blotting
The antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. Collected cells or homogenized kidney tissue samples were lysed. Lysates (50–80 μg) were separated by 10% or 12% SDS‐PAGE and electrotransferred to nitrocellulose membranes. The membranes were pre‐incubated for 1.5 hr at room temperature in Tris‐buffered saline, pH 7.6, containing 0.05% Tween 20 and 5% non‐fat milk. The membrane was then incubated with specific antibodies. Immunoreactive bands were detected by incubating with secondary antibody conjugated with HRP and were visualized using enhanced chemiluminescence reagents (Bio‐Rad, Hercules, CA, USA). The bands were analysed using Image J analysis software version 1.38e (RRID:SCR_003070) and normalized to their respective controls.
2.4. Immunoprecipitation
Lysates (300–500 μg) were incubated with anti‐MD2 or anti‐TLR4 antibodies for 1 hr. Proteins were immunoprecipitated with protein G‐Sepharose beads on a shaker at 4°C overnight. The supernatants were used for the detection of TLR4 or MyD88as co‐precipitated proteins, respectively, using western blotting. The density of the immunoreactive bands was analysed using Image J software (NIH, Bethesda, MD).
2.5. siRNA‐induced gene silencing
Silencing gene expression was achieved using the siRNA technique. MD2 or MyD88 siRNAs were purchased from Gene Pharma (Shanghai, China). Transfection of NRK‐52E cells with siRNA was carried out using LipofectAMINE™ 2000 (Invitrogen, Carlsbad, CA), according to the manufacturer's instruction. Some of the transfected cells were then treated with HG for the further experiments. Specific siRNA sequences were 5′‐CCAUCAUUCACCACCAUAATT‐3′ and 3′‐UUAUGGUGGUGAAUGAUGGTT‐5′ for rat MD2 and 5′‐GGAAUGUGACUUCCAGACCTT‐3′ and 3′‐GGUCUGGAAGUCACAUUCCTT‐5′ for rat MyD88.
2.6. Animal experiments
All animal care and experimental procedures were approved by the Wenzhou Medical University Animal Policy and Welfare Committee (Approval Document No. wydw2016‐0124), and all animals received humane care according to the National Institutes of Health (USA) guidelines. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath, Drummond, McLachlan, Kilkenny, & Wainwright, 2010) and with the recommendations made by the British Journal of Pharmacology.
Male C57BL/6 mice (totally n = 70) weighing 18–22 g were obtained from Animal Center of Wenzhou Medical University. Whole body MD2 knockout (KO) mice (B6.129P2‐Ly96 KO, MD2−/−) on C57BL/6 background (n = 16) were purchased from Riken BioResource Center (Tsukuba, Ibaraki, Japan; RRID:SCR_003250) and maintained in the Animal Centre of Wenzhou Medical University. Animals were housed at a constant room temperature with a 12:12 hr light–dark cycle and were fed with water and a standard rodent diet containing 10‐kcal.% fat, 20‐kcal.% protein, and 70‐kcal.% carbohydrate (MediScience Diets Co. LTD, Yangzhou, China, Cat. #MD12031). The animals were acclimatized to the laboratory for at least 2 weeks before initiating the studies. All animal experiments were performed and analysed by blinded experimenters. Treatment groups were assigned in a randomized fashion. Each mouse was assigned a temporary random number within the weight range. When mice were randomly divided in each group, they were given their permanent numerical designation in the cages. For each group, a cage was selected randomly from the pool of all cages.
To induce Type 1 diabetes, WT, and MD2−/− mice were treated with a single i.p. injection of streptozotocin (STZ; 100 mg·kg−1 in citrate buffer, pH 4.5), while the control animals were received the same volume of citrate buffer. The blood glucose level was monitored using a glucometer after 4 hr of fasting. One week after STZ injection, mice with fasting‐blood glucose >2.16 g·l−1 were considered as diabetic. The DN was developed in 2–4 months after STZ‐induced hyperglycaemia.
For MD2/TLR4 expression analysis, 14 C57BL/6 mice were treated with STZ injection to induce hyperglycaemia and then were fed with standard diet for 2 or 4 months, randomly, to develop nephropathy. Seven control mice were received the same volume of citrate buffer. Two months after STZ injection, seven STZ‐injected mice (STZ [2 M]) were killed using sodium pentobarbital anaesthesia. Four months after STZ injection, seven control mice (Ctrol) and seven STZ‐injected mice (STZ [4 M]) were killed using sodium pentobarbital anaesthesia (i.p. injection of 0.2‐ml sodium pentobarbital at 100 mg·ml−1).
For MD2 deficiency analysis, 24 C57BL/6 mice and 16 MD2−/− mice were randomly divided into five groups as WT‐Control, WT‐diabetes mellitus (DM), WT‐DM + Val, KO‐Control, and KO‐DM (n = 8 in each group) respectively. The randomized animal division results in five treatment groups in total. Mice in WT‐DM, WT‐DM + Val, and KO‐DM were treated with STZ injection to induce hyperglycaemia for 1 week and then were fed with standard diet for 15 weeks to develop nephropathy. Mice in WT‐Control and KO‐Control were received the same volume of citrate buffer. In WT‐DM + Val group, STZ‐induced diabetic mice were orally treated with valsartan (30 mg·kg−1 per 2 days) for 15 weeks. Body weight and blood glucose were recorded weekly. Sixteen weeks after STZ injection, animals were killed using sodium pentobarbital anaesthesia (i.p. injection of 0.2‐ml sodium pentobarbital at 100 mg·ml−1).
For L6H9 treatment analysis, 32 C57BL/6 mice (four groups, n = 8 per group) were used. One week after STZ injection, 24 mice with fasting‐blood glucose >216 mg·dl−1 were considered as diabetic. The control animals (n = 8) were received the same volume of citrate buffer. Randomly selected diabetic animals were divided into three groups: DM (n = 8), curcumin‐treated DM (DM + Cur, n = 8), and L6H9‐treated DM (DM + L6H9, n = 8). In the DM + Cur group and DM + L6H9 group, curcumin (50 mg·kg−1) and L6H9 (20 mg·kg−1) were administered as oral gavage once every 2 days respectively. The DM group and age‐matched control group (n = 8) received 1% CMC‐Na solution alone according to the same schedule. The randomized animal division results in four treatment groups in total. Body weight and blood glucose levels were recorded weekly. After 8 weeks of treatment, animals were killed under sodium pentobarbital anaesthesia (i.p. injection of 0.2‐ml sodium pentobarbital at 100 mg·ml−1).
At 6 hr before killing, the spontaneous urine was collected using the following method: (a) the mouse was placed in a clean, dry, and empty cage and allowed to roam around; (b) the mouse is returned to its home cage and the urine was aspirated using a pipette and transferred to a collection tube as soon as the mouse urinates. Blood and renal tissues were collected at the time of killing. The body weight and right kidney weight were recorded. Serum creatinine, urinary albumin, and urinary creatinine were detected using commercial kits (Nanjing Jiancheng, Jiangsu, China). The kidney tissues from all animals were fixed in 4% paraformaldehyde for paraffin embedding and histopathological analysis or were snap‐frozen in liquid nitrogen for gene and protein expression analysis. The fixed kidney tissues were sectioned at 5‐μm thickness. The sections were stained with haematoxylin and eosin for histopathological analysis. The slides were examined under light microscope (400× amplification; Nikon, Japan). Paraffin sections (5 μm) of the kidney tissues were stained using Masson's three‐colour staining kit, PAS, or sirius red staining kits (Beyotime Biotechnology, Nantong, China), respectively, according to the manufacturer's instructions. The slides were examined under light microscope (400× amplification; Nikon). To quantify the level of fibrosis in Masson staining or collagen deposition in sirius red staining, the Image J software (NIH) was used and 10 non‐overlapping fields in each tissue (n = 8) were scored on a semi‐quantitative scale (<5%, 5–10%, 10–25%, 25–50%, 50–75%, and 75–100%), relative to total tissue area in the field. For PAS scoring, semi‐quantitatively on 30–50 glomeruli per kidney in each group (n = 8), using a scale from 0 to 4 score for description: 0 indicates no mesangial matrix expansion, 1 for minimal, 2 for mild, 3 for moderate, and 4 for diffuse.
2.7. Ang II assay
The Ang II levels in cell culture medium, mouse serum, or kidney tissue lysates were determined using an ELISA kit (Bioscience, San Diego, CA) according to the manufacturer's instructions. The amount of Ang II was normalized to the total amount of protein in the same group.
2.8. Immunofluorescent double staining
For immunofluorescent double staining, frozen sections (5 μm) of renal tissues were washed three times with PBS at room temperature and each wash was carried out for 5 min. Slides were blocked using 5% BSA for 30 min and then incubated overnight at 4°C with both MD2 antibody and antibody of specific marker (anti‐WT1 from Novus; anti‐AQP1 from Santa Cruz; anti‐F4/80 from Abcam) respectively. Slides were then correspondingly incubated with two kinds of secondary antibody (TRITC labelled antibody from Abcam) at 37°C for 1 hr and washed four times with PBS. DAPI was used to stain nuclei for 10 min and all stained sections were viewed by fluorescence confocal microscopy (Nikon).
2.9. Data and statistical analysis
All experiments were randomized and blinded. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2015). In all in vitro experiments, data represented five independent experiments and expressed as means ± SEM. Statistical analysis was performed with GraphPad Prism 6.0 software (San Diego, CA, USA; RRID:SCR_002798). We used one‐way ANOVA followed by Dunnett's post hoc test when comparing more than two groups of data and one‐way ANOVA, non‐parametric Kruskal–Wallis test, followed by Dunn's post hoc test when comparing multiple independent groups. P values of ˂.05 were considered to be statistically significant. Post‐tests were run only if F achieved P < .05 and there was no significant variance in homogeneity.
2.10. Materials
Glucose, mannitol, STZ, valsartan, and curcumin were purchased from Sigma (St. Louis, MO). SB203580 (SB), SP600125 (SP), and PD98059 (PD) were purchased from Beyotime Biotechnology. The MD2 inhibitor L6H9, synthesized by our lab, with a purity of 98.9%, was dissolved in DMSO for in vitro experiments and was dissolved in 1% CMCNa for in vivo experiments. Antibodies for GAPDH, p‐IκBα, IκBα, p‐ERK, ERK, ACE, TGF‐β, collagen 4, TLR4, MD2 (RRID:AB_2138097), MyD88, MMP2, Bcl‐2, Bax, and CD68 were purchased from Santa Cruz (Santa Cruz, CA); and p‐JNK, JNK, p‐p38, TNF‐α, and p38 antibodies were obtained from Cell Signaling (Danvers, MA).
2.11. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017a, b; Alexander, Kelly et al., 2017a, b).
