

Abstract
Background and Purpose
Atherosclerosis is a chronic inflammatory disease, and retinoid X receptor‐α (RXRα) is an intriguing anti‐atherosclerosis target. This study investigated whether and how an RXRα modulator, K‐80003, derived from a non‐steroidal anti‐inflammatory drug attenuates atherosclerotic plaque progression and destabilization.
Experimental Approach
Our previously established ApoE−/− mouse model of carotid vulnerable plaque progression was treated with K‐80003 or vehicle for 4 or 8 weeks. Samples of carotid arteries and serum were collected to determine atherosclerotic lesion size, histological features, expression of related proteins, and lipid profiles. In vitro studies were carried out in 7‐ketocholesterol (7‐KC)‐stimulated macrophages treated with or without K‐80003.
Key Results
K‐80003 significantly reduced lesion size, plaque rupture, macrophage infiltration, and inflammatory cytokine levels. Plaque macrophages positive for nuclear p65 (RelA) NF‐κB subunit were markedly reduced after K‐80003 treatment. Also, K‐80003 treatment inhibited 7‐KC‐induced p65 nuclear translocation, IκBα degradation, and transcription of NF‐κB target genes. In addition, K‐80003 inhibited NF‐κB pathway mainly through the reduction of p62/sequestosome 1 (SQSTM1), probably due to promotion of autophagic flux by K‐80003. Mechanistically, cytoplasmic localization of RXRα was associated with decreased autophagic flux. Increasing cytoplasmic RXRα expression by overexpression of RXRα/385 mutant decreased autophagic flux in RAW264.7 cells. Finally, K‐80003 strongly inhibited 7‐KC‐induced RXRα cytoplasmic translocation.
Conclusions and Implications
K‐80003 suppressed atherosclerotic plaque progression and destabilization by promoting macrophage autophagic flux and consequently inhibited the p62/SQSTM1‐mediated NF‐κB proinflammatory pathway. Thus, targeting RXRα‐mediated autophagy‐inflammation axis by its noncanonical modulator may represent a promising strategy to treat atherosclerosis.
Abbreviations
- 7‐KC
7‐ketocholesterol
- Ad
adenovirus
- ApoE
apolipoprotein E
- BafA1
bafilomycin A1
- BMDMs
bone marrow‐derived macrophages
- CD68
cluster of differentiation 68
- LC3
microtubule‐associated protein light chain 3
- RXRα
retinoid X receptor‐α
- SQSTM1
sequestosome 1
What is already known
Retinoid X receptor‐α (RXRα) is an intriguing anti‐atherosclerosis target.
What this study adds
Targeting RXRα‐mediated autophagy‐inflammation axis may represent a promising strategy to treat atherosclerosis.
Treatment with K‐80003, a non‐canonical RXRα modulator, attenuates atherosclerotic plaque progression and destabilization.
What is the clinical significance
Targeting alternate binding sites on RXRα may represent a new strategy for combating atherosclerosis.
K‐80003 may represent a new drug lead that attenuates atherosclerotic plaque progression and destabilization.
1. INTRODUCTION
Atherosclerosis is considered to be not only a disorder of lipid accumulation in the arterial wall but also a chronic inflammatory disease (Libby, Tabas, Fredman, & Fisher, 2014). Inflammation occurs and contributes to all stages of atherosclerosis from initiation through progression and, ultimately, rupture (Fredman & Tabas, 2017; Hansson, Libby, & Tabas, 2015). The beneficial effects of targeting inflammation for prevention of atherosclerosis have been widely demonstrated in animal models (Charo & Taub, 2011). Importantly, two randomized placebo‐controlled phase 3 clinical trial testing anti‐inflammatory agents including canakinumab and methotrexate are being conducted in the United States and Canada (Everett et al., 2013; Ridker et al., 2017; Ridker, Thuren, Zalewski, & Libby, 2011). Thus, anti‐inflammation may represent a promising therapeutic strategy for combating atherosclerosis.
Retinoid X receptor‐α (RXRα) is a unique member of the nuclear receptor superfamily (Zhang et al., 2015), involved in the regulation of several cellular processes including inflammation (Desreumaux et al., 2001; Nunez et al., 2010). Numerous studies have reported the effects of RXRα ligands (rexinoids) on atherogenesis (Claudel et al., 2001; Lalloyer et al., 2006; Staels, 2001; Streb & Miano, 2003). The rexinoids LG100364 and LGD1069 (bexarotene) potently inhibited atherogenesis in ApoE−/− and apolipoprotein E2 knock‐in mice, respectively (Claudel et al., 2001; Lalloyer et al., 2006), suggesting that RXRα is a potential therapeutic target in atherosclerosis. However, treatment with these rexinoids consistently provokes some key unwanted side effects, such as hypertriglyceridaemia, hepatomegaly, and suppression of the thyroid hormone axis, which has hindered their further applications (Altucci, Leibowitz, Ogilvie, de Lera, & Gronemeyer, 2007; Dawson & Xia, 2012; Desvergne, 2007; Szanto et al., 2004). How RXRα mediates the anti‐atherosclerotic effect of its ligands also remains unknown. Like other nuclear receptors, RXRα is conventionally considered as a transcription factor that regulates target gene transcription by binding to DNA response elements. In addition, RXRα possesses extranuclear functions (Claudel et al., 2001; Lalloyer et al., 2006). For instance, RXRα can translocate from the nucleus to the cytoplasm in macrophages in response to stimuli associated with inflammation (Casas et al., 2003; Ghose, Zimmerman, Thevananther, & Karpen, 2004; Zimmerman, Thevananther, Ghose, Burns, & Karpen, 2006) and apoptosis (Cao et al., 2004), both of which are typical characteristics of progressive atherosclerotic lesions (Tabas, 2010). However, whether and how RXRα acts in the cytoplasm of macrophages during the development of atherosclerosis remains currently unknown.
The development of RXRα‐based drugs has been hampered partly due to the unwanted side effects associated with targeting its canonical ligand‐binding pocket to directly modulate its transcriptional activity (Zhang et al., 2015). Recent advances have revealed a new strategy of developing RXRα‐based drugs by targeting non‐canonical binding sites (Su et al., 2016; Tice & Zheng, 2016; Zhang et al., 2015). We previously reported that the non‐steroidal anti‐inflammatory drug sulindac could bind to RXRα to modulate its non‐transcriptional effect (Zhou et al., 2010). In addition, we identified several sulindac analogues including K‐80003, which preferentially bind RXRα but not to the COX enzymes, through a unique binding mechanism (Zhou et al., 2010). This is significant because of the potential adverse effect of the COX signalling pathway on vascular events (Fitzgerald, 2004). It is therefore of great interest to determine whether K‐80003 could exert anti‐atherosclerotic effects.
In the present study, we employed a mouse model of spontaneous plaque rupture with thrombus (Jin et al., 2012) to examine whether the sulindac‐derived RXRα modulator K‐80003 could inhibit atherosclerotic vulnerable plaque formation and progression. Our results demonstrated that K‐80003 could significantly suppress atherosclerotic plaque progression and destabilization by promoting autophagic flux and consequently suppressing the NF‐κB proinflammatory pathway.
2. METHODS
2.1. Cell culture
The RAW264.7 cells (mouse macrophage cell line; ATCC, Cat # TIB‐71; RRID:CVCL_0493) were seeded into 12‐well plates (2 × 105 cells per well) in DMEM (HyClone, Logan, UT, USA; SH30022.01B) containing 10% FBS in a humidified atmosphere containing 5% CO2 at 37°C. Subconfluent cells were used throughout the experiments. Cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). RAW264.7 stable cell lines overexpressing GFP and GFP–RXRα/385 were selected in 600 μg·ml−1 G418 for 3–4 weeks.
2.2. Isolation and culture of mouse bone marrow‐derived macrophages
Bone marrow cells were isolated from both femurs and tibiae of C57BL/6 mice. Cells were cultured in RPMI‐1640 (Hyclone) with 100 U·ml−1 penicillin/streptomycin (Gibco, Carlsbad, CA, USA), 10% FBS, and 20 ng·ml−1 colony‐stimulating factor for 5 days to generate bone marrow‐derived macrophages (BMDMs).
2.3. Animals and animal surgery
All animal care and experimental procedures conformed to the guidelines from the Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and were approved by the Medical Ethics Committee of Shanghai Jiaotong University, China. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology.
ApoE−/− mice (IMSR Cat# JAX:002052, RRID:IMSR_JAX:002052) on a background of C57BL/6 were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Animals were bred and housed under specific pathogen‐free conditions in the animal centre of the Shanghai Jiaotong University School of Medicine, China, and housed in a controlled environment (20 ± 2°C, 12/12‐hr light/dark cycle). A standard chow diet and water were provided ad libitum. At the age of 8 weeks (18–20 g), combined partial ligation of the left common carotid artery and left renal artery was performed in female mice as previously described (Jin et al., 2012), and the animals were then randomly divided into K‐80003 and control groups. K‐80003 was dissolved in Tween 80 and infused via a stomach tube at a dose of 30 mg·kg−1·day−1 after surgery for 2, 4, or 8 weeks. Control mice were infused with an equal volume of Tween 80. Ten mice from each group were killed at each time point, and the carotid arteries were collected for various assessments, which were performed by two investigators in a blinded manner.
