

Abstract
Neuropathic pain can arise from disease or damage to the nervous system. The most common symptoms of neuropathic pain include spontaneous pain, allodynia, and hyperalgesia. There is still limited knowledge about the factors that initiate and maintain neuropathic pain. However, ample evidence has proved the antinociceptive role of spinal α‐adrenoceptors following nerve injury. It is well‐documented that noradrenergic descending pathways from supraspinal loci exert an inhibitory influence on the spinal cord nociceptive neurons, mostly through the activation of spinal α2‐adrenoceptors. This, in turn, suppresses transmission of pain input and the hyperexcitability of spinal dorsal horn neurons. There is considerable evidence demonstrating that spinal application of α2‐adrenoceptor agonists leads to analgesic effects in animal models of neuropathic pain. Today, despite the recent rapid development of neuroscience and drug discovery, effective drugs with clear basic mechanisms have remained a mystery. Here, we give an overview of the cellular mechanisms through which brainstem adrenergic descending inhibitory processing can alter spinal pain transmission to the higher centres, and how these pathways change in neuropathic pain conditions focusing on the role of spinal α2‐adrenoceptors in the spinal dorsal horn. We then suggest that α2‐adrenoceptor agonist may be useful to treat neuropathic pain.
Linked Articles
This article is part of a themed section on Adrenoceptors—New Roles for Old Players. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.14/issuetoc
Abbreviations
- BDNF
brain‐derived neurotrophic factor
- I2
imidazoline‐2
- LC
locus coeruleus
- NA
noradrenaline
- PAG
periaqueductal grey
- RVM
rostral ventromedial medulla
- SNL
spinal nerve‐ligated
- TRPV1
transient receptor potential vanilloid type 1
- VPL
ventral posterolateral thalamus
1. INTRODUCTION
Many factors cause neuropathic pain states by producing lesions in the nervous system, these include metabolic disorders, viral infection, cancer, trauma, and nerve injuries (e.g., amputation and spinal cord injury; Baron, Binder, & Wasner, 2010; Zhuo, 2007). Neuropathic pain is a common and debilitating condition, which is characterized by spontaneous pain, allodynia (pain in response to normally innocuous stimuli), and hyperalgesia (exaggerated pain perception in response to noxious stimuli; Bahari, Manaheji, et al., 2015; Baron, 2006). Although the aetiology of neuropathic pain is not completely understood, the spinal adrenergic pathways have been recognized to be important in the process of pain development (Nakai et al., 2015). It is well accepted that spinal descending adrenergic processing pathways arise from the midbrain and brainstem structures and exert an inhibitory control over dorsal horn neuronal excitability (Malmberg, Hedley, Jasper, Hunter, & Basbaum, 2001; Ossipov, Morimura, & Porreca, 2014). These supraspinal adrenergic structures modulate the spinal cord neuron excitability through spinal α2‐adrenoceptors. Activation of spinal α2‐adrenoceptors suppresses transmission of pain inputs to higher centres and subsequently reduces the hyperexcitability of spinal dorsal horn neurons in neuropathic pain (Patel, Qu, Xie, Porreca, & Dickenson, 2018). It has been suggested that chronic neuropathic pain can induce plastic changes in the spinal α2‐adrenoceptors including increased efficacy of GPCRs as well as the potency of α2‐adrenoceptors (Bantel, Eisenach, Duflo, Tobin, & Childers, 2005). While trying to develop new ways of treating neuropathic pain, studies in both patients and animals have demonstrated the analgesic effects of spinal administration of α2‐adrenoceptor agonists (Bannister & Dickenson, 2016). The present review focuses on the cellular mechanisms by which the spinal adrenergic system may be involved in the modulation of pain perception in neuropathic pain. Emphasis is laid on the possible role of α2‐adrenoceptors’ activity in the spinal cord. We also address the latest electrophysiological evidence regarding α2‐adrenoceptor‐based mechanisms of analgesic action. We finally highlight basic and clinical proposals that indicate the analgesic effects of α2‐adrenoceptor agonists through various routes of administration in neuropathic pain states.
2. PAIN PERCEPTION AND SPINAL α2‐ADRENOCEPTORs
Following nerve damage, peripheral mechanisms are likely subjected to a filtering process through central mechanisms. Therefore, the relationship between the real peripheral nerve damage can become dissociated from the amount of pain perceived. Pain inputs within peripheral nerves are transmitted to the spinal cord and onwards to the pain matrix including the prefrontal cortex, insular cortex, anterior cingulate cortex, amygdala, and somatosensory cortex (Heidary, Sahraei, Afarinesh, Bahari, & Meftahi, 2018; Ossipov, Dussor, & Porreca, 2010). Pain perception does not result from activation of these ascending pathways alone, but rather from a communication between the pain matrix of the brain and the spinal cord. This top‐down processing descends from the brain back to the spinal cord (Bahari et al., 2014; Bahari, Sadr, et al., 2015; Bannister & Dickenson, 2016). The inhibitory pathways of top‐down processing of pain perception originate from the midbrain (periaqueductal grey [PAG]) and brainstem adrenergic structures (Di Cesare et al., 2017; Lopez‐Canul et al., 2015). It has been proposed that these descending inhibitory pathways can switch pain down. It is well established that noradrenaline (NA) is an important neurotransmitter, mainly implicated in the descending inhibitory control of pain. Synaptic contacts have been demonstrated between descending noradrenergic terminals of primary afferent nerve fibres or spinal dorsal horn neurons and interneurons (Wei & Pertovaara, 2006). Accumulating evidence has demonstrated that the pain‐modulatory‐related action of spinal NA is mediated by α‐adrenoceptors (Di Cesare et al., 2017; Eisenach, Zhang, & Duflo, 2005; Paqueron, Conklin, & Eisenach, 2003; Rahman, D'Mello, & Dickenson, 2008). α‐Adrenoceptors are classically divided into two main categories: α‐adrenoceptors type 1 (α1A, α1B, and α1D) and type 2 (α2A, α2B, and α2C; Alexander et al., 2013, 2015; Lee, Park, Park, & Lee, 2012). The brainstem adrenergic structures predominantly influence the spinal α2‐adrenoceptor subclass. Therefore, spinal α2‐adrenoceptor agonists are receiving increasing attention as analgesic agents.
Malmberg et al. (2001) examined the contribution of the different α2‐adrenoceptor subtypes to the development of neuropathic pain. They examined mice that carry either a point (α2A‐adrenoceptor) or null (α2B‐ and α2C‐adrenoceptor) mutation in the gene that encodes the α2‐adrenoceptor. The authors demonstrated that baseline mechanical and thermal thresholds were similar in all mutant and wild‐type mice; and, after peripheral nerve injury, all mice developed hypersensitivity (allodynia) to thermal and mechanical stimulation. Furthermore, Malmberg group found that antinociceptive effects of dexmedetomidine (α2‐adrenoceptor agonist) were intact in α2B‐ and α2C‐adrenoceptor, but absent in the α2A‐adrenoceptor mutant mice. They showed that the antinociceptive effects of dexmedetomidine are mediated via the activation of α2A‐adrenoceptors. The density and expression pattern of α2‐adrenoceptors is different in the spinal cord. Generally, α2A‐adrenoceptors are distributed across the spinal dorsal and ventral horn; α2B‐adrenoceptors were detected only in tiny amounts in the superficial dorsal horn; and α2C‐adrenoceptors were found in lower numbers across various layers of the dorsal horn (Fairbanks, Stone, & Wilcox, 2009). Immunohistochemical studies indicate that the localization of α2A‐adrenoceptors in the spinal dorsal horn is on the central terminals of nociceptive nerve fibres. In the spinal dorsal horn, the α2C‐adrenoceptors are located on the axon terminals of spinal interneurons. The expression of the α2B‐adrenoceptors is negligible in the spinal cord of postnatal animals (Pertovaara, 2006). Chronic neuropathic pain can induce neuroplasticity changes in the brainstem adrenergic pathways and spinal α2‐adrenoceptors. After nerve injury, several studies have indicated increased efficacy of G‐protein‐coupled‐α2‐adrenoceptors as well as potency of α2‐adrenoceptor agonists (Donaldson & Beazley‐Long, 2016; Oladosu, Maixner, & Nackley, 2015). It has also been reported that chronic neuropathic pain states can increase inhibitory noradrenergic innervations, the release of NA from descending adrenergic terminals, and the efficacy of α‐adrenoceptors, thereby causing analgesic effects of α‐adrenoceptor ligands. Hayashida, Clayton, Johnson, and Eisenach (2008) reported that nerve injury increases the expression of P2X4 receptors on the glial cells. Activation of P2X4 receptors via ATP augments the release of brain‐derived neurotrophic factor (BDNF) from glial cells. Then, the release of BDNF can induce descending adrenergic fibre sprouting and increased spinal NA content, thereby enhancing the analgesic effects of α2‐agonists in chronic neuropathic pain. Studies have revealed an up‐regulation of spinal α2‐adrenoceptors and increased spinal NA content in neuropathic pain. Therefore, nerve injury‐induced plasticity in the spinal α2‐adrenoceptors represents enhanced descending inhibition or sensitivity of the dorsal horn nociceptive neurons (Coull et al., 2005; Hayashida & Eisenach, 2011). It has been proposed that nerve injury‐induced neuroplastic changes are probably a homeostatic mechanism and attempt to counteract the nerve injury‐induced hyperexcitability of spinal neurons. Thus, the analgesia mediated by spinal delivery of α2‐adrenoceptor agonists is more effective for chronic neuropathic pain than for acute pain, due to plastic changes of the adrenergic pathways. In line with this, Parent et al. (2016) evaluated temporal changes in basal activity of the bulbo‐spinal adrenergic system, a novel double‐hit model of chronic/tonic pain, using LC coupled with tandem MS screening of CSF. They revealed that chronic neuropathic pain (chronic constriction injury model, first hit) leads to a transient excess of bulbo‐spinal descending adrenergic drive when that pathway has previously been primed using a tonic pain paradigm (formalin test, second hit). Specifically, neuropathic rats exhibited a reduced pain‐related behaviour response, particularly on Day 28 after neuropathy, when subjected to tonic pain (formalin test). This behavioural response was accompanied by a rapid rise in CSF NA content on Day 12 following neuropathy. However, recent animal studies have suggested that the analgesic efficacy of the α2‐adrenoceptor agonist declines following damage to the chronic phase of neuropathic pain, but not in the acute phase (Llorca‐Torralba, Borges, Neto, Mico, & Berrocoso, 2016).