3. RESULTS
3.1. MD2 silencing attenuated HG‐induced RAS activation and TGF‐β expression
In diabetic kidney, Ang II secretion and RAS activation have been reported to occur mainly in renal tubular cells (Vallon & Thomson, 2012). HG stimulates the expression of angiotensinogen, the precursor of Ang II in the proximal tubular epithelial cells (Feliers & Kasinath, 2010). To investigate whether MD2/TLR4 participates in the activation of the RAS induced by HG, we first performed MD2 knockdown experiment by using MD2 siRNA (Si‐MD2) in renal proximal tubular NRK‐52E cells. Figure 1a shows that 80% of MD2 expression was decreased after siMD2 treatment in NRK‐52E cells. MD2 knockdown and control cells were subsequently incubated in medium with HG (33 mM) for 12 hr. HG increased the expression of genes involved in the RAS in NRK‐52E cells (Figure 1b–f). The expression of genes in the RAS was not affected by treatment with 33‐mM mannitol, indicating that the osmotic effect did not induce RAS activation in renal cells (Figure 1b–f). Although Si‐MD2 did not affect the renin expression under HG conditions (Figure 1b), the HG‐induced elevations of ACE and AT1 receptors were prevented in the MD2 knockdown cells (Figure 1c–e). Si‐MD2 further decreased HG‐induced increased Ang II secretion into the cell culture medium (Figure 1f). The protein and mRNA analysis further showed that Si‐MD2 suppressed HG‐induced overexpression of TGF‐β (Figure 1c,g). As expected, the osmotic control, 33‐mM mannitol, did not affect TGF‐β gene transcription (Figure 1b–f). Taken together, these data demonstrated that MD2 silencing attenuates HG‐induced RAS activation and TGF‐β expression.
Figure 1.
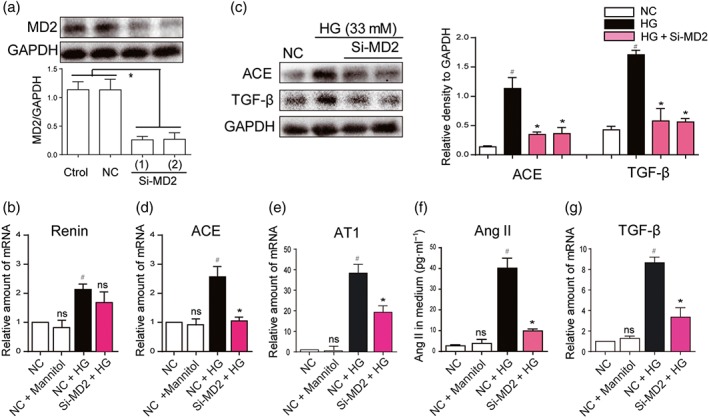
MD2 silencing attenuated the activation of the RAS, induced by high glucose concentrations. (a) MD2 knockdown in NRK‐52E cells by siRNA approach. Two si‐RNA sequences for MD2 gene (Si‐MD2) were transfected in NRK‐52E cells and the MD2 protein levels were measured by western blot analysis (Ctrol: non‐transfected cells; NC: non‐MD2 scrambled transfection cells; two different siRNAs for MD2 were used). (b–g) Effects of MD2 knockdown in macrophages stimulated by high glucose concentrations (HG; 33 mM) (b, d, e, and g) Si‐RNA transfected or NC NRK‐52E cells were incubated with HG or 33‐mM mannitol for 12 hr. The mRNA levels of renin, ACE, AT1 receptors (AT1) and TGF‐β were detected by real‐time quantitative PCR assay with β‐actin used as housekeeping gene. (c and f) Si‐RNA transfected or NC NRK‐52E cells were incubated with HG or 33‐mM mannitol for 24 hr. Shown are representative western blot analysis and densitometric results for ACE and TGF‐β protein levels in whole cell lysate with GAPDH as a loading control. Ang II level in cultured medium was detected by ELISA kit. n = 5 independent experiments; bar graph shows mean values ± SEM; # P < .05, significantly different from NC group; *P < .05, significantly different from HG group; ns = not significantly different from NC + HG group
3.2. The MD2 inhibitor L6H9 suppressed HG‐induced RAS activation and TGF‐β expression through inhibition of TLR4/MyD88 signalling pathway
Next, we used compound L6H9 (Figure 2a), a specific, low MW inhibitor of MD2 that blocked palmitic acid‐induced activation of MD2/TLR4 in cardiomyocytes (Wang et al., 2017), to further confirm these findings. Curcumin, a natural anti‐inflammatory compound which is known to inhibit activation of MAPKs by HG (Fang et al., 2015), was used as a positive control. NRK‐52E cells were pretreated with L6H9, followed by incubation with HG (33 mM). Transient HG challenge (5‐min incubation) rapidly activated MD2/TLR4/MyD88 signalling pathway, which was illustrated by stronger interaction between TLR4 and MD2 or MyD88 (Figures 2b and S1A–B). Pretreatment with L6H9, inhibiting the binding of MD2 to TLR4, resulted in reduced MyD88 recruitment to TLR4 (Figures 2b and S1A–B). For the activation of the MAPK signalling pathways, downstream ofTLR4, NRK‐52E cells were stimulated with HG for a slightly longer time (30 min). Pretreatment of L6H9 decreased HG‐induced phosphorylation of ERK, JNK, and p38 in a dose‐dependent fashion (Figures 2c and S1C–E). We then tested the effects of L6H9 on the expression of genes involved in the RAS and fibrosis in NRK‐52E cells. As expected, L6H9 had minimal effect on HG‐stimulated renin expression (Figure 2d). However, L6H9 dose dependently reduced the expression of ACE, AT1 receptors, TGF‐β, and collagen IV either at the mRNA (Figure 2e–g) or protein (Figure 2h) levels. As a result, the amount of Ang II released in the cultural medium from the cells was reduced following L6H9 pretreatment (Figure 2i). Activated MD2/TLR4 recruits two major adaptor molecules, MyD88 and TRIF. MyD88 activates downstream pro‐inflammatory cascade through NF‐κB and MAPK pathways, while TRIF causes phosphorylation and activation of IRF3 and the subsequent expression of IFN‐inducible genes (Park et al., 2012). Considering that MAPKs are downstream of MyD88, we only examined the MyD88 recruitment in this study. Collectively, these data showed that MD2 inhibitor L6H9 suppressed HG‐induced RAS activation and TGF‐β expression through inhibition of the TLR4/MyD88 signalling pathway.
Figure 2.
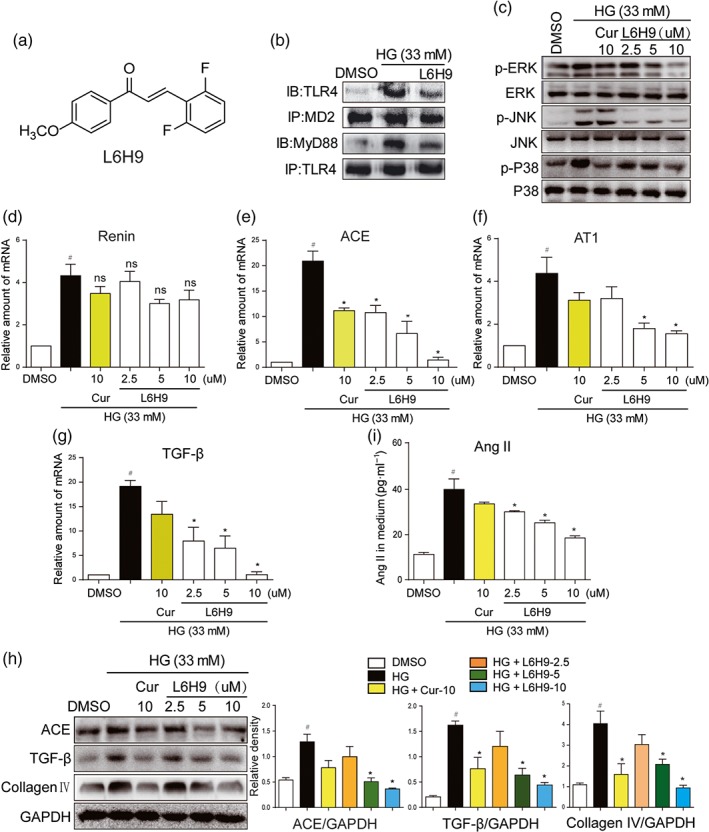
L6H9 suppressed HG‐induced MD2/TLR4‐MAPKs signalling and RAS activation. (a) Chemical structure of the MD2 inhibitor L6H9. (b–c) L6H9 suppressed the activation of MD2/TLR4/MyD88 signalling. (b) NRK‐52E cells were pretreated with L6H9 (10 μM) for 1 hr, followed by incubation with high glucose concentrations (HG; 33 mM) for 5 min, and cells were collected for co‐immunoprecipitation analysis. (c) L6H9 decreased the phosphorylation levels of MAPKs. NRK‐52E cells were pretreated with L6H9 and curcumin (Cur) 10 uM and then stimulated with HG for 30 min, total proteins were extracted for western blot detection. (d–g) The effects of L6H9 on mRNA expression of renin (d), ACE (e), AT1 receptors (AT1) (f), and TGF‐β (g). NRK‐52E cells were pretreated with L6H9 and Cur and then stimulated with HG for 12 hr, total mRNAs were extracted for real‐time qPCR assay. (h) The effects of L6H9 on production of ACE, TGF‐β, and collagen IV proteins. NRK‐52E cells were pretreated with L6H9 and Cur and then stimulated with HG for 24 hr. Total protein was extracted for western blot analysis. (i) L6H9 reduced HG‐increased Ang II level in cell medium. NRK‐52E cells were pretreated with L6H9 and Cur and then stimulated with HG for 24 hr. The media were collected for ELISA detection. n = 5 independent experiments; bar graph shows mean values ± SEM; # P < .05, significantly different from DMSO group; *P < .05, significantly different; ns = not significantly different from HG group
3.3. Both MyD88 silencing and MAPKs inhibition suppressed HG‐induced RAS activation and TGF‐β expression
As MD2/TLR4 mediates HG‐induced RAS activation, we assessed the involvement of MyD88 and MAPKs, the classic downstream molecules of MD2/TLR4 signalling, in RAS activation. Knockdown of MyD88 protein by siRNA in NRK‐52E cells showed no influence on HG‐increased renin mRNA level (Figure 3b). As expected, MyD88 knockdown significantly decreased HG‐induced overexpression of ACE, AT1 receptors, and TGF‐β both at the mRNA and the protein levels (Figure 3c–e). As a result, HG‐induced increased Ang II level in the media was significantly prevented by si‐MyD88 (Figure 3f). MAPKs constitute important downstream signalling pathways in the MD2/TLR4‐MyD88 signalling cascades. MyD88 knockdown also prevented activation of downstream MAPKs signalling pathways induced by HG (Figures 3g and S2A–C). To further characterize the roles of MAPKs in mediating MD2/TLR4‐dependent RAS activation, we pretreated NRK‐52E cells with specific inhibitors of MAPKs, including an ERK inhibitor PD98059, a p38 inhibitor SB203580 or a JNK inhibitor SP600125, and then stimulated cells with HG for 12 hr. All three MAPK inhibitors exhibited marked inhibition of ACE expression (Figure S3A–B), while only the ERK inhibitor showed significant inhibition of AT1 receptor expression (Figure S3C). All inhibitors suppressed HG‐induced expression of the fibrotic marker TGF‐β (Figure S3A,D). As expected, none of them showed any effects on HG‐induced renin mRNA production (Figure S3E), while all inhibitors blocked HG‐increased Ang II release into the cell culture medium (Figure S3F). These data further indicate that MyD88‐MAPKs mediates HG‐induced MD2/TLR4‐dependent RAS activation and TGF‐β expression.
Figure 3.