2.4. Tissue sample collection and processing
Mice were anaesthetized by isoflurane inhalation and then perfused with ice‐cold isotonic saline followed by 4% paraformaldehyde under physiological pressure. Left carotid arteries were isolated, embedded in optimal cutting temperature (OCT) compound (Sakura Finetechnical, Tokyo, Japan), snap frozen in liquid nitrogen, and stored at −80°C until use. Cryosections (5 μm) were cut every 200 μm over a 2‐mm length of the carotid artery from the distal bifurcation.
2.5. Blood lipid analysis
Blood samples were intracardially collected using a syringe after the mice were anaesthetized by isoflurane inhalation. Plasma was separated by centrifugation and stored at −80°C until use. Total plasma levels of cholesterol and triglycerides were measured using a Hitachi 7180 autoanalyzer (Hitachi High‐Technologies Corp., Japan) according to the manufacturer's instructions.
2.6. Histology
OCT‐embedded left common carotid artery sections were stained using haematoxylin and eosin, Oil Red O, or picrosirius red stains according to standard protocols. All images were captured using an Olympus digital camera (Tokyo, Japan). Lesion area, necrotic core area, Oil Red O‐positive area, and collagen area were analysed using ImagePro Plus software (Media Cybernetics, Rockville, MD, USA; RRID:SCR_016879). Necrotic core areas were defined as areas free of haematoxylin and eosin staining.
2.7. Western blotting
The antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. RAW264.7 cells and mouse BMDMs were treated as indicated in the figure legends. Cell lysates were prepared with NP‐40 lysis buffer (150‐mM NaCl, 1.0% NP‐40, and 50‐mM Tris [pH 8.0]), and equal amounts of lysates were separated by electrophoresis on a 10% SDS‐PAGE gel. PVDF membranes (Roche, Mannheim, Germany) were used for protein transfer. The membranes were then blocked with 5% nonfat milk in TBST (150‐mM NaCl, 50‐mM Tris–HCl [pH 7.4], and 0.1% Tween 20) for 1 hr, followed by incubation with appropriate primary antibodies at 4°C overnight. After primary antibody reaction, the membranes were washed with TBST three times and then incubated with HRP‐labelled secondary antibody at room temperature for 1 hr. After washing with TBST three times, the signals of secondary antibodies were detected by an enhanced chemiluminescence system. Dilutions of the primary antibodies were as follows: anti‐RXRα, 1:1,000; anti‐PARP, 1:1,000; anti‐GAPDH, 1:1,000; anti‐α‐tubulin, 1:1,000; anti‐p65, 1:1,000; anti‐p62/SQSTM1, 1:1,000; anti‐GFP, 1:1,000; anti‐LC3B, 1:3,000; and anti‐IκBα, 1:3,000. The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018).
2.8. siRNA and transfection
Small‐interfering RNA (siRNA) duplexes against mouse RXRα and p62/SQSTM1 were designed and synthesized by RiboBio (Guangzhou, China). A 5‐μl aliquot of 20‐μM siRNA per well was transfected into RAW264.7 cells seeded in six‐well plates with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
2.9. Human tissue specimens
The study conformed to the principles outlined in the Declaration of Helsinki and was approved by the Shanghai Jiaotong University Institute for Medical Ethics Committee, and all subjects gave their informed consent.
Arteries were dissected from patients with atherosclerosis who were undergoing lower extremity amputation and frozen immediately in liquid nitrogen until use. Tissue samples for in situ immunofluorescence were embedded within OCT, sectioned into 5‐μm‐thick slices, and fixed within 15 min in 4% formaldehyde in PBS. All specimens were classified as initial (Type I), fatty streak (Type II), intermediate (Type III), or advanced (Types IV–VI) lesions in terms of the American Heart Association classification (Sun et al., 2012).
2.10. Immunofluorescence
Immunohistochemistry staining was performed as described (Jin et al., 2012; Nie et al., 2014). Briefly, frozen sections were fixed with 4% paraformaldehyde at 4°C for 20 min, permeabilized with 0.2% Triton X‐100 for 10 min at room temperature and blocked with 5% BSA solution. Sections were stained overnight at 4°C in blocking buffer with the appropriate control IgG or with the appropriate primary antibody and detected by secondary antibodies labelled with green fluorescence (donkey anti‐rabbit IgG, 488 nm, Invitrogen), magenta fluorescence (goat anti‐rat, 647 nm, Invitrogen), or red fluorescence (donkey anti‐mouse IgG, 555 nm, Invitrogen). This was followed by costaining with DAPI (Beyotime, Shanghai, China) for 8 min to visualize the nuclei. The images were taken under an LSM 710 confocal laser scanning microscope system (Zeiss, Oberkochen, Germany)
2.11. Morphometry
For the analysis of the different plaque parameters (e.g., intimal surface area, macrophages, vascular smooth muscle cell, collagen, Oil Red O, MMPs, IL‐6, TNF‐α, and the chemokine CCL2, sections equally spaced over the entire length of the artery at 100‐μm intervals were used for analyses. For each type of analysis, 10 sections were used per animal. CD68+, SMA+, collagen, Oil Red O, MMPs+, IL‐6+, TNF‐α+, and CCL2+ areas were measured by image analysis software (ImageJ, National Institutes of Health, Bethesda, MD, USA; RRID:SCR_003070) and normalized to the total intimal surface area.
2.12. RNA extraction and quantitative real‐time PCR
Total RNAs were extracted by TRIzol (Invitrogen). The first‐strand cDNA synthesis was conducted with RevertAid First‐Strand cDNA Synthesis Kits (Fermentas, Madison, WI, USA). Quantitative real‐time PCR reactions were performed using SYBR Green dye and the Roche LightCycler® 480 II system. Relative gene expression (IL‐1α, IL‐1β, IL‐6, MMP13, TNF‐α, and CCL2) were normalized to β‐actin. The primer sequences are presented in Table S1. The resulting values were normalized to control to avoid unwanted sources of variation.
2.13. Adenoviral infection
Adenovirus (Ad)‐p62/SQSTM1 (VH858197) and Ad‐GFP (CV0001) were purchased from ViGene Biosciences Inc (Jinan, China). RAW264.7 cells were seeded in six‐well plates to approximately 75% confluence and infected with Ad‐p62/SQSTM1 or Ad‐GFP for 12 hr. The cells were then washed and incubated in fresh medium for an additional 24 hr before experimentation. These conditions produced an infection efficiency of at least 90%, as determined by GFP expression.
2.14. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Data are provided as mean ± SEM. Two‐tailed unpaired Student's t test (Gaussian distribution) or Mann–Whitney U test (non‐Gaussian distribution) was used to analyse significant differences between two groups. One‐way ANOVA with Tukey's multiple comparison post test (Gaussian distribution) or Kruskal–Wallis test with Dunn's multiple comparison post test (non‐Gaussian distribution) was used to compare variables among three or more groups. Differences in the classification and occurrence of adverse events were analysed with χ 2 tests. In each experiment, “n” means the number of mice and the number of independent experiments in in vivo and in vitro study respectively. Technical replicates were used to ensure the reliability of single values. P < .05 was considered significant. All tests were performed using GraphPad Prism version 6.00 (GraphPad Software, San Diego, CA, USA; RRID:SCR_002798
2.15. Materials
7‐Ketocholesterol (7‐KC; C2394), chloroquine (C6628), and bafilomycin A1 (B1793) were purchased from Sigma‐Aldrich (St. Louis, MO, USA), while Lipofectamine 2000 was obtained from Invitrogen. K‐80003 was dissolved in DMSO for cell‐based in vitro studies and dissolved in Tween 80 for animal study. Antibodies for RXR (sc‐553 and sc‐774), PARP‐1/2 (sc‐7150), GFP (sc‐8834), GAPDH (sc‐365062), p62/sequestosome 1 (SQSTM1; sc‐25575), α‐tubulin (sc‐8035), and p65 (sc‐109) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) (RRID:SCR_008987); antibodies for MMP8 (ab53017), α‐smooth muscle actin (ab21027), IL‐6 (ab6672), TNF‐α (ab6671), microtubule‐associated protein light chain 3 (LC3)‐II (ab51520), β‐actin (ab6276), and IκBα (32518) were from Abcam (Cambridge, MA, USA); antibody for CD68 (137001) was from BioLegend (San Diego, CA, USA); and antibody for MMP13 (NBP1‐45723) was from NOVUS (Cambridge, MA, USA)
2.16. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Cidlowski et al., 2017; Alexander, Fabbro et al., 2017, Alexander, Kelly et al., 2017).
3. RESULTS
3.1. Administration of K‐80003 suppresses atherosclerotic plaque progression and destabilization in ApoE−/− mice
ApoE−/− mice were divided into two groups after undergoing combined partial ligation of the left common carotid and left renal arteries (Jin et al., 2012). One group was treated with K‐80003, and the other group was treated with vehicle (K‐80003 and control groups, respectively; Figure 1a). Mice in the K‐80003 group developed significantly smaller lesions in the carotid arteries as compared with those in the control group after 4 or 8 weeks of treatment (Figure 1b). Further characterization of the lesion phenotype showed that 50% (5/10) of lesions in K‐80003 group and all (10/10) lesions in control group had vulnerable features, accompanied by a decreasing trend of multiple layers with discontinuity (80% vs. 40%), indicative of plaque instability. Meanwhile, there was a significant decrease of intraplaque haemorrhage in K‐80003 group in comparison with control group (80% vs. 20%, respectively;). Importantly, a marked decrease of plaque rupture with lumen thrombus was also observed in K‐80003 group, compared with control group (50% vs. 20%). The data are summarized in Table 1, and the representative images are shown in Figure S1. In addition, vehicle treatment did not change the vessel morphology of the mice without ligation (Figure S2). These data suggest that K‐80003 treatment protects ApoE−/− mice from vulnerable plaque formation and rupture.