An important clue regarding this lack of analgesic efficacy seems to be related to the fact that spinal α2‐adrenoceptor agonists also exert excitatory effects on the α2‐autoreceptors on the brainstem descending adrenergic terminals. This inhibits impulse discharge and causes further inhibition of NA release from adrenergic terminals (Gyires, Zádori, Török, & Mátyus, 2009; Taylor & Westlund, 2017). In fact, the term “autoreceptors” has been introduced for those receptors that are “sensitive to the neuron's own transmitter.” In contrast to autoreceptors, heteroreceptors are modulated by neurotransmitters derived from neighbouring neurons. It has been found that α2‐adrenoceptor heteroreceptors are located both in the peripheral sensory neurons and in the spinal neurons (Gilsbach & Hein, 2012). Autoreceptor activity is likely to negate the effectiveness of α2‐adrenoceptor ligands during chronic neuropathic pain (Gyires et al., 2009; Taylor & Westlund, 2017). The α2‐adrenoceptor autoreceptors are coupled to a Gi protein, reducing the activity of AC and PKA, then suppressing the release of NA from brainstem descending adrenergic terminals (Figure 1). In contrast, considering the potentiating tone of descending adrenergic pathways following nerve injury, chronic neuropathic pain has been proposed to be frequently associated with a loss of endogenous inhibition (Vaegter & Graven‐Nielsen, 2016). In this regard, enhancement of noradrenergic tone by α2‐adrenoceptor agonist or agents that block the reuptake of NA can be an effective treatment (Finnerup et al., 2015). Possibly, one reason for this contradictory data is the variety of neuropathy models in animal and human studies. Despite considerable progress in adrenergic biology, it is unclear whether the wide array of clinical functions of α2‐adrenoceptor agonists is mediated by the α2‐autoreceptors on adrenergic cells or whether and to what extent α2‐heteroreceptors on nonadrenergic cells are involved. To address this question, Gilsbach et al. (2009) crossed transgenic mice with an expression of α2A‐adrenoceptors under the control of the dopamine‐hydroxylase promoter to mice with a constitutive deletion of the α2A‐ and α2C‐adrenoceptor genes, thus generating mice that exclusively express α2A‐autoreceptors in adrenergic neurons. The authors provided a comprehensive phenotyping analysis of adrenergic cell versus nonadrenergic cell functions for α2‐adrenoceptors. In the baseline condition, they demonstrated that the latency time to withdrawal of the tail from the infrared light source did not differ between genotypes. Furthermore, administration of medetomidine (250 μg·kg−1 i.p.) markedly increased the latency time (analgesic effect) of tail withdrawal in wild‐type mice or in mice lacking α2C‐adrenoceptors, but not in mice with selective expression of α2A‐autoreceptors in adrenergic neurons. They suggest that α2A‐autoreceptors are not essential for the analgesic effect of medetomidine in the tail‐flick assay.
Figure 1.
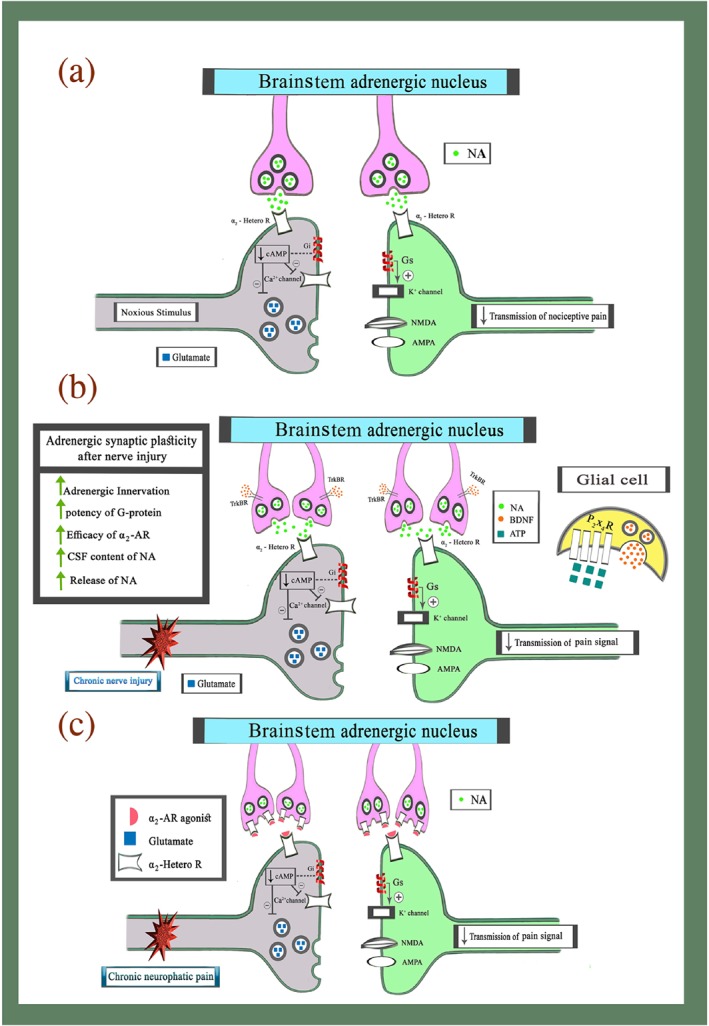
Brainstem inhibitory descending adrenergic modulation of pain in the physiological and the chronic neuropathic pain conditions. (a) Brainstem adrenergic inhibitory modulation of nociception in physiological condition. Application of noxious stimuli induces transmission of nociceptive information from ascending pathways to higher centres. At this time, noxious stimuli activate a brainstem adrenergic inhibitory pathways and increased the release of NA in dorsal horn from descending terminals. Activation of α2‐heteroreceptors on the presynaptic cell decreased entry of Ca2+ to cell and subsequently suppresses the release of glutamate and other stimulatory neurotransmitters via Gi‐protein activity. Additionally, the activity of α2‐heteroreceptors on the postsynaptic cell increased efflux of K+ from the cell and subsequently shift membrane potentials to a negative voltage via Gs‐protein activity. (b) Neuroplasticity changes in the brainstem descending adrenergic circuitries and the spinal cord in chronic neuropathic pain states. After nerve injury, up‐regulation of P2X4 receptors increased the release of BDNF from glial cells. The release of BDNF can induce descending adrenergic fibre sprouting and enhances analgesia effects of α2‐heteroreceptor agonists in chronic neuropathic pain. (c) Analgesic spinal effects of α2‐adrenoceptor (AR) agonist in chronic neuropathic pain. Although application of α2‐adrenoceptor agonist can induce analgesic effects via heteroreceptors on the pre and postsynaptic cells. However, in pharmacological studies α2‐adrenoceptor agonists can also decrease NA release from brainstem descending adrenergic terminals via autoreceptors and Gi‐protein activity in chronic neuropathic pain
3. BRAINSTEM DESCENDING ADRENERGIC CIRCUITS TO THE SPINAL CORD
A large body of basic research suggests that descending adrenergic pathways from brainstem structures diminish nociceptive transmission in the dorsal horn of the spinal cord (Bannister, Patel, Goncalves, Townson, & Dickenson, 2015; D'mello & Dickenson, 2008). The descending inhibitory pathways that diminish pain perception have two major efferent arms. The first efferent descending arm originates from the rostral ventromedial medulla (RVM), which includes the 5‐HT‐rich nucleus raphe magnus, modulating the maintenance of neuropathic pain. Neurons in the RVM then run to the dorsal horn, where they cause inhibition of nociceptive transmission. While neither the PAG nor the RVM contain noradrenergic neurons, both regions communicate with noradrenergic sites including the A5 (locus coeruleus [LC]), A6, and A7 nuclei. Electrical stimulation of the PAG or RVM elevates NA content in the CSF, and this effect is blocked by spinal adrenergic antagonists. Additionally, direct activation of the PAG and the raphe magnus nucleus generated spinal antinociception associated with spinal release of NA. This analgesic effect is reversed by spinal administration of α2‐adrenoceptor antagonists (Bitner et al., 1998; Budai, Harasawa, & Fields, 1998; de Novellis et al., 2008; Wang et al., 2014).