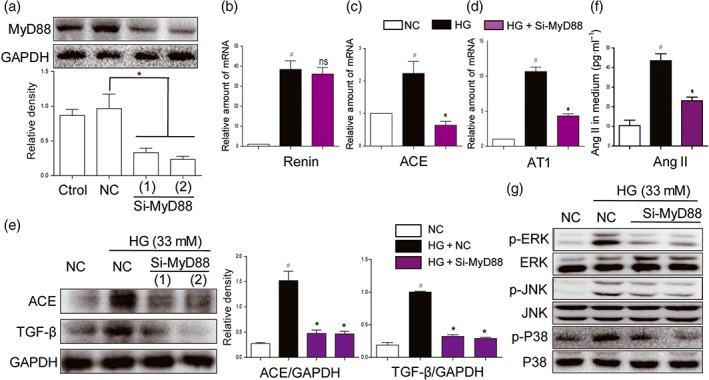
MyD88 silencing and inhibition of MAPKs suppressed activation of RAS and expression of fibrotic factors, induced by high glucose concentrations. (a) MyD88 knockdown in NRK‐52E cells by siRNA approach. Two si‐RNA sequences for MyD88 gene (si‐MyD88) were transfected in NRK‐52E cells and the MyD88 protein levels were measured by western blot analysis (Ctrol: non‐transfected cells; NC: non‐MD2 scrambled transfection cells; two different siRNAs for MyD88 were used). (b–g) MyD88 silencing attenuated activation of RAS and MAPKs, induced by high glucose ((HG; 33 mM) concentrations. NRK‐52E cells were firstly transfected with si‐MyD88 and then stimulated by HG conditions for different times. (b–d) The mRNA levels of ACE, renin, and AT1 receptors (AT1) were detected by real‐time qPCR assay after 12‐hr incubation of HG. (e) ACE and TGF‐β were detected by western blot with GAPDH as a loading control after 24‐hr incubation of HG. (f) The Ang II levels in cultural medium were detected by ELISA after 24‐hr HG challenge. (g) The phosphorylation levels of MAPKs were detected by western blot analysis after 30‐min HG incubation. n = 5 independent experiments; bar graph shows mean values ± SEM; # P < .05 significantly different from NC group; *P < .05, significantly different; ns, not significantly different from HG group
3.4. MD2 is up‐regulated and activated in diabetic mouse kidney
The results obtained so far indicated a relationship of MD2 with the pathogenesis of DN. We then evaluated the level of MD2 in the kidneys of mice with STZ‐induced diabetes. Mice were treated with a single i.p. injection of STZ to induce Type 1 diabetes and then were fed with standard diet for 2 or 4 months to develop nephropathy (see Section 2). The results showed that, compared with the non‐diabetic control mice, levels of MD2 and TLR4 protein in diabetic mouse kidney gradually increased with the development of DN (from 2 to 4 months; Figure 4a). Similar results were observed at the level of mRNA (Figure 4b,c). We next evaluated MD2 localization in kidney tissue. Tubular cells, podocytes, and infiltrated macrophages are very important in the development of DN. We carried out immunofluorescent double staining using MD2 antibody and cell‐type specific markers to identify MD2‐expressing renal cell types in diabetic mice. Consistent with our in vitro data, MD2 expression was detected mainly in AQP‐1‐positive tubular cells (Figure 4d), with much less in F4/80‐positive infiltrated macrophages and no expression seen in WT1‐positive podocytes (Figures S4 and S5). One of the limitations in this work is that we are unable to confirm all MD2‐expressing cell types in mouse kidneys. We performed the mechanistic studies only in cultured kidney epithelial cells. Although these data showed that renal tubular cells expressed MD2, we fully noted that analysis of other kidney cell types is important. We anticipate that other cell types in kidney tissues including mesangial cells and infiltrated T cells may also utilize this signalling pathway. However, this possibility requires confirmation in the future. In addition to protein expression, the formation of MD2/TLR4 complex reflects the activation of MD2/TLR4 signalling. Thus, we evaluated the interaction of MD2 and TLR4 in diabetic mouse kidney using the method of immunoprecipitation. As shown in Figure 4e, the amounts of MD2/TLR4 complex in diabetic kidney gradually increased with the development of DN. The findings indicated that the development of DN was associated with elevated and activated MD2/TLR4 level in kidney tissue, and the increased MD2 is mainly distributed in tubular cells.
Figure 4.
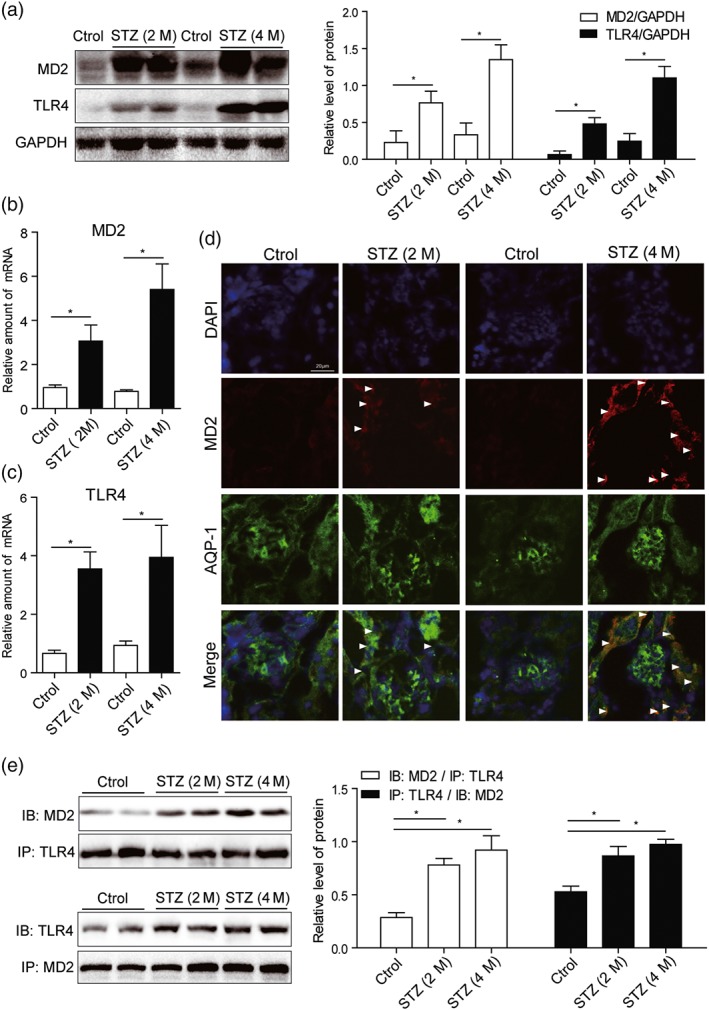
MD2 is overexpressed and activated in diabetic mouse kidney. Mice were treated with a single i.p. injection of STZ to induce Type 1 diabetes and then were fed with standard diet for 2 or 4 months to develop nephropathy. Kidney tissues were collected at the time of killing. (a) Representative western blot of MD2 and TLR4 proteins in kidney tissues. Densitometric quantification is shown in right (n = 7 mice per group). (b and c) Real‐time qPCR assay of MD2 and TLR4 mRNAs in kidney tissues (n = 7 mice per group). (d) Kidney tissue sections from each group were double‐stained with MD2 and AQP‐1 immunofluorescence antibodies (with DAPI staining for nuclei). Representative images from n = 7 per group were shown. Scale bar: 20 μm. White arrows indicate fluorescence‐positive MD2 protein. (e) MD2 is activated in diabetic kidney. Shown are representative western blots (IBs) from the co‐precipitation (IPs) studies for MD2/TLR4 complex formation. Densitometric quantification is shown in right (n = 7 mice per group). Bar graph shows mean values ± SEM; *P < .05, significantly different as indicated
3.5. MD2 KO inhibited RAS activation and attenuated renal dysfunction in diabetic mice
STZ was used to develop Type 1 diabetes both in the MD2‐KO mice and their wild‐type littermates (C57BL/6). The AT1 receptor antagonist, valsartan, was used as a comparison. During the 15‐week follow‐up, body weight loss and hyperglycaemia were recorded in diabetic groups. Neither MD2 KO nor valsartan affected serum glucose level and body weight (Figure S6A–B). Renal function was assessed by renal haemodynamics and biochemical markers. Either MD2‐KO or valsartan prevented the increases of kidney/body weight ratio, serum creatinine, urinary albumin, and urinary albumin/creatinine ratio in diabetic mice (Figure 5a–d). In MD2‐KO mice, both values of renal blood flow velocity (left kidney vein) and the systolic/diastolic pressure ratio were comparable to those of control WT mice (Figure S7A–B), indicating the preservation of normal renal haemodynamics. These data indicate improved renal function in the diabetic mice following MD2‐KO or RAS blockade.
Figure 5.
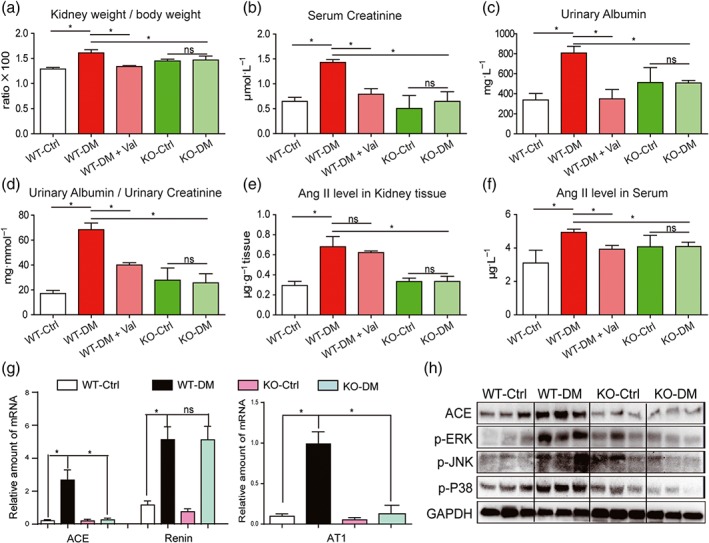
MD2 knockout attenuated diabetes‐induced renal damage and inhibited activation of RAS in mice. MD2 knockout mice (KO), valsartan (Val) ‐treated C57BL/6 mice, and their wild‐type control (WT) developed diabetes (DM), 4 months after STZ injection. (a–e) Kidney/body weight ratio (a), serum creatinine (b), urinary albumin (c), urinary albumin/creatinine ratio (d), Ang II level in kidney tissues (e), and Ang II level in serum (f) were detected by corresponding kits. (g) The kidney tissue mRNA for ACE, renin, and AT1 receptors were determined by real‐time qPCR assay and normalized to β‐actin. (h) Representative western blot analysis of ACE and phosphorylated MAPKs in kidney tissues. Bar graph shows mean values ± SEM; n = 8 mice per group, *P < .05, significantly different as indicated; ns = not significant. WT‐Ctrl, wild‐type non‐diabetic mice; WT‐DM, wild type diabetic mice; WT‐DM + Val valsartan‐treated diabetic mice; KO‐Ctrl, MD2 knockout nondiabetic mice; KO‐DM, MD2 knockout diabetic mice
We then determined the RAS profile in these animals. Figure 5e showed that the local Ang II level was significantly increased in the STZ‐induced diabetic mouse kidney, and this increase was prevented in the MD2‐KO mice. As an antagonist of AT1 receptors, valsartan did not affect local levels of Ang II in mouse kidney (Figure 5e). Hyperglycaemia induced only slight increase in serum Ang II level and MD2‐KO also reversed this change (Figure 5f). Further analysis demonstrated that kidneys from MD2‐KO mice with diabetes, had lower expression of RAS‐related transcripts, including ACE and AT1 receptors, compared to the WT diabetic mice (Figure 5g). As expected, MD2‐KO did not block diabetes‐induced renin production (Figure 5g). Similar results were obtained for expression of ACE protein, using western blot analysis (Figures 5h and S8A). Consistent with the in vitro data, the diabetes‐augmented phosphorylation and activation of ERK, JNK, and p38 were prevented in the MD2‐KO mice (Figures 5h and S8B–D). These data indicate the regulation of RAS activation by MD2 in the kidneys of diabetic animals.