Figure 1.
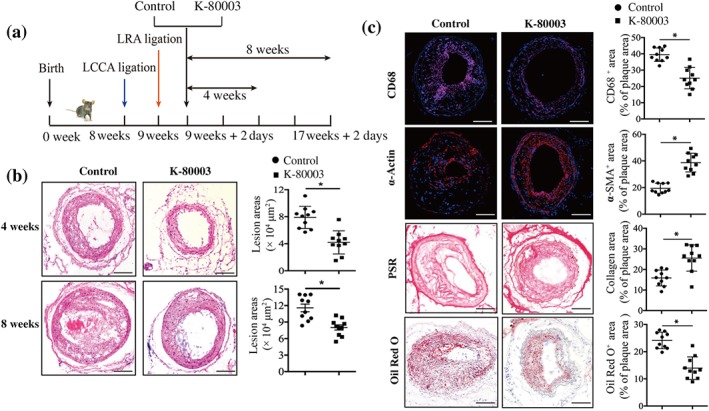
K‐80003 protects ApoE−/− mice from vulnerable plaque formation and rupture. (a) The development of a carotid vulnerable plaque in a mouse treated with K‐80003 or vehicle. (b) Representative images of haematoxylin and eosin staining and quantification of lesion areas of carotid arteries at 4 or 8 weeks for ApoE−/− mice treated with vehicle or K‐80003 (30 mg·kg−1) once a day by oral gavage. Values are mean ± SEM from 10 animals in each group. (c) Histological analysis of carotid arteries from ApoE−/− control and K‐80003 mice 8 weeks after surgery by staining with CD68, α‐smooth muscle actin (α‐SMA), Oil Red O, or picrosirius red (PSR). Quantification of CD68+, α‐SMA+, Oil Red O+, and collagen+ areas relative to total plaque area. Values are mean ± SEM from 10 animals in each group. Images for magnifications are 200× in (b, c). Scale bar: 100 μm. *P < .05, significantly different as indicated. LCCA, left common carotid artery; LRA, left renal artery
Table 1.
Effect of K‐80003 on lesion features in ApoE−/− mice
| Mice | Vulnerable phenotype | Intraplaque haemorrhage | Rupture with thrombus | Multilayer with discontinuity |
|---|---|---|---|---|
| Control (n = 10) | 100% (10) | 80% (8) | 50% (5) | 80% (8) |
| K‐80003 (n = 10) | 50% (5)* | 20% (2)* | 20% (2)* | 40% (4) |
Note. Lesion features of ApoE−/− control‐ and K‐80003‐treated mice 8 weeks after surgery, including the percentage of mice with a vulnerable phenotype, intraplaque haemorrhage, rupture with thrombus, and multilayer plaque with discontinuity.
P < .05, significantly different from control group.
We next characterized the histological features of the carotid artery lesions. As shown in Figure 1c, mice treated with K‐80003 showed larger α‐smooth muscle actin‐ and collagen‐positive areas and smaller CD68‐ and Oil Red O‐positive areas, compared with control mice (Figure 1c).
We further examined the effects of K‐80003 administration on serum lipid profiles. No significant differences were observed between control‐ and K‐80003‐treated mice for serum triglyceride and cholesterol levels (Figure S3a,b). Further, the levels of creatinine and alanine aminotransferase, and the body weights of the mice from both groups were similar after 8 weeks of treatment (Figure S3c–e), indicating the low toxicity of K‐80003. Together, these results suggest that K‐80003 protects ApoE−/− mice from plaque progression and destabilization without obvious toxicity and that the protective effects were most likely independent of serum lipid levels.
3.2. K‐80003 suppresses inflammation in ApoE−/− mice
Mounting evidence points to a role of inflammation in plaque progression and vulnerability (Kataoka, Puri, & Nicholls, 2015). We analysed the expression of inflammatory cytokines in carotid plaques in the control‐ and K‐80003‐treated mice. Immunostaining analysis showed that the expressions of MMP‐8, MMP‐13, IL‐6, TNF‐α, and CCL2 were significantly decreased in the K‐80003 group (Figure 2a). A similar tendency was observed for plasma levels of the inflammatory cytokines TNF‐α and CCL2 (Figure 2b). Further analysis showed that the administration of K‐80003 did not affect COX‐2 activity, as shown by the similar serum TXB2 and 6‐keto‐PGFα levels in both groups (Figure 2c). Thus, the administration of K‐80003 protected against inflammation in vivo, and this effect was probably independent of COX‐2 activity.
Figure 2.
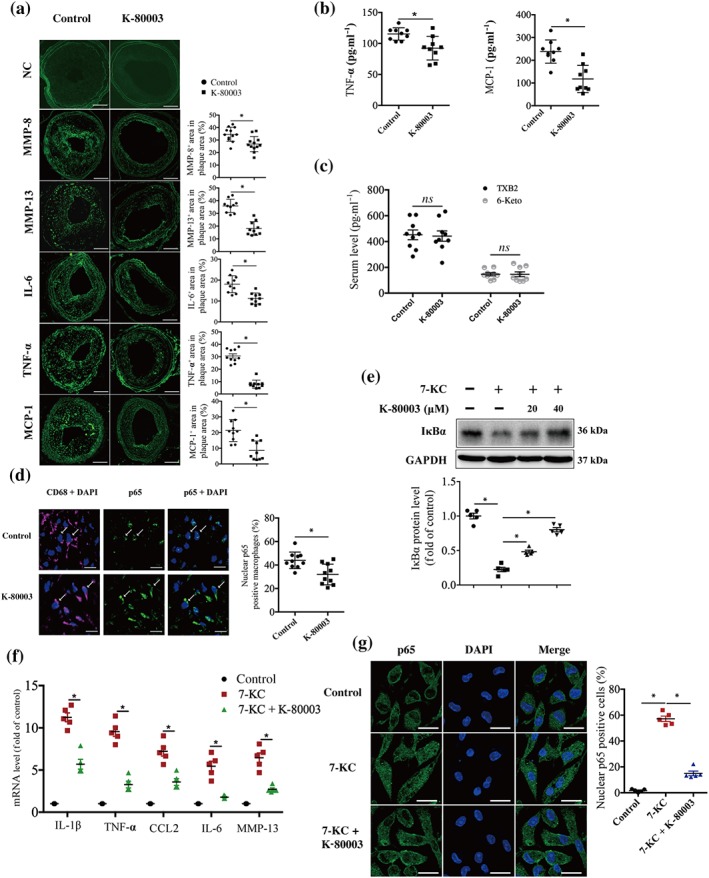
K‐80003 suppresses the inflammatory response in vivo and in vitro. (a) Representative images of immunofluorescence staining of MMP8, MMP13, IL‐6, TNF‐α, and CCL2 and quantification of their positive areas in plaques from ApoE−/− control‐ and K‐80003‐treated mice 8 weeks after surgery. Negative control (NC): Primary antibodies were omitted, and no specific immunostaining is visible after incubation with the secondary antibodies coupled with Alexa 488 (top two images). Images for magnifications are 200×. Scale bar: 100 μm. Values are mean ± SEM from 10 animals in each group. (b) TNF‐α and CCL2 protein levels were determined in plasma from ApoE−/− control and K‐80003 mice 8 weeks after surgery by elisa. Values are mean ± SEM from nine animals in each group. (c) TXB2 and 6‐keto‐PGFα protein levels were determined in plasma from ApoE−/− control and K‐80003 mice 8 weeks after surgery by elisa. Values are mean ± SEM from nine animals in each group. (d) Dual immunofluorescence staining of p65 (green) and CD68 (magenta) and quantification of the percentage of nuclear p65‐positive macrophages (CD68+) in lesions from ApoE−/− control and K‐80003 mice 8 weeks after surgery. Scale bar: 20 μm. Values are mean ± SEM from 10 animals in each group. (e–g) Bone marrow‐derived macrophages were treated with 7‐ketocholesterol (7‐KC; 40 μM) alone or together with K‐80003 (20 μM) for 16 hr. Immunoblotting was applied to examine IκBα expression (n = 5; e). The mRNA expression of the inflammatory markers (mean ± SEM, n = 5) was measured by quantitative real‐time PCR (f). p65 subcellular localization was analysed by confocal microscopy (n = 5). Scale bar: 20 μm. More than 200 cells were counted (g). *P < .05, significantly different as indicated
3.3. K‐80003 inhibits NF‐κB transcriptional activity in vivo and in vitro
Because NF‐κB is activated at sites of inflammation in atherosclerosis and can enhance intraplaque inflammatory response (Hansson et al., 2015; Kataoka et al., 2015; Tak & Firestein, 2001), we examined whether K‐80003 inhibited NF‐κB activation. Immunofluorescence staining showed that the percentage of macrophages positive for nuclear p65 was significantly decreased in mice treated with K‐80003 as compared with that in the control mice (Figure 2d). 7‐KC, a major component of oxidized lipoproteins in human atherosclerotic plaques (He et al., 2013; Sudo, Sato, Azechi, & Wachi, 2015), has been showed to activate numerous inflammatory pathways. Among those pathways, NF‐κB‐mediated cytokines production is the major pathway responding to 7‐KC‐induced inflammation. Therefore, we examined the impact of K‐80003 on 7‐KC‐induced NF‐κB activation in vitro. Our results showed that 20 μM K‐80003 treatment could effectively inhibit 7‐KC‐induced IκBα degradation, p65 nuclear translocation, and the transcription of NF‐κB target genes, including IL‐1β, TNF‐α, MCP‐1, IL‐6, and MMP13 in BMDMs (Figure 2e–g). Similar results were observed in RAW264.7 cells (Figure S4a–c). Pharmacokinetic studies showed that the half‐life of K‐80003 is about 2–3 hr. At 30 mg·kg−1, maximum K‐80003 concentration in mouse plasma is approximately 70 μM (data not shown), which is in its concentration range that inhibits inflammation. Together, these data indicate that K‐80003 can effectively suppress the NF‐κB proinflammatory pathway in vivo and in vitro.