The second descending inhibitory arm involves the release of NA in the spinal cord from brainstem nuclei, particularly the LC (A6 cell group), A5 nucleus, and A7 nucleus in the pons. The LC is a heterogeneous nucleus and located at the lateral edge of the pontine grey matter. LC neurons project to specific supraspinal pain matrix including the thalamus, prefrontal cortex, anterior cingulate cortex, and amygdala. Additionally, axons of noradrenergic ponto‐spinal neurons in the LC project to the spinal dorsal horn. It is well accepted that an important neuropathic pain‐related role of the LC is to promote the feedback inhibition of the spinal nociceptive transmission following nerve injury. For example, ablation of descending noradrenergic neurons by i.t. administration of antiDbH‐saporin induced an increase in mechanical hypersensitivity following nerve injury (Hayashida, Peters, Gutierrez, & Eisenach, 2012). Similarly, the spinal delivery of α2‐adrenoceptor agonists could reduce the signs of neuropathic pain in both animals and humans (Baba, Shimoji, & Yoshimura, 2000; Kawasaki, Kumamoto, Furue, & Yoshimura, 2003). Therefore, the brainstem adrenergic nuclei constitute a pain‐control inhibitory system originating from the brainstem and descending onto the spinal cord.
4. CELLULAR MECHANISMS FOR ANALGESIC EFFECTS OF α2‐ADRENOCEPTOR AGONIST
Several mechanisms have been well established for the analgesic effect of α2‐adrenoceptor agonists (Figure 2). The α2‐adrenoceptors on the C‐fibre terminals are coupled to a Gi protein, which reduces the activity of AC, then suppressing both the production of cAMP and the activity of PKA. Therefore, activation of Gi protein inhibits the presynaptic voltage‐gated Ca2+ channels and suppresses the release of glutamate and substance P from primary C‐fibre terminals in the spinal cord (Philipp, Brede, & Hein, 2002). This phenomenon is related to the inhibition of N‐type Ca2+ channels in the presynaptic membrane. Additionally, at the same time, G‐protein‐coupled conductance of inwardly rectifying K+ channels is facilitated in the postsynaptic dorsal horn neurons by α2‐adrenoceptors. In this way, the membrane potentials of dorsal horn neurons are hyperpolarized, while excitability diminishes. Recently, using whole‐cell voltage clamp method in rats, Matsushita, Manabe, Kitamura, and Shibuya (2018) revealed that clonidine and dexmedetomidine (a highly selective agonist of α2‐adrenoceptors) and NA reduced the amplitudes of the inward capsaicin currents, in a manner that was prevented by yohimbine (α2‐adrenoceptor antagonist). In addition to changes of pre or postsynaptic ion channel conductance, the α2‐adrenoceptors’ activity can also influence the expression and activity of several neurotransmitters, receptors, and proteins. For example, Yeo et al. (2016) noted that the antiallodynic effect of clonidine might be related to inhibiting phosphorylated p38 MAPK induced by oxaliplatin. Spinal phosphorylated p38 MAPK plays an important role in the induction and maintenance of neuropathic pain (Morioka, Tanabe, Inoue, Dohi, & Nakata, 2009). Zhou, Wu, Chen, Liu, and Miao (2014) stated that dexmedetomidine attenuates the nerve injury‐induced allodynia via down‐regulation the expression of P2X4 receptors, a subtype of ionotropic purinoceptor, and BDNF in microglia of spinal dorsal horn in rats. All these are essential for the maintenance of pain hypersensitivity following nerve damage (Trang, Beggs, Wan, & Salter, 2009). Matsushita et al. (2018) concluded that delivery of NA reduces the activity of transient receptor potential vanilloid type 1 (TRPV1) channels in dorsal sensory neurons. TRPV1 is a well‐known nociception‐mediating ion channel and is mainly expressed in peripheral Aδ‐ and C‐fibres as well as throughout the CNS, which overloads Ca2+ entry through the channels involved in neuropathic pain (Chen et al., 2015; Ren et al., 2015; Uslusoy, Nazıroğlu, & Çiğ, 2017). The contribution of TRPV1 has also been evaluated in several rodent models of neuropathic pain. Peripheral nerve injury can elicit an increase in TRPV1 expression in uninjured dorsal root ganglion neurons (Zakir et al., 2012). TRPV1 antisense oligodeoxy nucleotides attenuate the mechanical hyperalgesia after spinal nerve ligation (Christoph et al., 2007). Therefore, it seems that TRPV1 channels play a role in the development and maintenance of neuropathic pain. Recently, it has been reported that the application of NA and clonidine, α2‐adrenoceptor agonists, reduces the activity of TRPV1 in sensory neurons. This inhibitory effect of NA was mimicked by clonidine and antagonized by yohimbine, an α2‐adrenoceptor antagonist. Furthermore, the inhibitory effect of NA on the activity of TRPV1 channels was dependent on calcium influx as well as calcium/calmodulin‐dependent PK II (CaMKII) activity (Chakraborty, Elvezio, Kaczocha, Rebecchi, & Puopolo, 2017). It has been suggested as an additional mechanism of inhibitory feedback of spinal α2‐adrenoceptors. Another mechanism proposed for the spinal analgesic effects of α2‐adrenoceptors involves its interaction with the cholinergic interneurons via BDNF. Peripheral nerve injury increased BDNF release from terminals of primary afferents and spinal glia cells. It is highly likely that BDNF plays a critical role in synaptogenesis and plasticity in cholinergic interneurons. Additionally, the increase in BDNF content enhanced spinal ACh release by α2‐adrenoceptor stimulation after nerve injury. Furthermore, the enhancement of ACh release caused an analgesic effect, directly or indirectly via GABA signalling (Figure 2; Hayashida & Eisenach, 2010; Obata, 2017). Therefore, it is suggesting that functional alterations of α2‐adrenoceptors and actions of BDNF on cholinergic interneurons after nerve damage are critical for the analgesia mediated by α2‐adrenoceptor agonists in neuropathic pain. For example, i.t. administration of dexmedetomidine inhibited mechanical hyperalgesia by the release of ACh to the spinal cord and stimulation of muscarinic receptors (Kimura, Saito, & Obata, 2012). However, further studies are required to clarify whether BDNF affects the expression or Gs‐coupling of α2‐adrenoceptors on the cholinergic interneurons.
Figure 2.
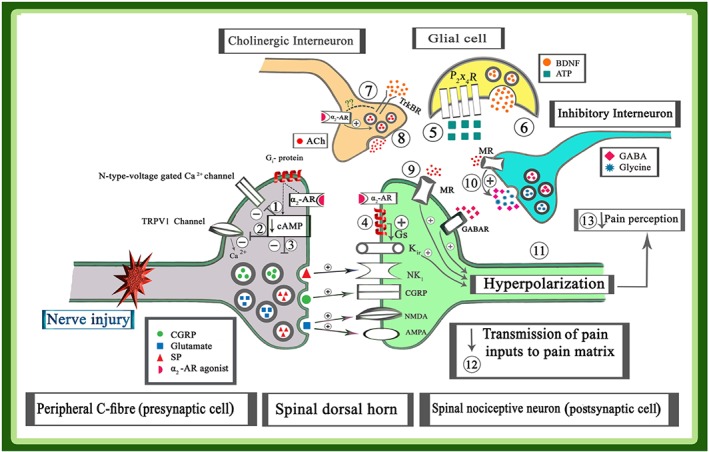
Cellular mechanisms for analgesic effects of α2‐adrenoceptor (AR) agonists; α2‐adrenoceptors located both in the peripheral sensory neurons and spinal neurons. The activation of α2‐adrenoceptors on presynaptic cells, using an α2‐adrenoceptor agonist, reduces the activity of AC, suppressing both the production of the cAMP and the activity of PKA. Then, suppressing the activity of PKA inhibited the presynaptic N‐type voltage‐gated Ca2+ channels (1) and TRPV1 channel activity (2); subsequently, the influx of Ca2+ ions to presynaptic cell and the release of stimulators neurotransmitters such as glutamate, substance P, and CGRP from primary fibre terminals in the spinal cord are decreased (3). Furthermore, application of α2‐adrenoceptor agonist increases the activity of Gs proteins in the postsynaptic cells. Activation of Gs protein, increased both the production of the cAMP and the activity of PKA, and then the activity of K+ ir channels and efflux of K+ ions are increased. Efflux of K+ ions induced hyperpolarization in the postsynaptic neurons (4). Nerve injury increased the release of ATP from the peripheral sensory neuron and also induced an up‐regulation of P2X4 receptors on glial cells (5). The activity of P2X4 receptors increased the release of BDNF from glial cells (6). At the same time, activation TrkB receptors on cholinergic interneurons (7) and its interaction with α2‐adrenoceptors via unknown mechanisms elicited the release of ACh (8). Activation of muscarinic receptors directly on the postsynaptic cells (9) or indirectly on the inhibitory GABAergic or glycinergic interneurons (10) can also induce hyperpolarization in the postsynaptic cells (11). Therefore, the application of an α2‐adrenoceptor agonist elicited hyperpolarization in the postsynaptic cell via above‐mentioned mechanisms. Hyperpolarization of the neuron decreased transmission of pain inputs to higher centres (12) and suppressed pain perception (13)
5. ELECTROPHYSIOLOGICAL EVIDENCE FOR SPINAL ADRENERGIC PAIN INHIBITION
Hyperalgesia and allodynia are common clinical symptoms of neuropathic pain and therefore hyperexcitability must be a major component of pain. The rapid advances made, generated through modern pharmacological, molecular, and cellular techniques have provided ample evidence on the underlying mechanisms involved in pain pathophysiology. Allodynia suggests the presence of a mechanism that amplifies nociceptive inputs in the central nociceptive system. A synaptic amplifier of nociception has been identified at the synapses between primary afferent C‐fibres and neurons in the spinal dorsal horn (Dubin & Patapoutian, 2010; Sandkühler & Gruber‐Schoffnegger, 2012). Although the mechanisms underlying the pathology of pain have remained unknown, it is widely accepted that LTP of nociceptive synaptic transmission in the spinal dorsal horn might be involved in cellular mechanism of chronic pain. LTP is a form of synaptic plasticity, which enhances the efficacy of nociceptive synaptic transmission. Previous studies demonstrated the existence of LTP at synapses between primary nociceptive C‐fibre afferents and spinal neurons following nerve injury (Chirila et al., 2014; Kronschläger et al., 2016; Ruscheweyh, Wilder‐Smith, Drdla, Liu, & Sandkühler, 2011). Therefore, neuropathic pain augments the efficacy of spinal nociceptive transmission by long‐lasting neuronal hypersensitivity to peripheral stimuli. Under this assumption, preventing and reversing established LTP may help prevent the development of pain‐related behaviours and may be helpful in treating patients who have an LTP component to their chronic pain. It was shown that spinal LTP is typically induced by noxious inputs such as high‐frequency stimulation of primary afferent fibres about two decades ago.