3.6. MD2 KO attenuated diabetic renal fibrosis in mice
Histological analysis of kidney tissue showed histological abnormalities, fibrosis, and mesangial expansion in WT diabetic mice (Figures 6a and S9A–C). However, these changes were absent in MD2‐KO mice with diabetes (Figures 6a and S9A–C). Histological abnormalities and fibrosis in diabetic mice were also ameliorated by valsartan treatment (Figures 6a and S9A–C). These results were further confirmed by the analysis of fibrotic markers such as collagen IV, CTGF, TGF‐β, and MMP2 at either protein or at mRNA levels. MD2 KO significantly reduced STZ‐induced overexpression of fibrotic proteins in mouse kidney (Figures 6b–c, and S10A–C). In addition, MD2 KO attenuated Bax expression and increased Bcl‐2 in the kidneys in diabetes (Figures 6c and S10D–E). Immunohistochemical staining confirmed reduction of TNF‐α and CD68 in kidneys from MD2‐KO mice with diabetes, compared with those from WT diabetic mice, suggesting prevention of diabetes‐induced macrophage infiltration into the renal tissue (Figure S11A). The mRNA analysis of renal tissue lysate demonstrated that MD2‐KO mice were resistant to the increased expression of inflammatory cytokines (TNF‐α and IL‐6) and adhesion molecule (ICAM‐1; Figure S11B–D), induced by diabetes. These results showed that MD2 gene KO effectively inhibited activation of the renal TLR4/MAPKs‐RAS pathway and attenuated renal injury in Type 1 diabetic mice.
Figure 6.
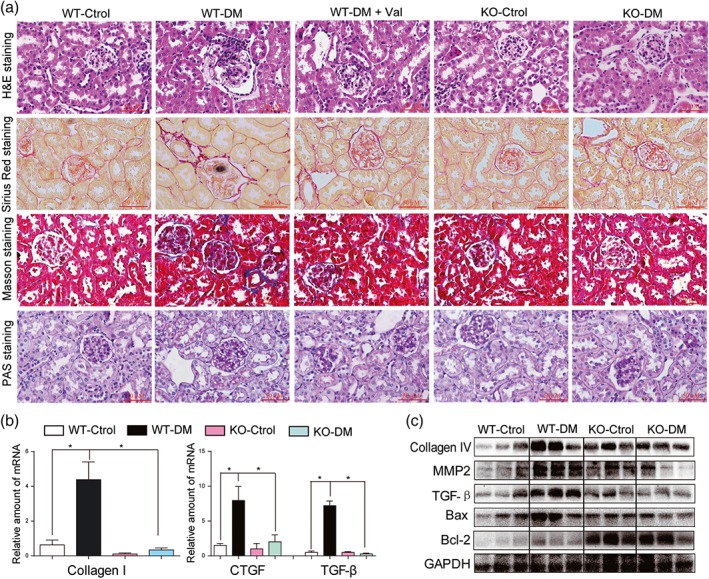
MD2 knockout decreased diabetes‐induced fibrosis in mouse kidney. MD2 knockout mice (KO), valsartan (Val)‐treated C57BL/6 mice, and their wild‐type control (WT) developed diabetes (DM), 4 months after STZ injection. (a) Representative light micrograph of histochemical assessment of kidney tissues: haematoxylin and eosin (H&E) staining, PAS for glycogen (purple), Masson's trichrome stain (Blue) for detection of connective tissue, and Sirius red staining were used for the detection of fibrosis (Red); 400× magnification; scale bar: 50 μm. (b) Kidney tissue content of fibrotic genes was determined by real‐time quantitative‐PCR for collagen 1, CTGF, and TGF‐β. Bar graph shows mean values ± SEM; n = 8 mice per group, *P < .05, significantly different as indicated. (c) Representative western blot analysis of indicated proteins in kidney tissues
3.7. Treatment with the MD2 inhibitor L6H9 attenuated DN in mice
To further validate MD2 as a therapeutic target for the treatment of DN in vivo, we studied the pharmacological effects of the MD2 inhibitor, L6H9, in Type 1 diabetic mice. Administration of L6H9 for 4 months in normal mice did not induce tubular injury, inflammation, serum creatinine up‐regulation, or renal dysfunction, indicating the safety of L6H9 itself (Figure S12A–F). In diabetic mice, L6H9 treatment did not alter body weight and blood glucose levels, but the kidney to body weight ratio was significantly reduced (Figure S13A–C). Histological analysis showed that diabetes‐induced structural alterations and glycogen accumulation were prevented by the treatment with L6H9 (Figure 7a). L6H9 decreased the phosphorylation and activation of MAPKs, including ERK, JNK, and p38, in diabetic kidneys (Figures 7b and S14A–C). Co‐immunoprecipitation revealed that L6H9 blocked the interaction between TLR4 and MD2 in vivo, which led to less MyD88 recruited to TLR4 (Figures 7c and S14D–E). As a result, L6H9 inhibited RAS activation in kidneys by reducing diabetes‐induced Ang II elevation (Figure 7d) and renal expression of ACE/AT1 receptors (Figures 7e–f and S14F). As expected, L6H9 did not suppress renin expression during diabetes (Figure 7e). Also, L6H9 decreased the inflammatory responses in kidneys of diabetic mice (Figure S15A–F). NF‐κB is a downstream transcriptional factor of the MD2/TLR4 pro‐inflammatory signalling pathway and we have therefore measured IκBα phosphorylation, as a marker of NF‐κB activation. The results showed that either L6H9 administration or MD2 KO prevented IκBα phosphorylation in STZ‐induced diabetic mouse kidneys (Figure S16).
Figure 7.
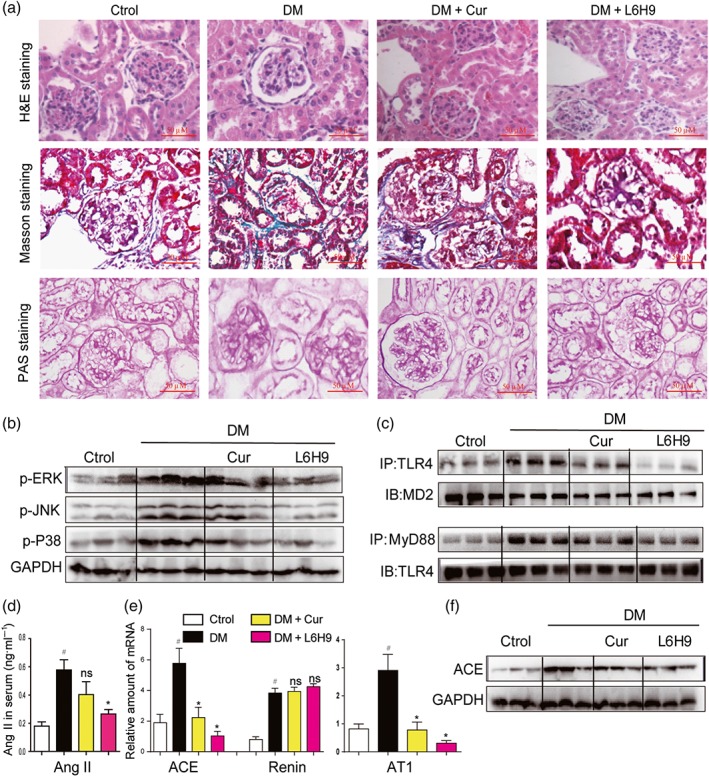
Oral administration of the MD2 inhibitor L6H9 ameliorated diabetic (DM)‐induced kidney damage in C57BL/6 mice. (a) Representative light micrograph of histochemical assessment of kidney tissues: haematoxylin and eosin (H&E) staining, Masson's trichrome staining, and PAS staining for detection of connective tissue; 400× magnification. (b) Representative western blot analysis of phosphorylated MAPKs in kidney tissues; n = 3 mice in each group. (c) Representative co‐immunoprecipitation analysis of TLR4‐MD2 and TLR4‐MyD88 bindings in kidney tissues; n = 3 mice in each group. (d) Serum Ang II level was detected by corresponding kit. (e) Kidney tissue content of RAS‐related genes was determined by real‐time quantitative‐PCR assay. (f) Representative western blot analysis of ACE in kidney tissues; n = 3 mice in each group. Bar graph shows mean values ± SEM; n = 8 in four groups, # P < .05, significantly different from Control group; *P < .05, significantly different from DM group; ns = not significantly different from DM group
4. DISCUSSION
Although DN is known to be strongly linked to the local activation of the RAS, less is known about the regulation of RAS components by hyperglycaemia. Our study documented for the first time the role of MD2/TLR4 in up‐regulating renal ACE/ AT1 receptor expression and Ang II production and renal dysfunction driven by hyperglycaemia. Briefly, the major findings of this work demonstrate that MD2/TLR4 complex is activated under HG conditions, which trigger MyD88‐dependent activation of MAPKs, contributing to local ACE/ AT1 receptor gene expression and Ang II production and subsequent fibrosis in kidneys. Either genetic deletion or pharmacological inhibition of MD2 has a protective effect on diabetes‐induced renal RAS activation and nephropathy, accompanied with decreased inflammation (Figure 8).
Figure 8.
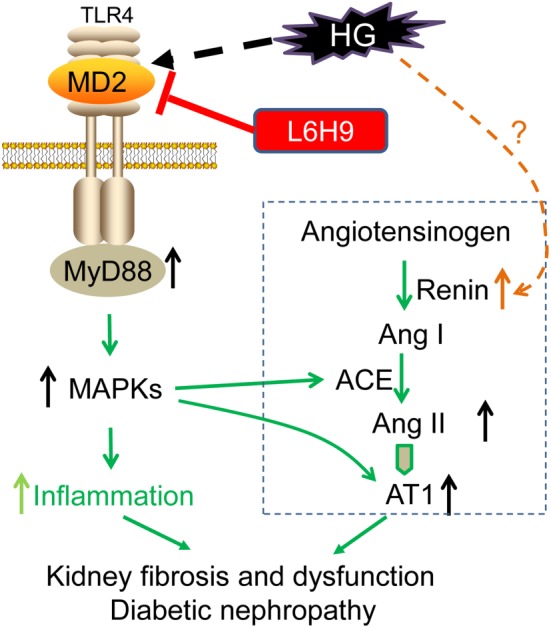
Scheme of the possible mechanisms of the involvement of MD2 in activation of the RAS and inflammation, induced by high‐glucose (HG) and the renal injury in diabetes. AT1, AT1 receptors
MD2 and the TLR4 signalling pathway are critically important for LPS‐induced inflammation in innate immunity. Recent studies have highlighted the critical role of TLR4 in the progression of DN (Kuwabara et al., 2012; Wada & Makino, 2016). TLR4 is highly expressed in human renal tubules and its expression is further up‐regulated by HG (Tang & Lai, 2012). HG‐induced TLR4 activation further triggers IL‐6 and CCL2 expression via MAPKs and NF‐κB activation, leading to interstitial macrophage infiltration, albuminuria, and renal dysfunction (Lin et al., 2012). TLR4‐KO mice displayed resistance to renal phenotypes in mice with STZ‐induced diabetes (Jialal, Major, & Devaraj, 2014). Although previous studies indicated that inhibition of the TLR4 signalling pathway has potential therapeutic implications in DN, few studies have focused on the function of the MD2 component, in diabetic renal diseases. It is expected that MD2, with TLR4 together, is able to regulate renal inflammation in diabetic mice. Our previous study has shown that L6H9 can prevent glucose‐induced inflammation in heart‐derived H9c2 cells (Zhong et al., 2015). In the present study, we found that MD2 was required for HG‐induced TLR4 activation and that HG induced the formation of the MD2/TLR4 complex and that MD2 inhibition significantly blocked the recruitment of MyD88 and MAPKs phosphorylation. Further, MD2 was significantly overexpressed and activated in diabetic mouse kidney tissues (Figure 4). MD2‐KO mice were resistant to diabetes‐induced renal inflammation and macrophage infiltration, and treatment with the MD2 inhibitor L6H9, significantly decreased inflammatory cytokine production in diabetic mouse kidney. Thus, the first finding of this work is the validation of the role of MD2 in mediating TLR4 pro‐inflammatory signalling activation in the pathogenesis of DN.