3.4. K‐80003 inhibits 7‐KC‐induced NF‐κB activation by reducing p62/SQSTM1 protein expression
There is evidence that p62/SQSTM1 acts as a crucial molecule in activating NF‐κB (Duran et al., 2008). We determined whether p62/SQSTM1 is necessary for 7‐KC to activate NF‐κB in macrophages by the knockdown of p62/SQSTM1. Our results showed that as compared with control siRNA, RAW267.4 cells transfected with p62/SQSTM1 siRNA showed decreased p62/SQSTM1 protein expression (Figure 3a). Concomitantly, the 7‐KC‐induced IκBα degradation (Figure 3a) and the transcription of NF‐κB target genes were markedly attenuated in the p62/SQSTM1 knockdown cells (Figures 3b and S5a). These data suggest that p62/SQSTM1 is required for 7‐KC‐mediated activation of the NF‐κB proinflammatory pathway.
Figure 3.
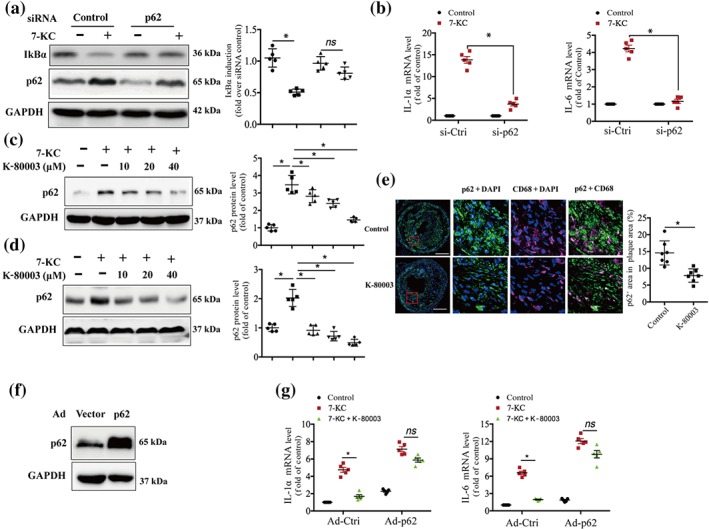
K‐80003 inhibits 7‐ketocholesterol (7‐KC) activation of NF‐κB by decreasing p62/sequestosome 1 (SQSTM1) expression. (a) RAW264.7 cells transfected with p62/SQSTM1 siRNA or scrambled control siRNA were treated with 7‐KC (40 μM) for 16 hr. Lysates were prepared and analysed by immunoblotting (n = 5). (b) siRNA of control or p62/SQSTM1 was transfected into RAW264.7 cells for 36 hr and then treated with 7‐KC (40 μM) for 16 hr. The mRNA expression of the inflammatory markers was measured by quantitative real‐time PCR (mean ± SEM, n = 5). Bone marrow‐derived macrophages (c) or RAW264.7 cells (d) were treated with 7‐KC (40 μM) in the absence or presence of the indicated concentrations of K‐80003 for 16 hr. Lysates were prepared and analysed by immunoblotting using appropriate antibodies (n = 5). (e) Dual immunofluorescence staining of p62/SQSTM1 (green) and CD68 ( magenta) and quantification of relative fluorescence intensity in lesions from ApoE−/− control and K‐80003 mice 8 weeks after surgery. Images for magnifications are 200×. Scale bar: 100 μm. Values are mean ± SEM from seven animals in each group. (f, g) RAW264.7 cells were infected with control (Ad‐vector) or p62/SQSTM1 (Ad‐p62/SQSTM1) adenovirus and then treated with 7‐KC (40 μM) alone or combined with K‐80003 (20 μM) for 16 hr. The mRNA expressions of IL‐1α and IL‐6 were measured by quantitative real‐time PCR (mean ± SEM, n = 5). *P < .05, significantly different as indicated; ns, P > .05
To determine whether K‐80003 inhibits 7‐KC activation of NF‐κB by decreasing p62/SQSTM1 expression, we first examined the effect of K‐80003 on p62/SQSTM1 protein expression. Our results showed that K‐80003 strongly decreased 7‐KC‐induced p62/SQSTM1 expression in a dose‐dependent manner in BMDMs (Figure 3c) and RAW264.7 cells (Figure 3d). Moreover, immunostaining data showed that p62/SQSTM1 expression in intraplaque macrophages (CD68+ cells) was significantly decreased in the mice treated with K‐80003 as compared with that in control mice (Figure 3e). We next determined whether the overexpression of p62/SQSTM1 could reverse the inhibitory effect of K‐80003 on 7‐KC‐induced NF‐κB activation. RAW264.7 cells were infected with Ad‐p62/SQSTM1 or Ad control. The expression of p62/SQSTM1 was examined by Western blotting (Figure 3f), and the NF‐κB activation was determined by analysing the transcription of its target genes. Our data showed that K‐80003 treatment markedly reduced the transcription of NF‐κB target genes IL‐1α and IL‐6 in the cells infected with control Ad (Figure 3g). The inhibitory effects of K‐80003 on 7‐KC‐induced IL‐1α and IL‐6 mRNA expression were decreased in the cells overexpressing p62/SQSTM1 (Figure 3g). Similar results were observed in the RAW264.7 cells stably expressing Tet‐on HA‐p62/SQSTM1, in which the expression of HA‐p62/SQSTM1 was turned on by the addition of doxycycline. As shown in Figure S5b,c, the inhibitory effects of K‐80003 on 7‐KC‐induced IL‐1α and IL‐6 mRNA expression were reversed in the cells treated with doxycycline. These data suggest that K‐80003 inhibits 7‐KC activation of the NF‐κB signalling pathway by decreasing p62/SQSTM1 expression.
3.5. K‐80003 reduces p62/SQSTM1 expression through autophagy
Apart from activating the transcription factor NF‐κB, p62/SQSTM1 shuttles cytoplasmic protein aggregates into autophagosomes for degradation including p62/SQSTM1 itself. In other words, p62/SQSTM1 levels are negatively regulated by autophagy (Figure S6a; Moscat & Diaz‐Meco, 2009; Moscat & Diaz‐Meco, 2012; Zotti et al., 2014). Therefore, we assessed whether K‐80003 decreased p62/SQSTM1 expression through autophagy, by the introduction of autophagy inhibitors. Our data showed that administration of autophagy inhibitors chloroquine or bafilomycin A1 (BafA1) effectively reversed the K‐80003‐induced reduction of p62/SQSTM1 protein expression in BMDMs (Figure 4a) and RAW264.7 cells (Figure 4b), suggesting that K‐80003 reduces p62/SQSTM1 protein expression through autophagy.
Figure 4.