Using immunofluorescence it has been shown that α2‐adrenoceptors are localized; α2‐adrenoceptors were detected on the central terminals of C‐fibre afferents and spinal excitatory neurons in superficial dorsal horn neurons (Gassner, Ruscheweyh, & Sandkühler, 2009; Millan, 2002). Indeed, activation of α2‐adrenoceptors suppresses excitatory neurotransmitter release from C‐fibre afferents and subsequently spinal synaptic transmission and pain amplification. Ohnami et al. (2012) revealed that the spinal application of milnacipran (NA reuptake inhibitor) suppressed C‐fibre‐mediated spinal nociceptive synaptic transmission along with the maintenance phase of LTP of C‐fibre‐evoked field potentials in nerve‐injured animals. These inhibitory effects of milnacipran were blocked by spinal administration of yohimbine or idazoxan (α2‐adrenoceptor antagonists). This provides novel evidence regarding the analgesic actions of NA reuptake inhibitors in neuropathic pain. Similarly, the electrophysiological field potential recorded from the dorsal horn neurons in rats demonstrated that application of a COMT inhibitor (OR 486) reduced spinal nociceptive responses to painful stimuli and attenuated the expression of spinal LTP. COMT is an enzyme that metabolizes catecholamines and thus modulates adrenergic and noradrenergic neuronal signalling. Accordingly, the observed reduction of dorsal horn nociceptive responses might result from an increased level of NA (Jacobsen, Eriksen, Pedersen, & Gjerstad, 2010). Additionally, in an in vitro whole‐cell voltage‐clamp study, a specific agonist of α2‐adrenoceptors (UK14304) inhibited the frequency of EPSCs and the amplitude of evoked monosynaptic and polysynaptic EPSCs of spinal lamina II neurons in diabetic neuropathy. Furthermore, administration of UK14304 increased the amplitude of postsynaptic G‐protein‐coupled inwardly rectifying K+ channel currents in diabetic neuropathy. The effects of UK14304 were completely antagonized by the α2‐adrenoceptor antagonist yohimbine (Chen, Chen, Yuan, & Pan, 2011). Meanwhile, an inhibitory G protein is known to be coupled with presynaptic α2‐adrenoceptors. Peripheral nerve injury leads to activation of presynaptic α2‐adrenoceptors, thus inhibiting AC. This, in turn, reduces PKA, protein phosphorylation, activation and inhibits voltage‐gated calcium channels while enhancing voltage‐gated potassium channels (Li et al., 2013; Unterberger, Moskvina, Scholze, Freissmuth, & Boehm, 2002; Willoughby, Wachten, Masada, & Cooper, 2010). All of these effects would eventually reduce the likelihood of spinal synaptic nociceptive transmission. Similarly, there is evidence that nerve injury may strengthen the coupling between α2‐adrenoceptors and inhibitory G proteins, leading to diminished synaptic transmission. Activating the α2‐adrenoceptors with clonidine, dexmedetomidine, or NA can reduce glutamate release from dorsal root ganglion neurons in response to capsaicin, an effect reversed by an α2‐adrenoceptor antagonist (BRL44408; Li & Eisenach, 2001). Furthermore, clonidine has been clinically successful in the treatment of neuropathic pain patients. The Food and Drug Administration approved clonidine for the treatment of cancer pain in the United States (Miljanich, Rauck, & Saulino, 2013). Recently, using in vivo electrophysiology, Patel et al. (2018) recorded the firing rate of a wide dynamic range of neurons in ventral posterolateral thalamus (VPL). The authors demonstrated that spinal block of α2‐adrenoceptors with atipamezole resulted in enhanced stimulus‐evoked and spontaneous firing in the VPL in the sham group. However, atipamezole enhanced only spontaneous firing in the VPL in spinal nerve‐ligated (SNL) rats. Potentiating spinal adrenergic tone in neuropathic rats with reboxetine, a selective noradrenergic reuptake inhibitor, modestly restored inhibitory control of evoked responses in the VPL, although it had no effect on spontaneous firing. However, the application of clonidine inhibited both evoked and spontaneous firing in SNL rats as compared with the sham group. It has been suggested that descending adrenergic inhibitory pathways are tonically active in sham rats. Also, descending inhibitory control is attenuated, but not completely absent, in neuropathic pain and differentiates between spontaneous and evoked neuronal activity.
6. α2‐ADRENOCEPTORs AS A TARGET FOR NEUROPATHIC PAIN TREATMENT; BEHAVIOURAL EVIDENCE
Neuropathic pain is now considered as a clinical entity, regardless of the underlying aetiology (Attal & Bouhassira, 2015). The difficulty in treating this condition is mainly due to insufficient knowledge about the underlying mechanisms. Although, ample studies reflect the interest in the α2‐adrenoceptor receptor as a target for the treatment of neuropathic pain. A number of α2‐adrenoceptor agonists or chemical compounds that block the reuptake of NA have been developed. In this context, we briefly present the major pharmacological treatments studied for the management of neuropathic pain. Clonidine, the first congener of the α2‐adrenoceptor agonist, is mainly used as an analgesic agent in the rodent and clinical studies. There is increasing behavioural evidence suggesting that clonidine may be effective in the management of neuropathic pain (Bharti, Dontukurthy, Bala, & Singh, 2013; Engelman & Marsala, 2012; Ydemann et al., 2018). Clonidine is approved by the United States Food and Drug Administration for epidural administration as a treatment for neuropathic cancer pain. The target receptors for clonidine include peripheral and postsynaptic α2‐adrenoceptors of spinal descending noradrenergic pathways, with an affinity predilection of 200:1 for α2‐ versus α1‐adrenoceptors respectively (Giovannitti, Thoms, & Crawford, 2015). In addition, clonidine acts as a ligand for an imidazoline‐binding site (I2‐BS). I2‐BS located in peripheral nerve terminals has proved to be a new drug target for analgesics. Therefore, the activation of I2‐BS may be considered as a secondary mechanisms for the analgesic action of clonidine (Bektas, Nemutlu, & Arslan, 2015). An i.p. injection of clonidine reduces mechanical allodynia in oxaliplatin‐induced neuropathic mice. Oxaliplatin is a platinum‐based agent, used as a therapy for cancer patients, which can also induce neuropathic pain (Yeo et al., 2016). Additionally, i.t. delivery of clonidine significantly reduced mechanical allodynia and thermal hyperalgesia in a rat nerve injury‐induced neuropathic pain model (Roh et al., 2008, 2010). An in vitro study of human cadaver trunk skin revealed that clonidine can be successfully delivered transdermally for analgesic effects when incorporated into transdermal lipoderm compound bases. Furthermore, clonidine, approximately 1 hr after application on trunk skin, had a rapid penetration to peak flux and a secondary peak at approximately 40 hr (Bassani & Banov, 2016). Although the systemic delivery of clonidine has attenuated hyperalgesia in several pain models, the clinical use of clonidine is dose limited, because of its adverse side effects such as drowsiness, hypotension, and sedation. In this context, some scientific research suggests that localized cutaneous neuropathic pain can be managed safely with topical medications (Casale, Symeonidou, & Bartolo, 2017; Zur, 2014). For example, the topical application of clonidine gel 0.1% significantly relieved pain in diabetic peripheral neuropathy patients, potentially contributing to pain relief with no systemic absorption (Kiani, Sajedi, Nasrollahi, & Esna‐Ashari, 2015; Wrzosek, Woron, Dobrogowski, Jakowicka‐Wordliczek, & Wordliczek, 2015). Several clinical studies suggest a combination drug therapy results in better pain relief and also reduced drug associated complications in patients (Holbech et al., 2017; Vorobeychik, Gordin, Mao, & Chen, 2011). Yoon, Kang, Kim, Kim, and Roh (2015) observed that coadministration of clonidine with BD‐1047, a sigma non‐opioid intracellular receptor 1 antagonist, increased low dose clonidine‐induced analgesic effects in a tonic pain model, without impairing motor coordination and BP. Furthermore, clonidine is occasionally coadministered as an adjuvant to opioids for chronic pain management. It may be effective for the treatment of chronic neuropathic pain in patients who have developed morphine tolerance (Stone, German, Kitto, Fairbanks, & Wilcox, 2014). Coadministration of opioid and α2‐adrenoceptor agonists such as clonidine can induce synergistic analgesic effects. This α2‐adrenoceptor–opioid synergy can potentially improve analgesic efficacy or reduce side effects, thus improving the therapeutic index (Chabot‐Doré, Schuster, Stone, & Wilcox, 2015). In support of an α2‐adrenoceptor–opioid receptor interaction, α2‐adrenoceptor stimulation enhances the antinociceptive effect of μ receptors, while α2‐adrenoceptor antagonists such as yohimbine or idazoxan attenuate morphine analgesia. It is likely that pharmacokinetic effects of α2‐adrenoceptor agonists like clonidine contribute to opioid–adrenoceptor synergy at the level of the spinal cord. Also, spinal application of clonidine exerts a vasoconstrictive effect, leading