Second, we demonstrated a new correlation between MD2/TLR4 innate immunity signalling and RAS activation in diabetic kidney. In HG‐stimulated tubular epithelial cells, MD2/MyD88‐MAPKs seem to serve as important upstream regulators of ACE and AT1 receptor expression. Blockage of MD2/TLR4 signalling pathway significantly suppressed HG‐induced ACE and AT1 receptor expression and Ang II production both in vitro and in vivo, which contributes to the attenuation of DN. A large number of studies have revealed a forward crosstalk between Ang II and TLR4 (Dange et al., 2014; Lv, Chen, Shao, Chen, & Shi, 2015). Ang II is able to activate MD2/TLR4 as well as its downstream signalling cascade (Nair, Ebenezer, Saini, & Francis, 2015). TLR4 has been considered a receptor‐like protein for Ang II, mediating the pro‐inflammatory and pro‐oxidative effects of Ang II in cardiovascular (Dange et al., 2014; Nakashima et al., 2015) and kidney diseases (Lv et al., 2015; Lv, Jia, Yang, Zhu, & Ding, 2009). Our previous study also found that MD2 mediated Ang II‐induced inflammation through direct interaction with Ang II in cardiomyocytes (Han et al., 2017). However, the MD2/TLR4 innate immunity system and the RAS may be mutually regulated in DN. Here, we identified the MD2‐TLR4/MyD88 pathway as an important upstream signalling pathway for the production of Ang II under hyperglycaemic conditions. This is the second finding of this work, indicating a reverse crosstalk between Ang II and the MD2/TLR4 signalling pathway.
During diabetes, regulation of the renal RAS seems to be independent of the circulating RAS, and local renal Ang II levels has been found to be several‐fold higher than that in plasma (Seikaly, Arant, & Seney, 1990). The pathway starts with pro‐renin production in the juxtaglomerular cells of kidney (Nguyen & Muller, 2010). Renin catalyses the conversion of angiotensinogen into the decapeptide angiotensin I which is then converted into the octapeptide Ang II by ACE. In diabetes, the increased renal levels of Ang II stimulate TGF‐β expression and fibrosis in kidney to enhance the development of DN (Kinoshita et al., 2011; Morsy, Heeba, & Mahmoud, 2015). Although the RAS is classically activated by reduced glomerular blood flow, which may be caused by loss of sodium and water or by renal artery narrowing, local RAS can be directly activated by hyperglycaemia, independent of a haemodynamic stimulus (Singh, Singh, Alavi, & Leehey, 2003; Vidotti et al., 2004; Xiao, Wang, Yang, Liu, & Sun, 2013). In keeping with previous studies, we found that HG treatment increases renin, angiotensinogen, ACE, and Ang II levels in cultured rat glomerular mesangial cells (Singh et al., 2003; Vidotti et al., 2004). However, the mechanism for hyperglycaemia‐induced generation of Ang II remains unaddressed. Here, we observed that inhibition of MD2/TLR4 signalling blocked HG‐increased expression of ACE/ AT1 receptor genes, but not renin, suggesting that the MD2/TLR4 pathway regulates part of the RAS cascade without affecting upstream renin production and secretion. Renin is a cAMP‐inducible gene. The cAMP response element‐binding protein cascade and members of the nuclear receptor superfamily have been reported to regulate renin transcription (Castrop et al., 2010). Under the condition of HG or diabetes, Visavadiya et al. showed that HG induces renin overexpression via phosphorylation of cAMP response element‐binding protein in human renal proximal tubular HK‐2 cells (Visavadiya, Li, & Wang, 2011). Studies also suggest that the GPR91 (succinate) receptor may be of relevance for the stimulation of renin release in response to HG levels (Toma et al., 2008) or in experimental diabetes (Vargas, Toma, Kang, Meer, & Peti‐Peterdi, 2009). However, these suggestions await further work to confirm their relevance.
Given the important role of MD2/TLR4 in innate immunity and inflammation, this study provides a new link between the RAS and immunity, and presents MD2 as an important target that regulates both local RAS and inflammation in diabetic kidneys. It still remains unclear to as how hyperglycaemia activates MD2. Apart from LPS, several other ligands have been recently reported to bind MD2 and activate the MD2/TLR4 pathway (Adanitsch et al., 2014; Aranda et al., 2014; Hussein, Liu, Skwarczynski, & Toth, 2014). Thus, an important and unsolved question remains as to the specific mechanism by which hyperglycaemia promotes MD2/TLR4 complex formation and TLR4 activation under hyperglycaemic conditions. Delineation of such pathways will contribute to a better understanding of the pathogenesis of DN.
In summary, we have demonstrated that hyperglycaemia can activate the MD2/TLR4‐MyD88‐MAPKs signalling cascade to induce ACE and AT1 receptor expression and Ang II production, as well as inflammation, in both tubular cells and diabetic mouse kidneys (Figure 8). We have identified a role of MD2 in modulating local RAS activation in kidney to stimulate renal dysfunction and fibrosis in diabetes. Our findings provide evidence that pharmacological inhibition of MD2 may be considered as a therapeutic approach to prevent or mitigate DN and that the low MW inhibitor L6H9 could be a potential candidate for such therapy.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Q.F., Y.J., C.Z., W.Y., W.L., G.L., Z.K., and Y.W. are responsible for the conception and design. Q.F., Y.J., C.Z., W.Y., W.L., Y.W., X.S., and R.C. are responsible for the collection of data. Q.F., Y.J., C.Z., W.Y., W.L., Y.W., X.S., and R.C. analysed the data. Q.F., Y.J., C.Z., W.Y., W.L., Y.W., X.S., R.C., G.L., Z.K., and Y.W. interpreted the data. Q.F., Y.J., C.Z., W.Y., and W.L. wrote the manuscript. G.L., Z.K., and Y.W. revised the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1:
Primers used for real‐time qPCR assay
Figure S1: Densitometric quantifications for Figure 2B and 2C. Panel A and B, for Figure 2B; Panel C‐E, for Figure 2C. (n = 5, #P < 0.05 vs the DMSO group; *P < 0.05 and ns = not significant, vs the HG group).
Figure S2: Densitometric quantifications for Figure 3G. (n = 5, #P < 0.05 and ns = not significant, vs the NC group; *P < 0.05, vs the HG + NC group).
Figure S3. MAPKs inhibitors blocked HG‐induced activation of RAS and expression of fibrotic factors. NRK‐ 52E cells were firstly pretreated with SB203580 (SB), SP600125 (SP), or PD98059 (PD) for 1 h and then stimulated by HG for 12 h (mRNA detection) or 24 h (protein detection), respectively. ACE and TGF‐β protein levels were measured by western blot assay (A). The mRNA levels of ACE (B), AT‐1 (C), TGF‐β (D), and Renin (E) were measured by real‐time qPCR assay. The cultural medium was collected for Ang II detection by ELISA (F). n = 5 independent experiments; Bar graph shows mean values ± SE; *p < 0.05, **p < 0.01, ***p < 0.001, and ns = not significant, versus HG group.
Figure S4: Co‐immofluorescence staining analysis for MD2 and WT‐1 in kidney tissues of diabetic mice. 14 mice were treated with a single intraperitoneal injection of STZ (100 mg/kg in citrate buffer, pH 4.5), while 14 control mice were received the same volume of citrate buffer. Seven days after STZ injection, mice with fastingblood glucose >216 mg/dL were considered as diabetic. All mice were fed with standard animal low‐fat diet. After 2 months, randomly 7 control mice and 7 diabetic mice were killed under ether anesthesia. After another 2 months, 7 control mice and 7 diabetic mice were killed under ether anesthesia. Renal tissues were collected at the time of sacrifice. Kidney tissue from each group was evaluated for distribution of MD2 and WT‐1 (a marker for podocytes) by double immunofluorescence staining (the blue are DAPI staining for nuclei). Representative images were shown. The merged images showed no overlap of MD2 and WT‐1 expression, indicating that podocytes do not express MD2 protein. Scale bar: 20 μm.
Figure S5: Co‐immofluorescence staining analysis for MD2 and F4/80 in kidney tissues of diabetic mice. 14 mice were treated with a single intraperitoneal injection of STZ (100 mg/kg in citrate buffer, pH 4.5), while 14 control mice were received the same volume of citrate buffer. Seven days after STZ injection, mice with fasting‐blood glucose >216 mg/dL were considered as diabetic. All mice were fed with standard animal low‐fat diet. After 2 months, randomly 7 control mice and 7 diabetic mice were killed under ether anesthesia. After another 2 months, 7 control mice and 7 diabetic mice were killed under ether anesthesia. Renal tissues were collected at the time of sacrifice. Kidney tissue from each group was evaluated for distribution of MD2 and F4/80 (a marker for infiltrated macrophages) by double immunofluorescence staining (the blue are DAPI staining for nuclei). Representative images were shown. The merged images showed slight overlap of MD2 and F4/80 expression, indicating that infiltrated macrophages express less MD2 protein. Scale bar: 20 μm.
Figure S6: Glucose and body weight monitoring of animals. MD2 knockout mice (KO) and their wild type control (C57BL/6, B6) were induced diabetes by STZ injection (as described in Materials and Methods). MD2 knockout had no effect on A) serum glucose levels and B) body‐weight (wild type non‐diabetic = WT‐Ctrol, wild type diabetic = WT‐DM, MD2 knockout non‐diabetic = KO‐Ctrl and MD2 knockout diabetic = KO‐DM, wild type diabetic mice with Valsartan treatment = WT‐DM + Val) . Values are reported as means ± SEM; n = 8 per group.
Figure S7: Ultrasound kidney function analysis showed that MD2 knockout attenuated kidney dysfunction in diabetic mice. MD2 knockout mice (KO) and their wild type control (C57BL/6, B6) were induced diabetes by STZ injection (as described in Materials and Methods). Before sacrifice, ultrasonography assay was performed on anesthetized mice at rest using a high‐resolution imaging system for small animals (Vevo 770, Visual Sonics, Canada), equipped with a high‐frequent ultrasound probe (RMV‐707B). All hair was removed from the lower back using a chemical hair remover and the aquasonic clear ultrasound gel (Parker Laboratories, Fairfield, NJ) was applied to the lower back to optimize the visibility of the renal artery. Mice were placed in the supine position on a heating pad to maintain body temperature at 36–37°C. Left kidney blood flow velocity (LKV), and systolic and diastolic pressure ratio (S/D) from four experimental groups were detected by ultrasonographic measurement. Wild type non‐diabetic = WT‐Ctrl, wild type diabetic = WT‐DM, MD2 knockout non‐diabetic = KO‐Ctrol and MD2 knockout diabetic = KO‐DM; Values are reported as means ± SEM; n = 8 per group. #P < 0.05 vs the WT‐Ctrl group; *P < 0.05 vs the WT‐DM group.