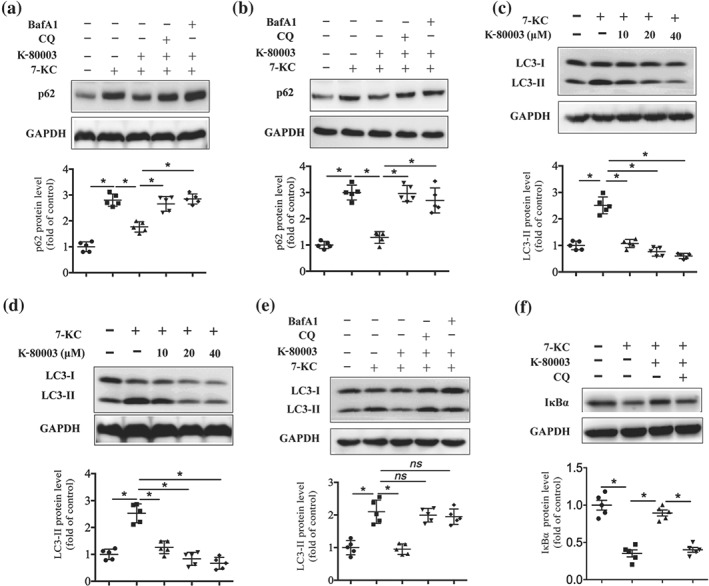
K‐80003 decreases p62/sequestosome 1 expression through promoting autophagic flux. Bone marrow‐derived macrophages (BMDMs; a) or RAW264.7 cells (b) were treated with 7‐keto‐cholesterol (7‐KC; 40 μM) together with or without K‐80003 (20 μM) for 16 hr, and bafilomycin A1 (BafA1; 50 nM) or chloroquine (CQ; 15 μg·ml−1) was added to cultures for the final 4 hr. Lysates were prepared and examined by immunoblotting (n = 5). BMDMs (c) or RAW264.7 cells (d) were treated with 7‐KC (40 μM) in the absence or presence of the indicated concentrations of K‐80003 for 16 hr. Lysates were prepared and analysed by immunoblotting using appropriate antibodies (n = 5). (e) BMDMs were treated with 7‐KC (40 μM) with or without K‐80003 (20 μM) for 16 hr, and BafA1 (50 nM) or chloroquine (15 μg·ml−1) was added to cultures for the final 4 hr. Lysates were prepared and examined by immunoblotting (n = 5). (f) BMDMs were pretreated with 7‐KC (40 μM) alone or together with K‐80003 (20 μM) for 12 hr and then co‐treated with chloroquine (15 μg·ml−1) for another 4 hr. Immunoblotting was applied to examine IκBα expression (n = 5). * P < .05, significantly different as indicated; ns, P > .05
We further analysed the effect of K‐80003 on autophagy. Autophagy is a dynamic process mainly comprising three steps: formation of autophagosomes, the fusion of autophagosomes with lysosomes, and degradation (Loos, du Toit, & Hofmeyr, 2014; Figure S6a). As the initiation of autophagosome formation results in the conversion of LC3‐I to LC3‐II, the expression of LC3‐II is widely used as a marker of autophagosome formation (Mizushima & Yoshimori, 2007; Zhang, Chen, Huang, & Le, 2013). To investigate whether K‐80003 could regulate autophagy, we treated BMDMs and RAW264.7 cells with K‐80003 and examined its effect on the expression of LC3‐II. Western blotting data showed no obvious differences in the LC3‐II protein levels after K‐80003 treatment (Figure S6b,c), indicating that K‐80003 does not induce the formation of autophagosome in macrophages. However, K‐80003 potently reduced 7‐KC‐induced LC3‐II expression in both BMDMs (Figure 4c) and RAW264.7 cells in a dose‐dependent manner (Figure 4d). As autophagy is a highly dynamic process and the final degradation of autophagosome by lysosome leads to the degradation of autophagosomal content and proteins, including LC3‐II, the reduction of LC3‐II could indicate either the inhibition of autophagosome formation or the promotion of lysosome‐mediated autophagosome degradation (Loos et al., 2014; Mizushima & Yoshimori, 2007; Zhang et al., 2013; Figure S6a). The term “autophagic flux” often refers to the autophagic degradation activity, and it can be illustrated by presenting a higher level of LC3‐II in the presence of chloroquine or BafA1 (Liao et al., 2012), both of which inhibit autophagic flux by preventing the fusion of autophagosome with lysosome and further lysosomal‐mediated protein degradation (Mizushima & Yoshimori, 2007; Shintani & Klionsky, 2004; Yamamoto et al., 1998; Figure S6a). Our data showed that K‐80003‐induced LC3‐II reduction was significantly reversed by the treatment of either chloroquine or BafA1in BMDMs (Figure 4e) and RAW264.7 cells (Figure S6d), suggesting that the reduction of LC3‐II upon K‐80003 treatment could be attributed to the promotion of autophagic flux rather than the inhibition of autophagosome formation. Moreover, the inhibitory effect of K‐80003 on 7‐KC‐induced IκBα degradation was also reversed by chloroquine in BMDMs (Figure 4f) and RAW264.7 cells (Figure S6e). In addition, 7‐KC‐induced the transcription of NF‐κB target genes, including, TNF‐α, IL‐1β, and Ccl2 were also reversed by chloroquine treatment (Figure S6f). Together, these data suggest that K‐80003 inhibits NF‐κB proinflammatory pathway and reduces p62/SQSTM1 expression through the promotion of autophagic flux.
3.6. RXRα is required for K‐80003‐mediated promotion of autophagic flux
We next studied whether the promotion of autophagic flux by K‐80003 was RXRα dependent. Our data showed that K‐80003 treatment reduced LC3‐II expression in the cells transfected with control siRNA (Figure 5a, left three lanes). However, when the cells were transfected with RXRα siRNA, K‐80003 could not reduce 7‐KC‐induced LC3‐II up‐regulation any more (Figure 5a, right three lanes). Interestingly, the 7‐KC‐induced expression of LC3‐II was also reduced in RXRα knockdown cells (Figure 5a). We next investigated if the reduction of LC3‐II in RXRα knockdown cells was due to the promotion of autophagic flux or the inhibition of autophagosome formation. As the promotion of autophagic flux reduces p62 expression, while inhibition of autophagosome formation results in the accumulation of p62 (Loos et al., 2014; Zhang et al., 2013), we examined the total cellular levels of p62/SQSTM1. Immunoblotting analysis showed that p62/SQSTM1 levels were significantly decreased in RAW264.7 cells transfected with RXRα siRNA as compared with those transfected with control siRNA under the stimulation of 7‐KC (Figure 5b), indicating that knockdown of RXRα promotes autophagic flux. Together, these results suggest that RXRα is a negative regulator of autophagic flux and that the expression of RXRα is necessary for K‐80003 to promote autophagic flux in macrophages.
Figure 5.
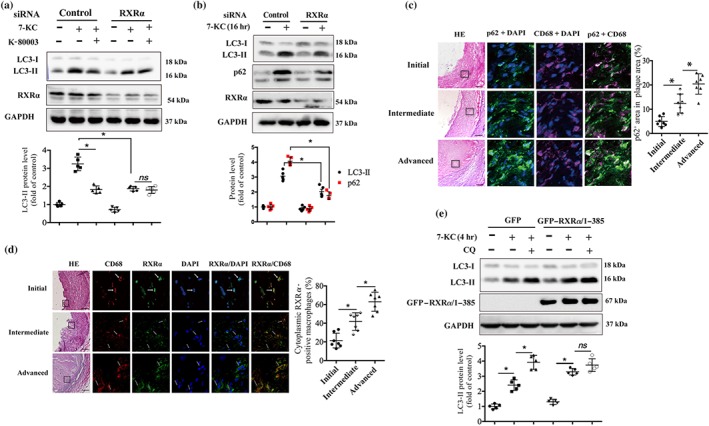
Retinoid X receptor‐α (RXRα) is required for K‐80003 to promote autophagic flux. (a) RAW264.7 cells transfected with siRNA of control or RXRα for 36 hr were treated with 7‐ketocholesterol (7‐KC; 40 μM) alone or together with K‐80003 (20 μM) for 16 hr. Lysates were prepared and analysed by immunoblotting (n = 5). (b) RAW264.7 cells transfected with RXRα siRNA or scrambled control siRNA for 36 hr were treated with 7‐KC (40 μM) for 16 hr. Lysates were prepared and analysed by immunoblotting (n = 5). Dual immunofluorescence staining of p62/sequestosome 1 (green) with CD68 (magenta; c) or RXRα (green) with CD68 (red; d) in initial, intermediate, and advanced human atherosclerotic lesions. Magnifications are 200×. Scale bar: 100 μm. Nuclei were visualized by DAPI staining (blue; n = 10). (e) Stable RAW264.7 cell lines expressing GFP or GFP–RXRα/385 were treated with 7‐KC (40 μM) in the presence or absence of chloroquine (CQ; 15 μg·ml−1) for 4 hr, and then cell lysates were prepared and analysed by immunoblotting. GAPDH was used as loading control (n = 5). *P < .05, significantly different as indicated; ns, P > .05. HE, haematoxylin and eosin
There is a decreased autophagic flux in macrophages from advanced plaques in mice, as shown by the accumulation of P62/SQSTM (Liao et al., 2012; Razani et al., 2012). Here, we have analysed the autophagic flux of human atherosclerotic lesions. Our data showed an increased p62/SQSTM1 expression in the macrophages of human advanced atherosclerotic lesions, compared with those from initial plaques (Figure 5c), indicating a decreased autophagic flux in human advanced plaque macrophages. Meanwhile, the immunofluorescence data showed that RXRα localized to the nucleus in macrophages from initial lesions, whereas these receptors translocated to the cytoplasm in macrophages from intermediate and advanced lesions (Figure 5d). Similar results were also observed in macrophages from mouse plaques. Dual immunofluorescence staining showed a progressive increase in p62/SQSTM1 expression and cytoplasmic RXRα in the intimal macrophages along with the progression of carotid vulnerable plaques at 2 and 8 weeks postoperatively (Figure S7a,b). In vitro data also showed that 7‐KC treatment induced the cytoplasmic translocation of RXRα in a dose‐dependent manner in RAW264.7 cells (Figure S7c). These results suggest that cytoplasmic translocation of RXRα may be associated with decreased autophagic flux in macrophages.
To study the relationship between the increased cytoplasmic RXRα and the decreased autophagic flux in macrophages, we constructed two stable cell lines expressing GFP or GFP–RXRα/385, a RXRα C‐terminal truncated mutant predominantly localized in the cytoplasm (Cao et al., 2004), in RAW264.7 cells, and examined the autophagic flux in the resulting stable cell lines. After stimulation with 7‐KC, both cell lines showed increased LC3‐II expression levels (Figure 5f). Moreover, GFP–RXRα/385‐expressing cells showed higher LC3‐II levels in comparison with GFP‐expressing cells after 7‐KC stimulation (Figure 5f). The higher expression of LC3‐II in GFP–RXRα/385 cell could result from increased autophagosome formation or impaired autophagosome degradation and these possibilities were assessed by using chloroquine. As showed in Figure 5f, LC3‐II levels increased to ~1.5‐fold of 7‐KC‐treated cells in the presence of chloroquine in the GFP‐expressing cells, while no significant difference was observed in the cells expressing GFP–RXRα/385, indicating that the increased LC3‐II in GFP–RXRα/385‐expressing cell is due to the inhibition of autophagic flux. Thus, these data suggest that impaired autophagic flux in advanced plaque macrophages may be attributed to the cytoplasmic localization of RXRα.