to reduced drug clearance from the site of administration (Iida, Ohata, Iida, Watanabe, & Dohi, 1999). Furthermore, it seems that the route of drug administration is an important determinant of the α2‐adrenoceptor–opioid receptor interaction. Ossipov, Harris, Lloyd, and Messineo (1990) for the first time evaluated opioid–adrenoceptor interactions, using isobolographic analysis. They observed that the synergistic effect of clonidine with morphine, fentanyl, and meperidine was stronger when the drugs were administered i.t. compared with the i.v. route. Therefore, it is likely that the opioid–adrenoceptor interaction took place at the spinal level. There is little evidence suggesting that synergy occurs outside the spinal cord. For example, despite an extensive coexpression of μ receptors and α2‐adrenoceptors in LC neurons, electrophysiological recordings revealed only an additive interaction, and not a synergistic interaction, among these receptors (Stone & Wilcox, 2004). Additionally, the use of different genetically altered mouse models has allowed the identification of a synergistic opioid–adrenoceptor interaction. For example, genetically altered mice lacking NA showed a significant attenuation of the analgesic effect induced by morphine (Jasmin et al., 2002; Meske et al., 2014). Analgesic synergism of α2‐adrenoceptor agonists with opioids was lost in the α2‐adrenoceptor‐knockout mice, supporting a central role of α2‐adrenoceptors in this effect (Özdoǧan, Lähdesmäki, Hakala, & Scheinin, 2004). Riedl et al. (2009) demonstrated that both opioid receptors and α2‐adrenoceptors colocalized on primary afferent nociceptive fibres, which represents anatomical evidence for a possible interaction between the two systems. Thus, it seems that activation of this α2‐adrenoceptor–opioid interaction is a key strategy to treat neuropathic pain. However, the development of therapeutic approaches that exploit the combination of opioids and clonidine is limited, in part, by the lack of mechanistic knowledge on how these drugs interact (Chabot‐Doré et al., 2015). Dexmedetomidine is another highly selective α2‐adrenoceptor agonist similar to clonidine, but with a greater affinity for α2‐adrenoceptors (Giovannitti et al., 2015). The main clinical application of dexmedetomidine is for sedation and anaesthesia (Naaz & Ozair, 2014). However, like clonidine, several clinical studies have also demonstrated the analgesic effects of dexmedetomidine on pain management. For example, Guneli, Yavasoglu, Apaydin, Uyar, and Uyar (2007) revealed that a systemically administered combination of tramadol and dexmedetomidine provides a potent synergistic analgesic effect at a low drug dose in neuropathic pain. This analgesic effect seems to be associated with a spinal mechanism since dexmedetomidine crosses the blood–brain barrier. Similarly, Lin et al. (2008) suggested that combining an i.v. injection of dexmedetomidine with morphine could improve analgesia while reducing opioid‐related side effects. They revealed that dexmedetomidine reduced the required dosage of morphine to 29% 0–24 hr after abdominal total hysterectomy. Nevertheless, the data about the analgesic effect of systemic dexmedetomidine on neuropathic pain in human and rodent models are generally limited. In a basic research programme, i.p. delivery of dexmedetomidine indicated a dose‐dependent antiallodynic effect on mechanical and cold stimuli in vincristine‐evoked neuropathic rat models (Park et al., 2012). Solanki et al. (2013) also revealed that dexmedetomidine (5 μg), when added to i.t. bupivacaine (15 mg), yielded longer post‐operative analgesia while causing no undesirable side effects among trauma patients undergoing lower limb surgery. Moreover, coadministration of dexmedetomidine with bupivacaine prolonged the duration of sensory and motor block and reduced analgesic requirements. A meta‐analysis of 28 clinical trials revealed that intraoperative application of dexmedetomidine reduced post‐operative pain perception and opioid consumption. Meanwhile, the post‐operative administration of dexmedetomidine did not yield a considerable effect on pain perception (Schnabel, Reichl, et al., 2013; Schnabel, Meyer‐Friessem, Reichl, Zahn, & Pogatzki‐Zahn, 2013). Peng, Jin, Liu, and Ji (2015) revealed that intraoperative infusion of dexmedetomidine was effective for reducing pain score and possible adverse events in patients after craniotomy.
Tizanidine is another α2‐adrenoceptor agonist, which is similar to clonidine and dexmedetomidine with some critical differences. As with clonidine and dexmedetomidine, it has analgesic effects, but it has a shorter duration of action and less effect on heart rate and BP (Giovannitti et al., 2015). It has been shown that i.t. injection of tizanidine can produce dose‐dependent analgesic effects in neuropathy rats (Ou‐Yang et al., 2008; Yekkirala, Roberson, Bean, & Woolf, 2017). Semenchuk and Sherman (2000) in an open‐label study assessed the effectiveness and tolerability of tizanidine in neuropathy patients. Patients with neuropathic pain received 1 to 4 mg of tizanidine once daily for 7 days; 68% of patients reported pain relief with tizanidine therapy. It has also been suggested that brimonidine (also known as UK14304) and fadolmidine, as with other α2‐adrenoceptor agonists, have analgesic effects. Brimonidine's binding affinity differs only slightly among the three receptor subtypes (α2A, α2B, and α2C) with a moderate preference (20‐fold) for α2A‐adrenoceptor versus α2C‐adrenoceptors (Kalapesi, Coroneo, & Hill, 2005; Sallinen et al., 2007). Intrathecally administered brimonidine was also used as antinociceptive in both rat and mouse. Kim, Park, and Hwang (2009) revealed that i.t. delivery of brimonidine provides an antiallodynic effect on L5/L6 SNL rats. Fadolmidine is highly selective for α2‐adrenoceptors. Due to its pharmacokinetic properties, it only poorly crosses the blood–brain barrier. Intrathecal administration of fadolmidine leads to antinociceptive effects in neuropathic pain, while producing no haemodynamic depression with considerably less sedation compared to dexmedetomidine. Systemic administration of fadolmidine had no or only a weak antinociceptive action, except following nerve injury, particularly that of the postganglionic sympathetic nerve fibres (Pertovaara, 2004). Nasirinezhad, Hosseini, and Salari (2015) observed that i.t. injection of NA (1 μg) significantly alleviated the cold and thermal allodynia in a rodent neuropathic pain model. As mentioned above, NA is implicated in enhancing endogenous analgesic mechanisms via the descending inhibitory pain pathways in the spinal cord (Suzuki, Rygh, & Dickenson, 2004). This is consistent with the clinical success of chemicals that enhance spinal monoaminergic activity, such as 5‐HT/NA reuptake inhibitors, in the treatment of chronic neuropathic pain. Iyengar, Webster, Hemrick‐Luecke, Xu, and Simmons (2004) identified that i.p. administration of duloxetine, a potent dual 5‐HT and NA reuptake inhibitor (3–15 mg·kg−1), suppressed late phase paw‐licking behaviour in a dose‐dependent manner in formalin‐induced pain. Furthermore, oral administration of duloxetine (5–30 mg·kg−1) attenuated mechanical allodynia in L5/L6 spinal nerve ligation model of neuropathic pain. However, oral administration of duloxetine (3–30 mg·kg−1) was minimally efficacious in the tail‐flick model of pain. The authors suggest that duloxetine may have utility in treating neuropathic pain states. Subsequently, Obata, Saito, Koizuka, Nishikawa, and Goto (2005) identified that i.t. administration of milnacipran, an NA reuptake inhibitor, (3–100 μg) yielded dose‐dependent antiallodynic effects in L5 and L6 spinal nerve ligated (SNL) rats. The antiallodynic effect of milnacipran was attenuated by i.t. coadministration of yohimbine (α2‐adrenoceptor antagonist, 30 μg). I.p administration of milnacipran had no antiallodynic effects at doses of 3 to 30 mg·kg−1. Furthermore, antiallodynic effects were not produced by i.t. administration of maprotiline, selective NA reuptake inhibitor, (10 to 100 μg). They suggested that simultaneous inhibition of 5‐HT and NA reuptake in the spinal cord is essential to mediate analgesic effects. Nakajima, Obata, Iriuchijima, and Saito (2012) showed that i.p. injection of milnacipran (3–30 mg·kg−1) induced a dose‐dependent antihyperalgesic effect in L5 and L6 SNL rats. The effect was reversed by i.t. injection of idazoxan (α2‐adrenoceptor antagonist, 30 μg). Delivery of maprotiline (10 and 30 mg·kg−1) generated an antihyperalgesic effect, which was reversed by i.t. injection of idazoxan. At the same time, following the injection of milnacipran, NA and 5‐HT concentrations increased in the spinal dorsal horn, while only NA rose after an injection of maprotiline. Hoshino, Obata, and Saito (2015) revealed that the i.p. administration of duloxetine (3, 10, and 30 mg·kg−1) dose‐dependently suppressed hyperalgesia induced by spinal nerve ligation. They further found that NA and 5‐HT concentrations increased after i.p. administration of duloxetine (10 mg·kg−1) using in vivo microdialysis in the spinal dorsal horn.