Figure S8: Densitometric quantifications for Figure 5H. (n = 7 per group, #P < 0.05 and ns = not significant, vs the WT‐Ctrl group; *P < 0.05, vs the WT‐DM group).
Figure S9: Quantification for staining results in Figure 6A. Data from 8 mice per group; values reported as mean + SEM; *P < 0.01; ns = no significance.
Figure S10: Densitometric quantifications for Figure 6C. (n = 7 per group, #P < 0.05 and ns = not significant, vs the WT‐Ctrl group; *P < 0.05, vs the WT‐DM group).
Figure S11:MD2 knockout mice reduced inflammatory changes in diabetes. MD2 knockout mice (KO) and their wild type control (C57BL/6, B6) were induced diabetes by STZ injection (as described in Materials and Methods). A) Immunohistochemical analysis showing diabetes induced increased TNFα and CD68 expression are prevented in the MD2 knockout mice. After deparaffinization and rehydration, slides were treated with 3% H2O2 for 10 min and with 1% bovine serum albumin in phosphate‐buffered saline (PBS) for 30 min. Slides were then incubated overnight at 4°C with anti‐CD68 and anti‐TNF‐α antibodies (1:200) followed by incubation with horseradish peroxidase‐conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:500) for 1 h at room temperature. Hematoxylin was used as nuclear stain. The sections were dehydrated with graded alcohol and xylene and mounted with neutral resins. The slides were examined microscopically (×200, Nikon, Tokyo, Japan). B‐D) In parallel diabetes induced increased renal B) TNF‐α, C) IL‐6 and D) ICAM‐1 mRNA expressions were prevented in the MD2 knockout mice. (mRNA data are normalized to β‐actin, wild type nondiabetic = B6‐Ctrol, wild type diabetic = B6‐DM, MD2 knockout non‐diabetic = KO‐Ctrol and MD2 knockout diabetic = KO‐DM). Values are reported as means ± SEM; n = 8 per group; ###P < 0.001, ** P < 0.01, and ***P < 0.001.
Figure S12:L6H9 administration showed no renal side‐effect in mice. 7 male C57BL/6 mice were administrated with L6H9 (20 mg/kg) by oral gavage once every 2 days for 4 months. The age‐matched control group (n = 7) received 1% CMC‐Na solution alone according to the same schedule. No death and no difference in body weight changes between these two groups were observed during the experiment. After 4 months, animals were killed under ether anesthesia. Blood, urine, and renal tissues were collected at the time of sacrifice. A) Representative light micrograph of histochemical assessment of kidney tissues: hematoxylin and eosin (H&E) staining, PAS for glycogen (purple), Masson's trichrome stain (Blue) for detection of connective tissue, and Sirius Red staining were used for the detection of fibrosis (Red); 400 x magnification (The methodology for histopathological analysis was referred the description in Materials and Methods). B) Immunohistochemical analysis showing L6H9 did not increase TNF‐α and F4/80 expression in mouse kidney (The methodology for histopathological analysis was referred the description in Figure S6A). C) Serum creatinine level was detected by corresponding kits. D) The ratio of kidney weight to body weight was recorded. E) Urinary albumin level was detected by corresponding kit. F) Urinary creatinine level was detected by commercial kit and the ratio of urinary albumin to urinary creatinine was calculated. Mean values ± SE; n = 7 per group; ns = not significant.
Figure S13. Phenotypic changes in L6H9‐treated type 1 diabetic mice. Curcumin, which has been reported to show protective effect on diabetic nephropathy, was used as a positive comparison. Method: Male C57BL/6 mice weighing 18–22 g were obtained from the Animal Centre of Wenzhou Medical University. Animals were housed at a constant room temperature with a 12:12 h light–dark cycle and were fed with a standard rodent diet and water. All animal care and experimental procedures were approved by the Wenzhou Medical University Animal Policy and Welfare Committee. To induce type 1 diabetes, mice were treated with a single intraperitoneal injection of STZ (100 mg/kg in citrate buffer, pH 4.5), while the control animals were received the same volume of citrate buffer. The blood glucose level was monitored using glucometer. One week after STZ injection, mice with fasting‐blood glucose >216 mg/dL were considered as diabetic. Randomly selected diabetic animals were divided into three groups: DM (n = 8), curcumin‐treated DM (DM + Cur, n = 8) and L6H9‐treated DM (DM + L6H9, n = 8). In the DM + Cur group and DM + L6H9 group, curcumin (50 mg/kg) and L6H9 (20 mg/kg) were administered as oral gavage once every 2 days, respectively. The DM group and age‐matched control group (n = 8) received 1% CMCNa solution alone according to the same schedule. Bodyweight and blood glucose levels were recorded weekly. After 8 weeks of treatment, animals were killed under ether anesthesia. Blood and renal tissues were collected at the time of sacrifice. A portion of renal tissues from all animals were fixed in 4% paraformaldehyde for paraffin embedding and histopathological analysis (The methodology for histopathological analysis was referred the description in Materials and Methods). Remaining renal tissues were snap‐frozen in liquid nitrogen for gene and protein expression analysis. A and B. L6H9 or curcumin treatment had no effect on body weight change (A) and serum glucose level (B). C. Both treatments however reduced kidney/body ratio, which indicated less kidney injury. Values are reported as means ± SEM; n = 8 in four groups; # P < 0.05, versus Ctrl group; * P < 0.05, versus DM group. Neither L6H9 nor curcumin (used as a positive control) treatment altered body weight or blood glucose levels in the diabetic mice. However, kidney to body weight ratio was significantly reduced following L6H9 treatment.
Figure S14: Densitometric quantifications for Figure 7B, 7C, and 7F. Panel A‐C, for Figure 7B; Panel D and E, for Figure 7C; Panel F, for Figure 7F. (n = 7 per group, #P < 0.05, vs the Ctrl group; *P < 0.05, vs the DM group).
Figure S15:Oral administration of L6H9 inhibited diabetic‐induced inflammation in C57BL/6 mouse kidney. The methodology of animal experiment was described in Figure S13. L6H9 lowered inflammatory response in kidneys in diabetes as evidenced by decreased hyperglycemia‐induced TNF‐α production, mRNA expression of inflammatory cytokines (TNF‐α, IL‐6, IL‐1β and IFN‐γ), adhesion molecules (ICAM‐1 and VCAM‐ 1) and chemokine (MCP‐1) at a level comparable to those of curcumin treatment. A) Immunohistochemical analysis showing diabetes induced increased TNFα protein expression is prevented following L6H9 and curcumin treatments. In parallel diabetes induced increased renal B) TNFα, C) IL‐1β D) IFN‐α, E) ICAM‐1 and VCAM‐1and F) MCP‐1 mRNA expressions were prevented following curcumin and L6H9 treatment (mRNA data are normalized to β‐actin, DM = diabetic). Values are reported as means ± SEM; n = 8 in four groups; # P < 0.05 versusCtrl group; * P < 0.05 versus DM group.
Figure S16: Either L6H9 administration or MD2 knockout inhibited diabetic‐induced IκBα phosphorylation in C57BL/6 mouse kidney. The methodology of animal experiments was described in Methods section. The mouse kidney tissues were performed to determine IκBα phosphorylation using Western blot assay. Below is the densitometric quantification. (n = 8 per group, #P < 0.05 and ns = no significance, vs the WT‐Ctrl group; *P < 0.05, vs the WT‐DM group).
ACKNOWLEDGEMENTS
This work was supported by the National Key Research Project (Grant 2017YFA0506000), the National Natural Science Foundation of China (Grants 81670768, 81770850, 81600659, and 81603180), and Natural Science Foundation of Zhejiang Province (Grants LR18H160003, LR16H310001, LY18H310012, LY18H290009, and LY17H050007).
Wang Y, Fang Q, Jin Y, et al. Blockade of myeloid differentiation 2 attenuates diabetic nephropathy by reducing activation of the renin‐angiotensin system in mouse kidneys. Br J Pharmacol. 2019;176:2642–2657. 10.1111/bph.14687
Yi Wang, Qilu Fang, and Yiyi Jin contributed equally to this work.
Contributor Information
Yi Wang, Email: yi.wang1122@wmu.edu.cn.
Guang Liang, Email: wzmcliangguang@163.com.