We further hypothesized that K‐80003 might promote autophagic flux by inhibiting the cytoplasmic translocation of RXRα. As expected, the treatment of RAW264.7 cells with K‐80003 significantly inhibited the 7‐KC‐induced cytoplasmic translocation of RXRα (Figure S7d). Together, these results suggest that increased cytoplasmic RXRα expression is involved in the impairment of autophagic flux of advanced plaque macrophages and that the effects of K‐80003 in promoting autophagic flux may occur through the inhibition of RXRα cytoplasmic translocation (Figure 6).
Figure 6.
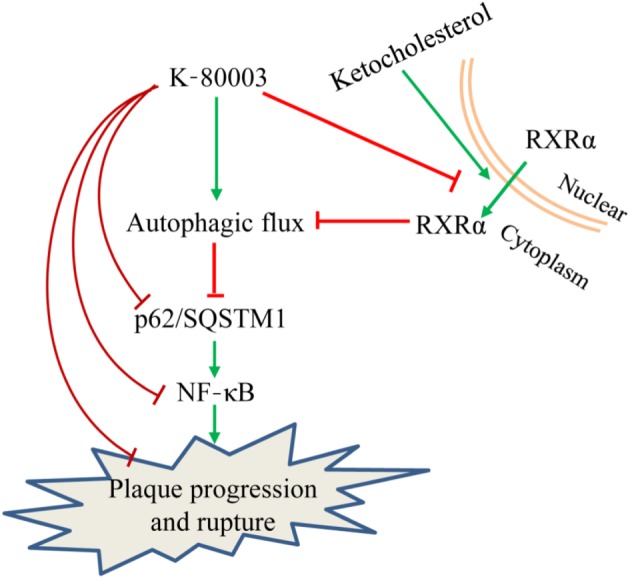
Diagram of the proposed underlying mechanism of anti‐atherosclerotic effect of K‐80003. Retinoid X receptor‐α (RXRα) inhibits macrophage autophagic flux by translocating from the nucleus to the cytoplasm under the stimulation of 7‐keto‐cholesterol in advanced plaque macrophages. Such an extranuclear effect of RXRα was inhibited by K‐80003, leading to an enhanced autophagic flux with reduced p62 expression and consequently inhibition of the p62‐mediated NF‐κB proinflammatory pathway and plaque progression and rupture. SQSTM1, sequestosome 1
4. DISCUSSION
Although RXRα has been recognized as an intriguing drug target for many years, the side effects associated with regulating its transcriptional activity have hampered the development of RXRα‐based drugs (Zhang et al., 2015). Here, we reported that the sulindac‐derived RXRα modulator K‐80003 significantly mitigated the progression and destabilization of carotid atherosclerotic plaques in ApoE−/− mice without any apparent toxicity to animals. Mechanistically, administration of K‐80003 potently suppressed the intraplaque inflammatory response through promoting autophagy‐mediated p62/SQSTM1 degradation and consequently inhibition of the p62/SQSTM1‐mediated NF‐κB proinflammatory pathway.
A significant finding presented here is that targeting alternative binding sites on RXRα to regulate its activities may represent a new strategy to develop RXRα‐based atheroprotective drugs. RXRα is an intriguing and unusual target for therapeutic applications in atherosclerosis. This is supported by the fact that RXRα ligands can effectively decrease atherosclerosis‐related risk factors such as inflammation, apoptosis, and plasma lipid concentrations and that administration of the rexinoids LG100363 and LGD1069 could significantly inhibit atherosclerosis progression in the mouse model (Claudel et al., 2001; Lalloyer et al., 2006). However, the side effects caused by targeting its cognate ligand‐binding pocket and activating its transcriptional activity hampered the further pharmacological development of these rexinoids. K‐80003, a newly synthesized low MW compound derived from sulindac, can bind to the RXRα ligand‐binding domain at a site different from the canonical ligand‐binding pocket and regulate its non‐transcriptional actions (Chen et al., 2014; Zhou et al., 2010). By using our newly constructed murine model of spontaneous plaque rupture (Jin et al., 2012), we found that the long‐term administration of K‐80003 significantly suppressed the progression of carotid artery plaques. More importantly, the percentage of vulnerable plaques and plaque rupture with thrombus was significantly reduced in the K‐80003‐treated mice. In addition, the serum triglyceride, cholesterol, and COX‐2 activity levels were similar in the control‐ and K‐80003‐treated mice. Furthermore, we observed that the administration of K‐80003 at a dose that effectively inhibited atherosclerotic plaque progression and destabilization did not show any apparent toxicity to animals. Thus, our new compound could be clinically useful.
An additional important finding is the cytoplasmic action of RXRα in attenuating autophagic flux. Previous studies showed that RXRα could act in the cytoplasm to induce apoptosis (Cao et al., 2004) and differentiation (Katagiri et al., 2000). In response to some apoptotic stimuli, RXRα translocates from the nucleus to the cytoplasm where it targets mitochondria, leading to cytochrome c release and apoptosis (Cao et al., 2004). Our current findings demonstrated that cytoplasmic RXRα could also decrease autophagic flux. In support of this notion, we showed that the cytoplasmically localized RXRα mutant GFP–RXRα/385 could decrease autophagic flux. These data indicated that RXRα‐mediated autophagic flux dysfunction was independent of its transcriptional function. It has been known that activated Bax could migrate to the lysosomal membrane to induce lysosomal membrane permeabilization in Parkinson's disease (Bove et al., 2014). Cytoplasmic RXRα has the potential to activate Bax (Cao et al., 2004; Liu et al., 2008). Additionally, we found that the overexpression of GFP–RXRα/385 significantly reduced LysoTracker Red intensity (decrease in LysoTracker intensity indicates lysosomal dysfunction) as compared with GFP controls (data not shown). Whether the impairment of autophagic flux by cytoplasmic RXRα occurs via the activation of Bax and the subsequent alteration of lysosomal membrane permeabilization warrants further investigation.
In the present study, we have demonstrated that cytoplasmic RXRα mutant RXRα/385 could impair autophagic flux in vitro. Several pieces of evidence reported here indicate that cytoplasmic RXRα participated in decreased autophagic flux in the pathological condition. First, we found that RXRα translocated from the nucleus to the cytoplasm in macrophages from advanced but not initial plaques in humans, as well as mice. In addition, we found that an oxysterol such as 7‐KC, which is a major component of oxidized lipoprotein in human atherosclerotic plaques, could induce cytoplasmic migration of RXRα in a dose‐dependent manner. Furthermore, a reduction in RXRα expression by siRNA significantly reversed the 7‐KC‐induced impairment of autophagic flux. Thus, targeting RXRα for restoring autophagic flux may hold promise for attenuating the development and destabilization of atherosclerotic plaques.
There is mounting evidence that autophagy has crucial effects on the regulation of inflammatory response. The best studied aspect of the interaction between autophagy and inflammation is probably represented by the regulation of NF‐κB signalling pathway by autophagy (Trocoli & Djavaheri‐Mergny, 2011). In the presence of heat shock protein 90 inhibitor, geldanamycin, the selective degradation of NF‐κB activators IKKα, IKKβ, and IKKγ is mediated through an autophagy pathway (Qing, Yan, & Xiao, 2006; Yan et al., 2007). Autophagy can also suppress NF‐κB signalling pathway through degradation of IKKβ in a Kelch‐like ECH‐associated protein 1‐ or E3 ubiquitin ligase Ro52‐dependent manner (Kim et al., 2010; Niida, Tanaka, & Kamitani, 2010). Aside from the direct degradation of NF‐κB signalling components like IKKs, autophagy can also inhibit NF‐κB activity through p62/SQSTM1. In addition of these catabolic roles, p62/SQSTM1 also activates NF‐κB in respond to several stimuli such as TNF‐α, IL‐1α, and nerve growth factor (Sanz, Diaz‐Meco, Nakano, & Moscat, 2000; Wooten et al., 2005; Zotti et al., 2014). In the present study, we showed that p62/SQSTM1 is necessary for 7‐KC‐induced activation of NF‐κB proinflammatory pathway and further identified a sulindac‐derived RXRα modulator K‐80003, which significantly suppressed 7‐KC activation of NF‐κB through autophagy‐mediated reduction of p62/SQSTM1, thus providing a new strategy for combating intraplaque inflammation.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
B.H., X.Z., L.S., Z.S., and P.N. conceived and designed the experiments. L.S., Z.S., P.N., R.Y., Z.C., C.W., L.H., S.J., and H.Z. performed the experiments. B.H., X.Z., L.S., Z.S., and P.N. analysed the data. L.S., Z.S., and P.N. contributed the reagents/materials/analysis tools. B.H., X.Z., L.S., and Z.S. wrote the paper.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Table S1. Summary of the quantitative RT‐PCR primer sets used
Figures S1. Effect of K80003 on lesion features in ApoE−/− mice
Figure S2. The effect of vehicle on plaque progression
Figure S3. K‐80003 does not affect serum lipid profiles
Figure S4. Administration of K80003 suppresses inflammation
Figure S5. K‐80003 inhibits 7‐KC activation of NF‐κB by decreasing p62/SQSTM1 expression
Figure S6. K‐80003 promotes autophagic flux in macrophages
Figure S7. K‐80003 protects against 7‐KC‐induced RXRα cytoplasmic translocation
ACKNOWLEDGEMENTS
This work was supported by Grants 91539106, 81500266 81770428, 81830010, 81330006, 81370399, 81600337, 91129302, 91429306, and U1405229 from the National Natural Science Foundation of China; Grant 2017YFC 0909301 from the National Key R&D Program; Grant 15ZR1425800 from the Natural Science Foundation of Shanghai; and Grant 20154Y0036 from the Shanghai Municipal Commission of Health and Family Planning. We thank the members of the Department of Laboratory Animal Science, Shanghai Jiaotong University School of Medicine, for their technical support.