Among the 5‐HT receptors, the 5‐HT1A receptor has been one of the first to be pharmacologically characterized (Fargin et al., 1988). It is expressed in the raphe nucleus and in several postsynaptic areas involved in central nociceptive mechanisms (Kalipatnapu & Chattopadhyay, 2007). Full and partial 5‐HT1A receptor agonists have been shown to be beneficial in pain treatments (Nadeson & Goodchild, 2002) arousing great interest as future therapeutic agents (Muthuraman, Singh, Singh Jaggi, & Ramesh, 2014; Panczyk et al., 2015).
It has been demonstrated that the (S)‐(−) enantiomer of biphenyline, described as an α2 adrenoceptor (Gentili et al., 2004) and 5‐HT1A receptor (Del Bello et al., 2016) agonist, behaved as a potent and long‐lasting antinociceptive agent in algesiometric paradigms (Gentili et al., 2004). Considering the important role played by both 5‐HT1A receptors and α2 adrenoceptors in nociception, Di Cesare Mannelli et al. (2017) are presented with a novel pharmacodynamic approach for the treatment of neuropathic pain. They examined the combined activation of 5‐HT1A and α2 receptors by use of a rationally‐designed imidazoline ligand (S)‐1 ((S)‐2‐(1‐([1,1′‐biphenyl]‐2‐yloxy)ethyl)‐4,5‐dihydro‐1H‐imidazole) in a rat model of chronic constriction injury of the sciatic nerve. They observed that the acute administration p.o. of (S)‐(−)‐1 (0.1–1 mg·kg−1) significantly increased the pain threshold to mechanical noxious stimuli. (S)‐(−)‐1 efficacy was confirmed by the decrease in spontaneous pain evaluated as hindlimb weight bearing alterations. The administration of gabapentin (100 mg·kg−1 p.o.) induced an analgesic effect similar to (S)‐(−)‐1 administered at a 100‐fold lower dose. They also showed that the application of other selected analogues such as compound 5 (2‐(1‐(2‐cyclohexylphenoxy)ethyl)‐4,5‐dihydro‐1H‐imidazole; 1 mg·kg−1 p.o.) fully reversed the nerve injury‐induced hypersensitivity. The pain relieving activity of compound 5 was prevented by the WAY 100635 (selective 5‐HT1A receptor antagonist, 1 mg·kg−1, i.p.) and, to a lesser extent, by yohimbine (an α2 antagonist, 3 mg·kg−1, i.p.). Therefore, it is likely that the bioversatile 2‐imidazoline‐substituted scaffold might be a valuable structure in the building of novel analgesic agents.
Another new compound that has recently been considered in the treatment of neuropathic pain is tapentadol. It is indicated as a novel component for treatment of cancer‐related neuropathic pain. It was introduced as an atypical opioid with a unique mechanism of action, which includes acting as a μ receptor agonist, NA reuptake inhibitor, and α2‐adrenoceptor activator (Afilalo & Morlion, 2013; Rahman & Dickenson, 2011; Tzschentke et al., 2007). Therefore, it seems that the efficacy of tapentadol to treat chronic pain is expected to be better than that of other opioids. Tapentadol has been calculated to have a K i of 0.5 μM in rat synaptosomal NA reuptake assays and 0.1 μM in μ receptor‐binding assays (Tzschentke et al., 2007). Bee, Bannister, Rahman, and Dickenson (2011) performed in vivo electrophysiological study in SNL rats to show that systemic application of tapentadol (1 and 5 mg·kg−1) dose‐dependently decreased the responses of spinal dorsal horn neurons evoked by a range of peripheral stimuli, including brush, punctate mechanical, and thermal stimuli. Additionally, they showed that spinal application of the atipamezole (selective α2‐adrenoceptor antagonist), or naloxone (the μ receptor antagonist), reversed tapentadol's inhibitory effects, which suggests not only that the spinal cord is the key site of tapentadol's actions but also that tapentadol interacts with both α2‐adrenoceptors and μ receptors.
Sugiyama et al. (2018) investigated the effectiveness of oral tapentadol for relieving cancer‐related neuropathic pain in a retrospective study. Their study included 38 Japanese patients with advanced cancer. They revealed that oral administration of tapentadol decreased the pain score from 3.78 to 2.78. However, the performance status of their patients did not change after the introduction of tapentadol. The analgesic efficacy of tapentadol was investigated by Yadav, Jain, Samprathi, Baghel, & Singh, 2016, using a prospective study. They demonstrated that a single preemptive oral dose of tapentadol (75 mg) decreased perioperative analgesic requirements and also the post‐operative pain in 60 patients after laparoscopic cholecystectomy, without added complications. In a study that compared the side effects of morphine with tapentadol, it was found that tapentadol was less likely to cause vomiting. In addition, the dosage that was needed to elicit these responses was smaller for morphine than for tapentadol. These data suggest that tapentadol induces fewer gastrointestinal complications than morphine (Tzschentke et al., 2006). However, a report of cardiovascular complications following administration of tapentadol (100 mg) raised the need for a dose–response study to evaluate its safety and efficacy (Vachhani, Barvaliya, Naik, & Tripathi, 2014).
Recently, tapentadol has been demonstrated to be effective in the management of both diabetic‐ and chemotherapy‐induced neuropathic pain (Vadivelu et al., 2015, Galiè, Villani, Terrenato, & Pace, 2017). Similarly, Schwartz et al. (2015) evaluating the efficacy and tolerability of tapentadol extended its release for diabetic peripheral neuropathy. They indicate that treatment with tapentadol in an extended release tablet (100–250 mg, twice per day) markedly suppressed pain intensity. The effectiveness of tapentadol was maintained for patients who continued taking tapentadol extended release, while pain intensity increased for patients who switched to the placebo. Additionally, Baron et al., 2015 studied the effectiveness and tolerability of tapentadol prolonged release tablet (300 mg·day−1) in patients with severe chronic low back pain with a neuropathic component. The study demonstrated significant improvements in pain intensity scores, pain‐related symptoms, and quality of life. However, there is no clear standard for the use of tapentadol for cancer‐related neuropathic pain. Hence, further studies are needed to better understand tapentadol interactions with adrenoceptors and neuropathic pain modulation. Recently, several studies revealed that nitrogen‐containing heterocyclic compounds also have analgesic effects. Two nitrogen atoms containing heterocyclic compounds such as diazines, pyridazines, pyrimidines, and piperazines are an important structural feature of many biologically active compounds and possess antinociceptive properties (Asif, Singh, & Singh, 2011). Here, our interest has focused on diazines, because of their easy fictionalization, which makes them attractive synthetic compounds for designing and the development of the novel drugs in the future. Diazine is an important lead owing to its inherent properties and therapeutic actions. Many of the successful drugs have the diazine ring as a pharmacophore. Diazines have received much attention in the preparation of organic intermediates and physiologically active compounds. Recently, many pyridazine derivatives have been reported to possess interesting biological properties, such as analgesic. Additionally, various pharmacological studies have revealed that the analgesia induced by pyridazine and phthalazine derivatives is mediated through the adrenergic system (Biancalani et al., 2009; Cesari et al., 2006). The pyridazine and phatalzine and its 3‐oxo derivatives (pyridazinones and phthalazinone) have attracted a great deal of attention because of the wide spectrum of their pharmacological as well and agrochemical activities. Recently, in vitro radioligand‐binding studies were performed on a panel of adrenoceptors in order to define the pharmacological profile of pyridazine derivatives, using a prototypical compound of the series. Cesari et al. (2006) synthesized and tested the potential antinociceptive activity of the (5‐{[4‐(3‐chlorophenyl)piperazin‐1‐yl]‐propyl}‐3‐methyl‐7‐phenylisossazolo[4,5‐d]pyridazin‐4‐(5H)‐one; compound A) in the two different experimental models: the abdominal constriction test, in which a painful chemical stimulus was applied, and the hot plate test, in which a thermal stimulus was used. They observed that p.o. administration of compound A (20 mg·kg−1) significantly reduce the number of abdominal constrictions by more than 50% as compared with the control group. They also showed that pretreatment with yohimbine (an α2‐antagonist) prevents the analgesic effects of compound A. Additionally, they evaluated the effect of administration of the selective α2A‐adrenoceptor antagonist (BRL44408) and the selective α2C‐adrenoceptor antagonist (ARC‐239) on the analgesic effects of compound A. Only BRL44408 was able to completely antagonize the antinociceptive effects of compound A. Pretreatment with prazosin (α1‐adrenoceptor antagonist) did not modify the increased pain threshold produced by compound A, further confirming a α2‐mediated mechanism of action. They also revealed that compound A has a K i > 1 μM, using α2‐binding experiments in the rat cortical membranes. This indicates an absence of affinity for α2‐adrenoceptors. However, an increase in NA content was revealed after administration of compound A (1 and 20 mg·kg−1, i.p.). Also, they observed inhibition of the synaptosomal [3H]‐noradrenaline uptake in the presence of compound A, suggesting the analgesic action of compound A underlies an indirect activation of the noradrenergic system, via the inhibition of NA reuptake. Similarly, Biancalani et al., in 2009 synthesized and tested a number of pyridazinone derivatives that contain an arylpiperazinylalkyl chain in the tail‐flick and hot plate model of nociception in mice. The most potent of the pyridazinone derivatives was compounded 6a, which showed an ED50 = 3.5 μg, about threefold higher than that of morphine. The same compound also showed high potency in the hot plate test. Furthermore, the antinociceptive effect of 6a was completely reversed by pretreatment with yohimbine both in the hot plate test and in the tail‐flick tests. To further verify the pharmacological profile of compound 6a, the Biancalani group performed in vitro radioligand‐binding studies on the interaction between compound 6a and different subtypes of adrenoceptors. Compound 6a showed selectivity for α1A‐, α1B‐, and α2A‐adrenoceptors, with IC50 values 56–320 nM. By contrast, compound 6a showed low affinity for α2C‐adrenoceptors in comparison with the other subtypes examined. These data confirm an involvement of the α2‐adrenoceptors in the analgesia produced by compound 6a. Asif et al., in 2011 synthesized three 6‐phenyl‐4‐substituted benzylidene tetrahydro‐pyridazin‐3(2H)‐one derivatives from 6‐phenyl‐4,5‐dihydropyridazin‐3(2H)‐one. They observed that all three compounds exhibited markedly analgesic activity in a hot plate model and was less active than aspirin 100 mg·kg−1, as a reference drug. These results proved that different substituted pyridazinone compounds play a critical role in the analgesic activity. Alternatively, Vergelli et al. (2015) identified a large number of potent compounds, with a pyridazine scaffold and the most interesting compound was ET1 (4‐amino‐6‐methyl‐2‐[3‐(4‐p‐tolylpiperazin‐1‐yl)propyl]‐5‐vinylpyridazin‐3(2H)‐one). They synthesized and tested new pyridazine derivatives such as ET1 for their analgesic activity in the writhing test. Administration of ET1 (20–40 mg·kg−1 p.o.) reduced the number of abdominal constrictions by more than 47%. The analgesia induced by the ET1 was completely prevented by pretreatment with yohimbine (α2‐adrenoceptor antagonist), confirming the involvement of the adrenergic system in the mechanism of action of these new compounds. Similarly, a recent study demonstrated that application of ‐(3,4‐dichlorophenyl)‐6‐(3‐(piperidin‐1yl)propoxy)pyridazin‐3(2H)‐one (compound 54) exhibited dose‐dependent antiallodynic properties in the mouse formalin model and rat chronic constriction injury model of neuropathic pain. In addition, no motor impairments were found in the rotarod tests and no sedative side effect was evident in locomotor activity tests. This suggests compound 54 is a member of a novel class of candidate drugs for the treatment of neuropathic pain (Cao et al., 2016). To elucidate the mechanism of action of these compounds, further studies are needed.