REFERENCES
- Adanitsch, F. , Ittig, S. , Stockl, J. , Oblak, A. , Haegman, M. , Jerala, R. , … Zamyatina, A. (2014). Development of αGlcN(1<‐‐>1)αMan‐based lipid A mimetics as a novel class of potent Toll‐like receptor 4 agonists. Journal of Medicinal Chemistry, 57, 8056–8071. 10.1021/jm500946r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017a). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017b). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017a). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Other proteins. British Journal of Pharmacology, 174, S1–S16. 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017b). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Other ion channels. British Journal of Pharmacology, 174, S195–S207. 10.1111/bph.13881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda, F. , Vacchelli, E. , Obrist, F. , Eggermont, A. , Galon, J. , Sautes‐Fridman, C. , … Galluzzi, L. (2014). Trial Watch: Toll‐like receptor agonists in oncological indications. Oncoimmunology, 3, e29179 10.4161/onci.29179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett, A. H. , Bain, S. C. , Bouter, P. , Karlberg, B. , Madsbad, S. , Jervell, J. , & Mustonen, J. (2004). Angiotensin‐receptor blockade versus converting‐enzyme inhibition in type 2 diabetes and nephropathy. The New England Journal of Medicine, 351, 1952–1961. 10.1056/NEJMoa042274 [DOI] [PubMed] [Google Scholar]
- Castrop, H. , Hocherl, K. , Kurtz, A. , Schweda, F. , Todorov, V. , & Wagner, C. (2010). Physiology of kidney renin. Physiological Reviews, 90, 607–673. 10.1152/physrev.00011.2009 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Bond, R. A. , Spina, D. , Ahluwalia, A. , Alexander, S. P. , Giembycz, M. A. , … McGrath, J. C. (2015). Experimental design and analysis and their reporting: New guidance for publication in BJP. British Journal of Pharmacology, 172, 3461–3471. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dange, R. B. , Agarwal, D. , Masson, G. S. , Vila, J. , Wilson, B. , Nair, A. , & Francis, J. (2014). Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II‐induced hypertension. Cardiovascular Research, 103, 17–27. 10.1093/cvr/cvu067 [DOI] [PubMed] [Google Scholar]
- Denton, K. M. , Fennessy, P. A. , Alcorn, D. , & Anderson, W. P. (1992). Morphometric analysis of the actions of angiotensin II on renal arterioles and glomeruli. The American Journal of Physiology, 262, F367–F372. [DOI] [PubMed] [Google Scholar]
- Fang, Q. , Wang, J. , Wang, L. , Zhang, Y. , Yin, H. , Li, Y. , … Zheng, C. (2015). Attenuation of inflammatory response by a novel chalcone protects kidney and heart from hyperglycemia‐induced injuries in type 1 diabetic mice. Toxicology and Applied Pharmacology, 288, 179–191. 10.1016/j.taap.2015.07.009 [DOI] [PubMed] [Google Scholar]
- Feliers, D. , & Kasinath, B. S. (2010). Mechanism of VEGF expression by high glucose in proximal tubule epithelial cells. Molecular and Cellular Endocrinology, 314, 136–142. 10.1016/j.mce.2009.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J. , Zou, C. , Mei, L. , Zhang, Y. , Qian, Y. , You, S. , … Liang, G. (2017). MD2 mediates angiotensin II‐induced cardiac inflammation and remodeling via directly binding to Ang II and activating TLR4/NF‐κB signaling pathway. Basic Research in Cardiology, 112, 9 10.1007/s00395-016-0599-5 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res, 46 (D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein, W. M. , Liu, T. Y. , Skwarczynski, M. , & Toth, I. (2014). Toll‐like receptor agonists: A patent review (2011‐2013). Expert Opinion on Therapeutic Patents, 24, 453–470. 10.1517/13543776.2014.880691 [DOI] [PubMed] [Google Scholar]
- Jialal, I. , Major, A. M. , & Devaraj, S. (2014). Global Toll‐like receptor 4 knockout results in decreased renal inflammation, fibrosis and podocytopathy. Journal of Diabetes and its Complications, 28, 755–761. 10.1016/j.jdiacomp.2014.07.003 [DOI] [PubMed] [Google Scholar]
- Kawai, T. , & Akira, S. (2006). TLR signaling. Cell Death and Differentiation, 13, 816–825. 10.1038/sj.cdd.4401850 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & Group NCRRGW (2010). Animal research: reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, Y. , Kondo, S. , Urushihara, M. , Suga, K. , Matsuura, S. , Takamatsu, M. , … Kagami, S. (2011). Angiotensin II type I receptor blockade suppresses glomerular renin‐angiotensin system activation, oxidative stress, and progressive glomerular injury in rat anti‐glomerular basement membrane glomerulonephritis. Translational Research, 158, 235–248. 10.1016/j.trsl.2011.05.003 [DOI] [PubMed] [Google Scholar]
- Kuwabara, T. , Mori, K. , Mukoyama, M. , Kasahara, M. , Yokoi, H. , Saito, Y. , … Nakao, K. (2012). Exacerbation of diabetic nephropathy by hyperlipidaemia is mediated by Toll‐like receptor 4 in mice. Diabetologia, 55, 2256–2266. 10.1007/s00125-012-2578-1 [DOI] [PubMed] [Google Scholar]
- Lin, M. , Yiu, W. H. , Wu, H. J. , Chan, L. Y. , Leung, J. C. , Au, W. S. , … Tang, S. C. (2012). Toll‐like receptor 4 promotes tubular inflammation in diabetic nephropathy. Journal of the American Society of Nephrology, 23, 86–102. 10.1681/ASN.2010111210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv, J. , Chen, Q. , Shao, Y. , Chen, Y. , & Shi, J. (2015). Cross‐talk between angiotensin‐II and toll‐like receptor 4 triggers a synergetic inflammatory response in rat mesangial cells under high glucose conditions. Biochemical and Biophysical Research Communications, 459, 264–269. 10.1016/j.bbrc.2015.02.096 [DOI] [PubMed] [Google Scholar]
- Lv, J. , Jia, R. , Yang, D. , Zhu, J. , & Ding, G. (2009). Candesartan attenuates angiotensin II‐induced mesangial cell apoptosis via TLR4/MyD88 pathway. Biochemical and Biophysical Research Communications, 380, 81–86. 10.1016/j.bbrc.2009.01.035 [DOI] [PubMed] [Google Scholar]
- Ma, J. , Chadban, S. J. , Zhao, C. Y. , Chen, X. , Kwan, T. , Panchapakesan, U. , … Wu, H. (2014). TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS ONE, 9, e97985 10.1371/journal.pone.0097985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , Drummond, G. B. , McLachlan, E. M. , Kilkenny, C. , & Wainwright, C. L. (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. British Journal of Pharmacology, 160, 1573–1576. 10.1111/j.1476-5381.2010.00873.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsy, M. A. , Heeba, G. H. , & Mahmoud, M. E. (2015). Ameliorative effect of eprosartan on high‐fat diet/streptozotocin‐induced early diabetic nephropathy in rats. European Journal of Pharmacology, 750, 90–97. 10.1016/j.ejphar.2015.01.027 [DOI] [PubMed] [Google Scholar]
- Nair, A. R. , Ebenezer, P. J. , Saini, Y. , & Francis, J. (2015). Angiotensin II‐induced hypertensive renal inflammation is mediated through HMGB1‐TLR4 signaling in rat tubulo‐epithelial cells. Experimental Cell Research, 335, 238–247. 10.1016/j.yexcr.2015.05.011 [DOI] [PubMed] [Google Scholar]
- Nakashima, T. , Umemoto, S. , Yoshimura, K. , Matsuda, S. , Itoh, S. , Murata, T. , … Matsuzaki, M. (2015). TLR4 is a critical regulator of angiotensin II‐induced vascular remodeling: The roles of extracellular SOD and NADPH oxidase. Hypertension Research, 38, 649–655. 10.1038/hr.2015.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, G. , & Muller, D. N. (2010). The biology of the (pro)renin receptor. Journal of the American Society of Nephrology, 21, 18–23. 10.1681/ASN.2009030300 [DOI] [PubMed] [Google Scholar]
- Pan, Y. , Huang, Y. , Wang, Z. , Fang, Q. , Sun, Y. , Tong, C. , … Liang, G. (2014). Inhibition of MAPK‐mediated ACE expression by compound C66 prevents STZ‐induced diabetic nephropathy. Journal of Cellular and Molecular Medicine, 18, 231–241. 10.1111/jcmm.12175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. H. , Kim, N. D. , Jung, J. K. , Lee, C. K. , Han, S. B. , & Kim, Y. (2012). Myeloid differentiation 2 as a therapeutic target of inflammatory disorders. Pharmacology & Therapeutics, 133, 291–298. 10.1016/j.pharmthera.2011.11.001 [DOI] [PubMed] [Google Scholar]
- Roscioni, S. S. , Heerspink, H. J. , & de Zeeuw, D. (2014). The effect of RAAS blockade on the progression of diabetic nephropathy. Nature Reviews. Nephrology, 10, 77–87. 10.1038/nrneph.2013.251 [DOI] [PubMed] [Google Scholar]
- Ruster, C. , & Wolf, G. (2006). Renin‐angiotensin‐aldosterone system and progression of renal disease. Journal of the American Society of Nephrology, 17, 2985–2991. 10.1681/ASN.2006040356 [DOI] [PubMed] [Google Scholar]
- Seikaly, M. G. , Arant, B. S. Jr. , & Seney, F. D. Jr. (1990). Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat. The Journal of Clinical Investigation, 86, 1352–1357. 10.1172/JCI114846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, R. , Alavi, N. , Singh, A. K. , & Leehey, D. J. (1999). Role of angiotensin II in glucose‐induced inhibition of mesangial matrix degradation. Diabetes, 48, 2066–2073. 10.2337/diabetes.48.10.2066 [DOI] [PubMed] [Google Scholar]
- Singh, R. , Singh, A. K. , Alavi, N. , & Leehey, D. J. (2003). Mechanism of increased angiotensin II levels in glomerular mesangial cells cultured in high glucose. Journal of the American Society of Nephrology, 14, 873–880. 10.1097/01.ASN.0000060804.40201.6E [DOI] [PubMed] [Google Scholar]
- Tang, S. C. , & Lai, K. N. (2012). The pathogenic role of the renal proximal tubular cell in diabetic nephropathy. Nephrology, Dialysis, Transplantation, 27, 3049–3056. 10.1093/ndt/gfs260 [DOI] [PubMed] [Google Scholar]
- Toma, I. , Kang, J. J. , Sipos, A. , Vargas, S. , Bansal, E. , Hanner, F. , … Peti‐Peterdi, J. (2008). Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. The Journal of Clinical Investigation, 118, 2526–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon, V. , & Thomson, S. C. (2012). Renal function in diabetic disease models: The tubular system in the pathophysiology of the diabetic kidney. Annual Review of Physiology, 74, 351–375. 10.1146/annurev-physiol-020911-153333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas, S. L. , Toma, I. , Kang, J. J. , Meer, E. J. , & Peti‐Peterdi, J. (2009). Activation of the succinate receptor GPR91 in macula densa cells causes renin release. Journal of the American Society of Nephrology, 20, 1002–1011. 10.1681/ASN.2008070740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri, N. D. , Bai, Y. , Ni, Z. , Quiroz, Y. , Pandian, R. , & Rodriguez‐Iturbe, B. (2007). Intra‐renal angiotensin II/AT1 receptor, oxidative stress, inflammation, and progressive injury in renal mass reduction. The Journal of Pharmacology and Experimental Therapeutics, 323, 85–93. 10.1124/jpet.107.123638 [DOI] [PubMed] [Google Scholar]
- Verzola, D. , Cappuccino, L. , D'Amato, E. , Villaggio, B. , Gianiorio, F. , Mij, M. , … Garibotto, G. (2014). Enhanced glomerular Toll‐like receptor 4 expression and signaling in patients with type 2 diabetic nephropathy and microalbuminuria. Kidney International, 86, 1229–1243. 10.1038/ki.2014.116 [DOI] [PubMed] [Google Scholar]
- Vidotti, D. B. , Casarini, D. E. , Cristovam, P. C. , Leite, C. A. , Schor, N. , & Boim, M. A. (2004). High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. American Journal of Physiology. Renal Physiology, 286, F1039–F1045. 10.1152/ajprenal.00371.2003 [DOI] [PubMed] [Google Scholar]
- Visavadiya, N. P. , Li, Y. , & Wang, S. (2011). High glucose upregulates upstream stimulatory factor 2 in human renal proximal tubular cells through angiotensin II‐dependent activation of CREB. Nephron. Experimental Nephrology, 117, e62–e70. 10.1159/000320593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada, J. , & Makino, H. (2016). Innate immunity in diabetes and diabetic nephropathy. Nature Reviews. Nephrology, 12, 13–26. 10.1038/nrneph.2015.175 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Qian, Y. , Fang, Q. , Zhong, P. , Li, W. , Wang, L. , … Liang, G. (2017). Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nature Communications, 8, 13997 10.1038/ncomms13997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, L. , Wang, M. , Yang, S. , Liu, F. , & Sun, L. (2013). A glimpse of the pathogenetic mechanisms of Wnt/β‐catenin signaling in diabetic nephropathy. BioMed Research International, 2013, 987064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zain, M. , & Awan, F. R. (2014). Renin angiotensin aldosterone system (RAAS): Its biology and drug targets for treating diabetic nephropathy. Pakistan Journal of Pharmaceutical Sciences, 27, 1379–1391. [PubMed] [Google Scholar]
- Zhong, P. , Wu, L. , Qian, Y. , Fang, Q. , Liang, D. , Wang, J. , … Liang, G. (2015). Blockage of ROS and NF‐κB‐mediated inflammation by a new chalcone L6H9 protects cardiomyocytes from hyperglycemia‐induced injuries. Biochimica et Biophysica Acta, 1852, 1230–1241. 10.1016/j.bbadis.2015.02.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1:
Primers used for real‐time qPCR assay
Figure S1: Densitometric quantifications for Figure 2B and 2C. Panel A and B, for Figure 2B; Panel C‐E, for Figure 2C. (n = 5, #P < 0.05 vs the DMSO group; *P < 0.05 and ns = not significant, vs the HG group).
Figure S2: Densitometric quantifications for Figure 3G. (n = 5, #P < 0.05 and ns = not significant, vs the NC group; *P < 0.05, vs the HG + NC group).