Shen L, Sun Z, Nie P, et al. Sulindac‐derived retinoid X receptor‐α modulator attenuates atherosclerotic plaque progression and destabilization in ApoE−/− mice. Br J Pharmacol. 2019;176:2559–2572. 10.1111/bph.14682
Drs Shen, Sun, and Nie contributed equally to this work.
Contributor Information
Xiaokun Zhang, Email: xzhang@sbpdiscovery.org.
Ben He, Email: drheben@126.com.
REFERENCES
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. British Journal of Pharmacology, 174, S208–S224. 10.1111/bph.13880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. British Journal of Pharmacology, 174, S1–S16. 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altucci, L. , Leibowitz, M. D. , Ogilvie, K. M. , de Lera, A. R. , & Gronemeyer, H. (2007). RAR and RXR modulation in cancer and metabolic disease. Nature Reviews. Drug Discovery, 6, 793–810. 10.1038/nrd2397 [DOI] [PubMed] [Google Scholar]
- Bove, J. , Martinez‐Vicente, M. , Dehay, B. , Perier, C. , Recasens, A. , Bombrun, A. , … Vila, M. (2014). BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy, 10, 889–900. 10.4161/auto.28286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, X. , Liu, W. , Lin, F. , Li, H. , Kolluri, S. K. , Lin, B. , … Zhang, X. K. (2004). Retinoid X receptor regulates Nur77/TR3‐dependent apoptosis [corrected] by modulating its nuclear export and mitochondrial targeting. Molecular and Cellular Biology, 24, 9705–9725. 10.1128/MCB.24.22.9705-9725.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas, F. , Daury, L. , Grandemange, S. , Busson, M. , Seyer, P. , Hatier, R. , … Wrutniak‐Cabello, C. (2003). Endocrine regulation of mitochondrial activity: Involvement of truncated RXRα and c‐Erb Aα1 proteins. The FASEB Journal, 17, 426–436. 10.1096/fj.02-0732com [DOI] [PubMed] [Google Scholar]
- Charo, I. F. , & Taub, R. (2011). Anti‐inflammatory therapeutics for the treatment of atherosclerosis. Nature Reviews. Drug Discovery, 10, 365–376. 10.1038/nrd3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Wang, Z. G. , Aleshin, A. E. , Chen, F. , Chen, J. , Jiang, F. , … Su, Y. (2014). Sulindac‐derived RXRα modulators inhibit cancer cell growth by binding to a novel site. Chemistry & Biology, 21, 596–607. 10.1016/j.chembiol.2014.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudel, T. , Leibowitz, M. D. , Fievet, C. , Tailleux, A. , Wagner, B. , Repa, J. J. , … Auwerx, J. (2001). Reduction of atherosclerosis in apolipoprotein E knockout mice by activation of the retinoid X receptor. Proceedings of the National Academy of Sciences of the United States of America, 98, 2610–2615. 10.1073/pnas.041609298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, M. I. , & Xia, Z. (2012). The retinoid X receptors and their ligands. Biochimica et Biophysica Acta, 1821, 21–56. 10.1016/j.bbalip.2011.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desreumaux, P. , Dubuquoy, L. , Nutten, S. , Peuchmaur, M. , Englaro, W. , Schoonjans, K. , … Auwerx, J. (2001). Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator‐activated receptor γ (PPARγ) heterodimer: A basis for new therapeutic strategies. The Journal of Experimental Medicine, 193, 827–838. 10.1084/jem.193.7.827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvergne, B. (2007). RXR: From partnership to leadership in metabolic regulations. Vitamins and Hormones, 75, 1–32. 10.1016/S0083-6729(06)75001-4 [DOI] [PubMed] [Google Scholar]
- Duran, A. , Linares, J. F. , Galvez, A. S. , Wikenheiser, K. , Flores, J. M. , Diaz‐Meco, M. T. , & Moscat, J. (2008). The signaling adaptor p62 is an important NF‐κB mediator in tumorigenesis. Cancer Cell, 13, 343–354. 10.1016/j.ccr.2008.02.001 [DOI] [PubMed] [Google Scholar]
- Everett, B. M. , Pradhan, A. D. , Solomon, D. H. , Paynter, N. , Macfadyen, J. , Zaharris, E. , … Ridker, P. M. (2013). Rationale and design of the Cardiovascular Inflammation Reduction Trial: A test of the inflammatory hypothesis of atherothrombosis. American Heart Journal, 166, 199–207 e115. 10.1016/j.ahj.2013.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald, G. A. (2004). Coxibs and cardiovascular disease. The New England Journal of Medicine, 351, 1709–1711. 10.1056/NEJMp048288 [DOI] [PubMed] [Google Scholar]
- Fredman, G. , & Tabas, I. (2017). Boosting inflammation resolution in atherosclerosis: The next frontier for therapy. The American Journal of Pathology, 187, 1211–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose, R. , Zimmerman, T. L. , Thevananther, S. , & Karpen, S. J. (2004). Endotoxin leads to rapid subcellular re‐localization of hepatic RXRα: A novel mechanism for reduced hepatic gene expression in inflammation. Nuclear Receptor, 2, 4 10.1186/1478-1336-2-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson, G. K. , Libby, P. , & Tabas, I. (2015). Inflammation and plaque vulnerability. Journal of Internal Medicine, 278, 483–493. 10.1111/joim.12406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, C. , Zhu, H. , Zhang, W. , Okon, I. , Wang, Q. , Li, H. , … Xie, Z. (2013). 7‐Ketocholesterol induces autophagy in vascular smooth muscle cells through Nox4 and Atg4B. The American Journal of Pathology, 183, 626–637. 10.1016/j.ajpath.2013.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, S. X. , Shen, L. H. , Nie, P. , Yuan, W. , Hu, L. H. , Li, D. D. , … He, B. (2012). Endogenous renovascular hypertension combined with low shear stress induces plaque rupture in apolipoprotein E‐deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 32, 2372–2379. 10.1161/ATVBAHA.111.236158 [DOI] [PubMed] [Google Scholar]
- Katagiri, Y. , Takeda, K. , Yu, Z. X. , Ferrans, V. J. , Ozato, K. , & Guroff, G. (2000). Modulation of retinoid signalling through NGF‐induced nuclear export of NGFI‐B. Nature Cell Biology, 2, 435–440. 10.1038/35017072 [DOI] [PubMed] [Google Scholar]
- Kataoka, Y. , Puri, R. , & Nicholls, S. J. (2015). Inflammation, plaque progression and vulnerability: Evidence from intravascular ultrasound imaging. Cardiovasc Diagn Ther, 5, 280–289. 10.3978/j.issn.2223-3652.2015.05.06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. E. , You, D. J. , Lee, C. , Ahn, C. , Seong, J. Y. , & Hwang, J. I. (2010). Suppression of NF‐κB signaling by KEAP1 regulation of IKKβ activity through autophagic degradation and inhibition of phosphorylation. Cellular Signalling, 22, 1645–1654. 10.1016/j.cellsig.2010.06.004 [DOI] [PubMed] [Google Scholar]
- Lalloyer, F. , Fievet, C. , Lestavel, S. , Torpier, G. , van der Veen, J. , Touche, V. , … Tailleux, A. (2006). The RXR agonist bexarotene improves cholesterol homeostasis and inhibits atherosclerosis progression in a mouse model of mixed dyslipidemia. Arteriosclerosis, Thrombosis, and Vascular Biology, 26, 2731–2737. 10.1161/01.ATV.0000248101.93488.84 [DOI] [PubMed] [Google Scholar]
- Liao, X. , Sluimer, J. C. , Wang, Y. , Subramanian, M. , Brown, K. , Pattison, J. S. , … Tabas, I. (2012). Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metabolism, 15, 545–553. 10.1016/j.cmet.2012.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby, P. , Tabas, I. , Fredman, G. , & Fisher, E. A. (2014). Inflammation and its resolution as determinants of acute coronary syndromes. Circulation Research, 114, 1867–1879. 10.1161/CIRCRESAHA.114.302699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Zhou, W. , Li, S. S. , Sun, Z. , Lin, B. , Lang, Y. Y. , … Zeng, J. Z. (2008). Modulation of orphan nuclear receptor Nur77‐mediated apoptotic pathway by acetylshikonin and analogues. Cancer Research, 68, 8871–8880. 10.1158/0008-5472.CAN-08-1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos, B. , du Toit, A. , & Hofmeyr, J. H. (2014). Defining and measuring autophagosome flux—Concept and reality. Autophagy, 10, 2087–2096. 10.4161/15548627.2014.973338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. British Journal of Pharmacology, 172, 3189–3319. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N. , & Yoshimori, T. (2007). How to interpret LC3 immunoblotting. Autophagy, 3, 542–545. 