Although the primary goal of pain management in clinical practice is to relieve pain using a single agent, the reality is that monotherapy rarely provides adequate relief from chronic neuropathic pain. In these complex and refractory situations, combination therapy is needed. Combination therapy with two or more agents with different modes of action at suboptimal doses might provide synergistic effects required for optimal pain relief without compromising the side‐effect profile of each agent (Jackson, 2006).
7. CONCLUSION
Neuropathic pain treatment is a challenge for which only a limited number of effective therapeutic options are available. However, the α2‐adrenoceptor, a novel pharmacological target, has emerged as a key player in the pathophysiological nervous pathways underlying neuropathic pain. In the current review, we discussed the mechanisms of spinal adrenergic pain modulation, and how intrinsic adrenergic pathways and an α2‐adrenoceptor agonist at the spinal level have the ability to suppress pain perception. Nerve injury can induce neuroplasticity in the brainstem‐originated descending adrenergic pathway and spinal cord, including increased spinal NA content, to counteract these pathological states, but there are still some gaps that must be filled. Accordingly, groundbreaking studies have been trying to develop new approaches, where the methodological tools available will be important in helping to understand the neuroplasticity after nerve injury. We tried to explain how current α2‐adrenoceptor agonists as well as agents that block the reuptake of NA function in the treatment of chronic neuropathic pain states such as chronic postsurgical pain due to nerve damage. These agents may also be effective where opioids are of limited use due to inadequate pain relief or adverse effects. However, large randomized controlled trials are required to firmly identify the analgesic role of α2‐adrenoceptor agonist in neuropathic pain states. In addition to their spinal action, it should also be remembered that α2‐adrenoceptor agonists may also have a significant supraspinal mechanism of action, which may act on the psychological component of pain perception, thereby allowing patients to better cope with their pain. The heterogeneity of neuropathic pain conditions as well as the complexity and multiplicity of underlying pathophysiological mechanisms has made it difficult to identify tractable targets. Accordingly, targeting a single target may not lead to significant pain relief across different populations.
7.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017; Alexander, Striessnig et al., 2017).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENT
This study was supported by Neuroscience Sciences Research Center, Baqiyatallah University of Medical Sciences, Tehran, Iran.
Bahari Z, Meftahi GH. Spinal α2‐adrenoceptors and neuropathic pain modulation; therapeutic target. Br J Pharmacol. 2019;176:2366–2381. 10.1111/bph.14580
REFERENCES
- Afilalo, M. , & Morlion, B. (2013). Efficacy of tapentadol ER for managing moderate to severe chronic pain. Pain Physician, 16(1), 27–40. [PubMed] [Google Scholar]
- Alexander, S. P. H. , Benson, H. E. , Faccenda, E. , Pawson, A. J. , Sharman, J. L. , Spedding, M. , … CGTP Collaborators (2013). The concise guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. British Journal of Pharmacology, 170(8), 1459–1581. 10.1111/bph.12445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Davenport, A. P. , Kelly, E. , Marrion, N. , Peters, J. A. , Benson, H. E. , … CGTP Collaborators (2015). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. British Journal of Pharmacology, 172(24), 5744–5869. 10.1111/bph.13348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174(S1), S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174(S1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Striessnig, J. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. British Journal of Pharmacology, 174(S1), S160–S194. 10.1111/bph.13884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asif, M. , Singh, D. , & Singh, A. (2011). Analgesic activity of some 6‐phenyl‐4‐substituted benzylidene tetrahydro pyridazin‐3 (2H)‐ones. Global Journal of Pharmacology, 5(1), 18–22. [Google Scholar]
- Attal, N. , & Bouhassira, D. (2015). Pharmacotherapy of neuropathic pain: Which drugs, which treatment algorithms? Pain, 156, S104–S114. 10.1097/01.j.pain.0000460358.01998.15 [DOI] [PubMed] [Google Scholar]
- Baba, H. , Shimoji, K. , & Yoshimura, M. (2000). Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (Part 1) effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology: The Journal of the American Society of Anesthesiologists, 92(2), 473–473, 484. 10.1097/00000542-200002000-00030 [DOI] [PubMed] [Google Scholar]
- Bahari, Z. , Manaheji, H. , Dargahi, L. , Daniali, S. , Norozian, M. , Meftahi, G. H. , & Sadeghi, M. (2015). Time profile of nNOS expression in the spinal dorsal horn after L 5 spinal root transection in rats. Neurophysiology, 47(4), 287–294. 10.1007/s11062-015-9535-9 [DOI] [Google Scholar]
- Bahari, Z. , Manaheji, H. , Hosseinmardi, N. , Meftahi, G. H. , Sadeghi, M. , Danialy, S. , & Noorbakhsh, S. M. (2014). Induction of spinal long‐term synaptic potentiation is sensitive to inhibition of neuronal NOS in L5 spinal nerve‐transected rats. EXCLI Journal, 13, 751–760. [PMC free article] [PubMed] [Google Scholar]
- Bahari, Z. , Sadr, S. S. , Meftahi, G. H. , Ghasemi, M. , Manaheji, H. , Mohammadi, A. , & Mehranfard, N. (2015). Nerve injury‐induced plasticity in the nociceptive pathways. Archives of Neuroscience, 2(2), e18214. [Google Scholar]
- Bannister, K. , & Dickenson, A. H. (2016). What do monoamines do in pain modulation? Current Opinion in Supportive and Palliative Care, 10(2), 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister, K. , Patel, R. , Goncalves, L. , Townson, L. , & Dickenson, A. H. (2015). Diffuse noxious inhibitory controls and nerve injury: Restoring an imbalance between descending monoamine inhibitions and facilitations. Pain, 156(9), 1803–1811. [DOI] [PubMed] [Google Scholar]
- Bantel, C. , Eisenach, J. C. , Duflo, F. , Tobin, J. R. , & Childers, S. R. (2005). Spinal nerve ligation increases α2‐adrenergic receptor, G‐protein coupling in the spinal cord. Brain Research, 038, 76–82. [DOI] [PubMed] [Google Scholar]
- Baron, R. (2006). Mechanisms of disease: Neuropathic pain—A clinical perspective. Nature Reviews Neurology, 2(2), 95. [DOI] [PubMed] [Google Scholar]
- Baron, R. , Binder, A. , & Wasner, G. (2010). Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. The Lancet Neurology, 9(8), 807–819. 10.1016/S1474-4422(10)70143-5 [DOI] [PubMed] [Google Scholar]
- Baron, R. , Kern, U. , Müller, M. , Dubois, C. , Falke, D. , & Steigerwald, I. (2015). Effectiveness and tolerability of a moderate dose of tapentadol prolonged release for managing severe, chronic low back pain with a neuropathic component: An open‐label continuation arm of a randomized phase 3b study. Pain Practice, 15(5), 471–486. 10.1111/papr.12199 [DOI] [PubMed] [Google Scholar]
- Bassani, A. S. , & Banov, D. (2016). Evaluation of the percutaneous absorption of ketamine HCl, gabapentin, clonidine HCl, and baclofen, in compounded transdermal pain formulations, using the Franz finite dose model. Pain Medicine, 17(2), 230–238. 10.1111/pme.12899 [DOI] [PubMed] [Google Scholar]
- Bee, L. A. , Bannister, K. , Rahman, W. , & Dickenson, A. H. (2011). Mu‐opioid and noradrenergic α2‐adrenoceptor contributions to the effects of tapentadol on spinal electrophysiological measures of nociception in nerve‐injured rats. Pain, 152(1), 131–139. 10.1016/j.pain.2010.10.004 [DOI] [PubMed] [Google Scholar]