Figure S3. MAPKs inhibitors blocked HG‐induced activation of RAS and expression of fibrotic factors. NRK‐ 52E cells were firstly pretreated with SB203580 (SB), SP600125 (SP), or PD98059 (PD) for 1 h and then stimulated by HG for 12 h (mRNA detection) or 24 h (protein detection), respectively. ACE and TGF‐β protein levels were measured by western blot assay (A). The mRNA levels of ACE (B), AT‐1 (C), TGF‐β (D), and Renin (E) were measured by real‐time qPCR assay. The cultural medium was collected for Ang II detection by ELISA (F). n = 5 independent experiments; Bar graph shows mean values ± SE; *p < 0.05, **p < 0.01, ***p < 0.001, and ns = not significant, versus HG group.
Figure S4: Co‐immofluorescence staining analysis for MD2 and WT‐1 in kidney tissues of diabetic mice. 14 mice were treated with a single intraperitoneal injection of STZ (100 mg/kg in citrate buffer, pH 4.5), while 14 control mice were received the same volume of citrate buffer. Seven days after STZ injection, mice with fastingblood glucose >216 mg/dL were considered as diabetic. All mice were fed with standard animal low‐fat diet. After 2 months, randomly 7 control mice and 7 diabetic mice were killed under ether anesthesia. After another 2 months, 7 control mice and 7 diabetic mice were killed under ether anesthesia. Renal tissues were collected at the time of sacrifice. Kidney tissue from each group was evaluated for distribution of MD2 and WT‐1 (a marker for podocytes) by double immunofluorescence staining (the blue are DAPI staining for nuclei). Representative images were shown. The merged images showed no overlap of MD2 and WT‐1 expression, indicating that podocytes do not express MD2 protein. Scale bar: 20 μm.
Figure S5: Co‐immofluorescence staining analysis for MD2 and F4/80 in kidney tissues of diabetic mice. 14 mice were treated with a single intraperitoneal injection of STZ (100 mg/kg in citrate buffer, pH 4.5), while 14 control mice were received the same volume of citrate buffer. Seven days after STZ injection, mice with fasting‐blood glucose >216 mg/dL were considered as diabetic. All mice were fed with standard animal low‐fat diet. After 2 months, randomly 7 control mice and 7 diabetic mice were killed under ether anesthesia. After another 2 months, 7 control mice and 7 diabetic mice were killed under ether anesthesia. Renal tissues were collected at the time of sacrifice. Kidney tissue from each group was evaluated for distribution of MD2 and F4/80 (a marker for infiltrated macrophages) by double immunofluorescence staining (the blue are DAPI staining for nuclei). Representative images were shown. The merged images showed slight overlap of MD2 and F4/80 expression, indicating that infiltrated macrophages express less MD2 protein. Scale bar: 20 μm.
Figure S6: Glucose and body weight monitoring of animals. MD2 knockout mice (KO) and their wild type control (C57BL/6, B6) were induced diabetes by STZ injection (as described in Materials and Methods). MD2 knockout had no effect on A) serum glucose levels and B) body‐weight (wild type non‐diabetic = WT‐Ctrol, wild type diabetic = WT‐DM, MD2 knockout non‐diabetic = KO‐Ctrl and MD2 knockout diabetic = KO‐DM, wild type diabetic mice with Valsartan treatment = WT‐DM + Val) . Values are reported as means ± SEM; n = 8 per group.
Figure S7: Ultrasound kidney function analysis showed that MD2 knockout attenuated kidney dysfunction in diabetic mice. MD2 knockout mice (KO) and their wild type control (C57BL/6, B6) were induced diabetes by STZ injection (as described in Materials and Methods). Before sacrifice, ultrasonography assay was performed on anesthetized mice at rest using a high‐resolution imaging system for small animals (Vevo 770, Visual Sonics, Canada), equipped with a high‐frequent ultrasound probe (RMV‐707B). All hair was removed from the lower back using a chemical hair remover and the aquasonic clear ultrasound gel (Parker Laboratories, Fairfield, NJ) was applied to the lower back to optimize the visibility of the renal artery. Mice were placed in the supine position on a heating pad to maintain body temperature at 36–37°C. Left kidney blood flow velocity (LKV), and systolic and diastolic pressure ratio (S/D) from four experimental groups were detected by ultrasonographic measurement. Wild type non‐diabetic = WT‐Ctrl, wild type diabetic = WT‐DM, MD2 knockout non‐diabetic = KO‐Ctrol and MD2 knockout diabetic = KO‐DM; Values are reported as means ± SEM; n = 8 per group. #P < 0.05 vs the WT‐Ctrl group; *P < 0.05 vs the WT‐DM group.
Figure S8: Densitometric quantifications for Figure 5H. (n = 7 per group, #P < 0.05 and ns = not significant, vs the WT‐Ctrl group; *P < 0.05, vs the WT‐DM group).
Figure S9: Quantification for staining results in Figure 6A. Data from 8 mice per group; values reported as mean + SEM; *P < 0.01; ns = no significance.
Figure S10: Densitometric quantifications for Figure 6C. (n = 7 per group, #P < 0.05 and ns = not significant, vs the WT‐Ctrl group; *P < 0.05, vs the WT‐DM group).
Figure S11:MD2 knockout mice reduced inflammatory changes in diabetes. MD2 knockout mice (KO) and their wild type control (C57BL/6, B6) were induced diabetes by STZ injection (as described in Materials and Methods). A) Immunohistochemical analysis showing diabetes induced increased TNFα and CD68 expression are prevented in the MD2 knockout mice. After deparaffinization and rehydration, slides were treated with 3% H2O2 for 10 min and with 1% bovine serum albumin in phosphate‐buffered saline (PBS) for 30 min. Slides were then incubated overnight at 4°C with anti‐CD68 and anti‐TNF‐α antibodies (1:200) followed by incubation with horseradish peroxidase‐conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:500) for 1 h at room temperature. Hematoxylin was used as nuclear stain. The sections were dehydrated with graded alcohol and xylene and mounted with neutral resins. The slides were examined microscopically (×200, Nikon, Tokyo, Japan). B‐D) In parallel diabetes induced increased renal B) TNF‐α, C) IL‐6 and D) ICAM‐1 mRNA expressions were prevented in the MD2 knockout mice. (mRNA data are normalized to β‐actin, wild type nondiabetic = B6‐Ctrol, wild type diabetic = B6‐DM, MD2 knockout non‐diabetic = KO‐Ctrol and MD2 knockout diabetic = KO‐DM). Values are reported as means ± SEM; n = 8 per group; ###P < 0.001, ** P < 0.01, and ***P < 0.001.
Figure S12:L6H9 administration showed no renal side‐effect in mice. 7 male C57BL/6 mice were administrated with L6H9 (20 mg/kg) by oral gavage once every 2 days for 4 months. The age‐matched control group (n = 7) received 1% CMC‐Na solution alone according to the same schedule. No death and no difference in body weight changes between these two groups were observed during the experiment. After 4 months, animals were killed under ether anesthesia. Blood, urine, and renal tissues were collected at the time of sacrifice. A) Representative light micrograph of histochemical assessment of kidney tissues: hematoxylin and eosin (H&E) staining, PAS for glycogen (purple), Masson's trichrome stain (Blue) for detection of connective tissue, and Sirius Red staining were used for the detection of fibrosis (Red); 400 x magnification (The methodology for histopathological analysis was referred the description in Materials and Methods). B) Immunohistochemical analysis showing L6H9 did not increase TNF‐α and F4/80 expression in mouse kidney (The methodology for histopathological analysis was referred the description in Figure S6A). C) Serum creatinine level was detected by corresponding kits. D) The ratio of kidney weight to body weight was recorded. E) Urinary albumin level was detected by corresponding kit. F) Urinary creatinine level was detected by commercial kit and the ratio of urinary albumin to urinary creatinine was calculated. Mean values ± SE; n = 7 per group; ns = not significant.
Figure S13. Phenotypic changes in L6H9‐treated type 1 diabetic mice. Curcumin, which has been reported to show protective effect on diabetic nephropathy, was used as a positive comparison. Method: Male C57BL/6 mice weighing 18–22 g were obtained from the Animal Centre of Wenzhou Medical University. Animals were housed at a constant room temperature with a 12:12 h light–dark cycle and were fed with a standard rodent diet and water. All animal care and experimental procedures were approved by the Wenzhou Medical University Animal Policy and Welfare Committee. To induce type 1 diabetes, mice were treated with a single intraperitoneal injection of STZ (100 mg/kg in citrate buffer, pH 4.5), while the control animals were received the same volume of citrate buffer. The blood glucose level was monitored using glucometer. One week after STZ injection, mice with fasting‐blood glucose >216 mg/dL were considered as diabetic. Randomly selected diabetic animals were divided into three groups: DM (n = 8), curcumin‐treated DM (DM + Cur, n = 8) and L6H9‐treated DM (DM + L6H9, n = 8). In the DM + Cur group and DM + L6H9 group, curcumin (50 mg/kg) and L6H9 (20 mg/kg) were administered as oral gavage once every 2 days, respectively. The DM group and age‐matched control group (n = 8) received 1% CMCNa solution alone according to the same schedule. Bodyweight and blood glucose levels were recorded weekly. After 8 weeks of treatment, animals were killed under ether anesthesia. Blood and renal tissues were collected at the time of sacrifice. A portion of renal tissues from all animals were fixed in 4% paraformaldehyde for paraffin embedding and histopathological analysis (The methodology for histopathological analysis was referred the description in Materials and Methods). Remaining renal tissues were snap‐frozen in liquid nitrogen for gene and protein expression analysis. A and B. L6H9 or curcumin treatment had no effect on body weight change (A) and serum glucose level (B). C. Both treatments however reduced kidney/body ratio, which indicated less kidney injury. Values are reported as means ± SEM; n = 8 in four groups; # P < 0.05, versus Ctrl group; * P < 0.05, versus DM group. Neither L6H9 nor curcumin (used as a positive control) treatment altered body weight or blood glucose levels in the diabetic mice. However, kidney to body weight ratio was significantly reduced following L6H9 treatment.
Figure S14: Densitometric quantifications for Figure 7B, 7C, and 7F. Panel A‐C, for Figure 7B; Panel D and E, for Figure 7C; Panel F, for Figure 7F. (n = 7 per group, #P < 0.05, vs the Ctrl group; *P < 0.05, vs the DM group).
Figure S15:Oral administration of L6H9 inhibited diabetic‐induced inflammation in C57BL/6 mouse kidney. The methodology of animal experiment was described in Figure S13. L6H9 lowered inflammatory response in kidneys in diabetes as evidenced by decreased hyperglycemia‐induced TNF‐α production, mRNA expression of inflammatory cytokines (TNF‐α, IL‐6, IL‐1β and IFN‐γ), adhesion molecules (ICAM‐1 and VCAM‐ 1) and chemokine (MCP‐1) at a level comparable to those of curcumin treatment. A) Immunohistochemical analysis showing diabetes induced increased TNFα protein expression is prevented following L6H9 and curcumin treatments. In parallel diabetes induced increased renal B) TNFα, C) IL‐1β D) IFN‐α, E) ICAM‐1 and VCAM‐1and F) MCP‐1 mRNA expressions were prevented following curcumin and L6H9 treatment (mRNA data are normalized to β‐actin, DM = diabetic). Values are reported as means ± SEM; n = 8 in four groups; # P < 0.05 versusCtrl group; * P < 0.05 versus DM group.
Figure S16: Either L6H9 administration or MD2 knockout inhibited diabetic‐induced IκBα phosphorylation in C57BL/6 mouse kidney. The methodology of animal experiments was described in Methods section. The mouse kidney tissues were performed to determine IκBα phosphorylation using Western blot assay. Below is the densitometric quantification. (n = 8 per group, #P < 0.05 and ns = no significance, vs the WT‐Ctrl group; *P < 0.05, vs the WT‐DM group).