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- Moscat, J. , & Diaz‐Meco, M. T. (2009). p62 at the crossroads of autophagy, apoptosis, and cancer. Cell, 137, 1001–1004. 10.1016/j.cell.2009.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat, J. , & Diaz‐Meco, M. T. (2012). p62: A versatile multitasker takes on cancer. Trends in Biochemical Sciences, 37, 230–236. 10.1016/j.tibs.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie, P. , Li, D. , Hu, L. , Jin, S. , Yu, Y. , Cai, Z. , … He, B. (2014). Atorvastatin improves plaque stability in ApoE‐knockout mice by regulating chemokines and chemokine receptors. PLoS ONE, 9, e97009 10.1371/journal.pone.0097009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida, M. , Tanaka, M. , & Kamitani, T. (2010). Downregulation of active IKKβ by Ro52‐mediated autophagy. Molecular Immunology, 47, 2378–2387. 10.1016/j.molimm.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez, V. , Alameda, D. , Rico, D. , Mota, R. , Gonzalo, P. , Cedenilla, M. , … Ricote, M. (2010). Retinoid X receptor α controls innate inflammatory responses through the up‐regulation of chemokine expression. Proceedings of the National Academy of Sciences of the United States of America, 107, 10626–10631. 10.1073/pnas.0913545107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing, G. , Yan, P. , & Xiao, G. (2006). Hsp90 inhibition results in autophagy‐mediated proteasome‐independent degradation of IκB kinase (IKK). Cell Research, 16, 895–901. 10.1038/sj.cr.7310109 [DOI] [PubMed] [Google Scholar]
- Razani, B. , Feng, C. , Coleman, T. , Emanuel, R. , Wen, H. , Hwang, S. , … Semenkovich, C. F. (2012). Autophagy links inflammasomes to atherosclerotic progression. Cell Metabolism, 15, 534–544. 10.1016/j.cmet.2012.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker, P. M. , Everett, B. M. , Thuren, T. , MacFadyen, J. G. , Chang, W. H. , Ballantyne, C. , … CANTOS Trial Group . (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. The New England Journal of Medicine, 377, 1119–1131. 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- Ridker, P. M. , Thuren, T. , Zalewski, A. , & Libby, P. (2011). Interleukin‐1β inhibition and the prevention of recurrent cardiovascular events: Rationale and design of the Canakinumab Anti‐inflammatory Thrombosis Outcomes Study (CANTOS). American Heart Journal, 162, 597–605. 10.1016/j.ahj.2011.06.012 [DOI] [PubMed] [Google Scholar]
- Sanz, L. , Diaz‐Meco, M. T. , Nakano, H. , & Moscat, J. (2000). The atypical PKC‐interacting protein p62 channels NF‐κB activation by the IL‐1‐TRAF6 pathway. The EMBO Journal, 19, 1576–1586. 10.1093/emboj/19.7.1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani, T. , & Klionsky, D. J. (2004). Autophagy in health and disease: A double‐edged sword. Science, 306, 990–995. 10.1126/science.1099993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staels, B. (2001). Regulation of lipid and lipoprotein metabolism by retinoids. Journal of the American Academy of Dermatology, 45, S158–S167. 10.1067/mjd.2001.113718 [DOI] [PubMed] [Google Scholar]
- Streb, J. W. , & Miano, J. M. (2003). Retinoids: Pleiotropic agents of therapy for vascular diseases? Current Drug Targets. Cardiovascular & Haematological Disorders, 3, 31–57. 10.2174/1568006033337393 [DOI] [PubMed] [Google Scholar]
- Su, Y. , Zeng, Z. , Zhang, W. , Chen, Z. , Xu, D. , & Zhang, X. K. (2016). Recent progress in the design and discovery of RXR modulators targeting alternate binding sites of the receptor. Current Topics in Medicinal Chemistry, 17, 663–675. 10.2174/1568026616666160617092241 [DOI] [PubMed] [Google Scholar]
- Sudo, R. , Sato, F. , Azechi, T. , & Wachi, H. (2015). 7‐Ketocholesterol‐induced lysosomal dysfunction exacerbates vascular smooth muscle cell calcification via oxidative stress. Genes to Cells, 20, 982–991. 10.1111/gtc.12301 [DOI] [PubMed] [Google Scholar]
- Sun, Z. , Cao, X. , Jiang, M. M. , Qiu, Y. , Zhou, H. , Chen, L. , … Zhang, X. K. (2012). Inhibition of β‐catenin signaling by nongenomic action of orphan nuclear receptor Nur77. Oncogene, 31, 2653–2667. 10.1038/onc.2011.448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szanto, A. , Narkar, V. , Shen, Q. , Uray, I. P. , Davies, P. J. , & Nagy, L. (2004). Retinoid X receptors: X‐ploring their (patho)physiological functions. Cell Death and Differentiation, 11(Suppl 2), S126–S143. 10.1038/sj.cdd.4401533 [DOI] [PubMed] [Google Scholar]
- Tabas, I. (2010). Macrophage death and defective inflammation resolution in atherosclerosis. Nature Reviews. Immunology, 10, 36–46. 10.1038/nri2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak, P. P. , & Firestein, G. S. (2001). NF‐κB: A key role in inflammatory diseases. The Journal of Clinical Investigation, 107, 7–11. 10.1172/JCI11830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice, C. M. , & Zheng, Y. J. (2016). Non‐canonical modulators of nuclear receptors. Bioorganic & Medicinal Chemistry Letters, 26, 4157–4164. 10.1016/j.bmcl.2016.07.067 [DOI] [PubMed] [Google Scholar]
- Trocoli, A. , & Djavaheri‐Mergny, M. (2011). The complex interplay between autophagy and NF‐κB signaling pathways in cancer cells. American Journal of Cancer Research, 1, 629–649. [PMC free article] [PubMed] [Google Scholar]
- Wooten, M. W. , Geetha, T. , Seibenhener, M. L. , Babu, J. R. , Diaz‐Meco, M. T. , & Moscat, J. (2005). The p62 scaffold regulates nerve growth factor‐induced NF‐κB activation by influencing TRAF6 polyubiquitination. The Journal of Biological Chemistry, 280, 35625–35629. 10.1074/jbc.C500237200 [DOI] [PubMed] [Google Scholar]
- Yamamoto, A. , Tagawa, Y. , Yoshimori, T. , Moriyama, Y. , Masaki, R. , & Tashiro, Y. (1998). Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H‐4‐II‐E cells. Cell Structure and Function, 23, 33–42. 10.1247/csf.23.33 [DOI] [PubMed] [Google Scholar]
- Yan, P. , Qing, G. , Qu, Z. , Wu, C. C. , Rabson, A. , & Xiao, G. (2007). Targeting autophagic regulation of NFκB in HTLV‐I transformed cells by geldanamycin: Implications for therapeutic interventions. Autophagy, 3, 600–603. 10.4161/auto.4761 [DOI] [PubMed] [Google Scholar]
- Zhang, X. J. , Chen, S. , Huang, K. X. , & Le, W. D. (2013). Why should autophagic flux be assessed? Acta Pharmacologica Sinica, 34, 595–599. 10.1038/aps.2012.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. K. , Su, Y. , Chen, L. , Chen, F. , Liu, J. , & Zhou, H. (2015). Regulation of the nongenomic actions of retinoid X receptor‐α by targeting the coregulator‐binding sites. Acta Pharmacologica Sinica, 36, 102–112. 10.1038/aps.2014.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Liu, W. , Su, Y. , Wei, Z. , Liu, J. , Kolluri, S. K. , … Zhang, X. K. (2010). NSAID sulindac and its analog bind RXRα and inhibit RXRα‐dependent AKT signaling. Cancer Cell, 17, 560–573. 10.1016/j.ccr.2010.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman, T. L. , Thevananther, S. , Ghose, R. , Burns, A. R. , & Karpen, S. J. (2006). Nuclear export of retinoid X receptor α in response to interleukin‐1β‐mediated cell signaling: Roles for JNK and SER260. The Journal of Biological Chemistry, 281, 15434–15440. 10.1074/jbc.M508277200 [DOI] [PubMed] [Google Scholar]
- Zotti, T. , Scudiero, I. , Settembre, P. , Ferravante, A. , Mazzone, P. , D'Andrea, L. , … Stilo, R. (2014). TRAF6‐mediated ubiquitination of NEMO requires p62/sequestosome‐1. Molecular Immunology, 58, 27–31. 10.1016/j.molimm.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of the quantitative RT‐PCR primer sets used
Figures S1. Effect of K80003 on lesion features in ApoE−/− mice
Figure S2. The effect of vehicle on plaque progression
Figure S3. K‐80003 does not affect serum lipid profiles
Figure S4. Administration of K80003 suppresses inflammation
Figure S5. K‐80003 inhibits 7‐KC activation of NF‐κB by decreasing p62/SQSTM1 expression
Figure S6. K‐80003 promotes autophagic flux in macrophages
Figure S7. K‐80003 protects against 7‐KC‐induced RXRα cytoplasmic translocation