- Bektas, N. , Nemutlu, D. , & Arslan, R. (2015). The imidazoline receptors and ligands in pain modulation. Indian Journal of Pharmacology, 47(5), 472–478. 10.4103/0253-7613.165196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti, N. , Dontukurthy, S. , Bala, I. , & Singh, G. (2013). Postoperative analgesic effect of intravenous (iv) clonidine compared with clonidine administration in wound infiltration for open cholecystectomy. British Journal of Anaesthesia, 111(4), 656–661. 10.1093/bja/aet130 [DOI] [PubMed] [Google Scholar]
- Biancalani, C. , Giovannoni, M. P. , Pieretti, S. , Cesari, N. , Graziano, A. , Vergelli, C. , … Dal Piaz, V. (2009). Further studies on arylpiperazinyl alkyl pyridazinones: Discovery of an exceptionally potent, orally active, antinociceptive agent in thermally induced pain. Journal of Medicinal Chemistry, 52(23), 7397–7409. 10.1021/jm900458r [DOI] [PubMed] [Google Scholar]
- Bitner, R. S. , Nikkel, A. L. , Curzon, P. , Arneric, S. P. , Bannon, A. W. , & Decker, M. W. (1998). Role of the nucleus raphe magnus in antinociception produced by ABT‐594: Immediate early gene responses possibly linked to neuronal nicotinic acetylcholine receptors on serotonergic neurons. Journal of Neuroscience, 18(14), 5426–5432. 10.1523/JNEUROSCI.18-14-05426.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budai, D. , Harasawa, I. , & Fields, H. L. (1998). Midbrain periaqueductal gray (PAG) inhibits nociceptive inputs to sacral dorsal horn nociceptive neurons through α2‐adrenergic receptors. Journal of Neurophysiology, 80(5), 2244–2254. 10.1152/jn.1998.80.5.2244 [DOI] [PubMed] [Google Scholar]
- Cao, X. , Chen, Y. , Zhang, Y. , Lan, Y. , Zhang, J. , Xu, X. , … Zhang, G. (2016). Synthesis and biological evaluation of novel σ1 receptor ligands for treating neuropathic pain: 6‐Hydroxypyridazinones. Journal of Medicinal Chemistry, 59(7), 2942–2961. 10.1021/acs.jmedchem.5b01416 [DOI] [PubMed] [Google Scholar]
- Casale, R. , Symeonidou, Z. , & Bartolo, M. (2017). Topical treatments for localized neuropathic pain. Current Pain and Headache Reports, 21(3), 15 10.1007/s11916-017-0615-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesari, N. , Biancalani, C. , Vergelli, C. , Dal Piaz, V. , Graziano, A. , Biagini, P. , … Giovannoni, M. P. (2006). Arylpiperazinylalkylpyridazinones and analogues as potent and orally active antinociceptive agents: Synthesis and studies on mechanism of action. Journal of Medicinal Chemistry, 49(26), 7826–7835. 10.1021/jm060743g [DOI] [PubMed] [Google Scholar]
- Chabot‐Doré, A. J. , Schuster, D. J. , Stone, L. S. , & Wilcox, G. L. (2015). Analgesic synergy between opioid and α2‐adrenoceptors. British Journal of Pharmacology, 172(2), 388–402. 10.1111/bph.12695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, S. , Elvezio, V. , Kaczocha, M. , Rebecchi, M. , & Puopolo, M. (2017). Presynaptic inhibition of transient receptor potential vanilloid type 1 (TRPV1) receptors by noradrenaline in nociceptive neurons. The Journal of Physiology, 595(8), 2639–2660. 10.1113/JP273455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. L. , Li, H. , Xing, X. H. , Guan, H. S. , Zhang, J. H. , & Zhao, J. W. (2015). Effect of TRPV1 gene mutation on bronchial asthma in children before and after treatment. Allergy & Asthma Proceedings, 36(2), 29–36. 10.2500/aap.2015.36.3828 [DOI] [PubMed] [Google Scholar]
- Chen, S. R. , Chen, H. , Yuan, W. X. , & Pan, H. L. (2011). Increased presynaptic and postsynaptic α2‐adrenoceptor activity in the spinal dorsal horn in painful diabetic neuropathy. Journal of Pharmacology and Experimental Therapeutics, 337(1), 285–292. 10.1124/jpet.110.176586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirila, A. M. , Brown, T. E. , Bishop, R. A. , Bellono, N. W. , Pucci, F. G. and Kauer, J. A. (2014). Long‐term potentiation of glycinergic synapses triggered by interleukin 1β. Proceedings of the National Academy of Sciences, 201401013. [DOI] [PMC free article] [PubMed]
- Christoph, T. , Gillen, C. , Mika, J. , Grünweller, A. , Schäfer, M. K. H. , Schiene, K. , … Kurreck, J. (2007). Antinociceptive effect of antisense oligonucleotides against the vanilloid receptor VR1/TRPV1 . Neurochemistry International, 50(1), 281–290. 10.1016/j.neuint.2006.08.017 [DOI] [PubMed] [Google Scholar]
- Coull, J. A. , Beggs, S. , Boudreau, D. , Boivin, D. , Tsuda, M. , Inoue, K. , … De Koninc, Y. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature, 438(7070), 1017–1021. 10.1038/nature04223 [DOI] [PubMed] [Google Scholar]
- Del Bello, F. , Cilia, A. , Carrieri, A. , Fasano, D. C. , Ghelardini, C. , Di Cesare Mannelli, L. , … Pigini, M. (2016). The versatile 2‐substituted imidazoline nucleus as a structural motif of ligands directed to the serotonin 5‐HT1A receptor. ChemMedChem, 11(20), 2287–2298. 10.1002/cmdc.201600383 [DOI] [PubMed] [Google Scholar]
- Di Cesare, M. , Micheli, L. , Crocetti, L. , Paola Giovannoni, M. , Vergelli, C. , & Ghelardini, C. (2017). α2 Adrenoceptor: A target for neuropathic pain treatment. Mini Reviews in Medicinal Chemistry, 17(2), 95–107. [DOI] [PubMed] [Google Scholar]
- D'mello, R. , & Dickenson, A. H. (2008). Spinal cord mechanisms of pain. British Journal of Anaesthesia, 101(1), 8–16. 10.1093/bja/aen088 [DOI] [PubMed] [Google Scholar]
- Donaldson, L. F. , & Beazley‐Long, N. (2016). Alternative RNA splicing: Contribution to pain and potential therapeutic strategy. Drug Discovery Today, 21(11), 1787–1798. 10.1016/j.drudis.2016.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin, A. E. , & Patapoutian, A. (2010). Nociceptors: The sensors of the pain pathway. The Journal of Clinical Investigation, 120(11), 3760–3772. 10.1172/JCI42843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenach, J. C. , Zhang, Y. , & Duflo, F. (2005). α2‐Adrenoceptors inhibit the intracellular, Ca2+ response to electrical stimulation in normal and injured sensory neurons, with increased inhibition of calcitonin gene‐related peptide expressing neurons after injury. Neuroscience, 131, 189–197. 10.1016/j.neuroscience.2004.10.017 [DOI] [PubMed] [Google Scholar]
- Engelman, E. , & Marsala, C. (2012). Efficacy of adding clonidine to intrathecal morphine in acute postoperative pain: Meta‐analysis. British Journal of Anaesthesia, 110(1), 21–27. 10.1093/bja/aes344 [DOI] [PubMed] [Google Scholar]
- Fairbanks, C. A. , Stone, L. S. , & Wilcox, G. L. (2009). Pharmacological profiles of α2 adrenergic receptor agonists identified using genetically altered mice and isobolographic analysis. Pharmacology & Therapeutics, 123(2), 224–238. 10.1016/j.pharmthera.2009.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fargin, A. , Raymond, J. R. , Lohse, M. J. , Kobilka, B. K. , Caron, M. G. , & Lefkowitz, R. J. (1988). The genomic clone G‐21 which resembles a β‐adrenergic receptor sequence encodes the 5‐HT1A receptor. Nature, 335(6188), 358–360. 10.1038/335358a0 [DOI] [PubMed] [Google Scholar]
- Finnerup, N. B. , Attal, N. , Haroutounian, S. , McNicol, E. , Baron, R. , Dworkin, R. H. , … Wallace, M. (2015). Pharmacotherapy for neuropathic pain in adults: A systematic review and meta‐analysis. The Lancet Neurology, 14(2), 162–173. 10.1016/S1474-4422(14)70251-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiè, E. , Villani, V. , Terrenato, I. , & Pace, A. (2017). Tapentadol in neuropathic pain cancer patients: A prospective open label study. Neurological Sciences, 38(10), 1747–1752. 10.1007/s10072-017-3035-1 [DOI] [PubMed] [Google Scholar]
- Gassner, M. , Ruscheweyh, R. , & Sandkühler, J. (2009). Direct excitation of spinal GABAergic interneurons by noradrenaline. Pain, 145(1–2), 204–210. [DOI] [PubMed] [Google Scholar]
- Gentili, F. , Ghelfi, F. , Giannella, M. , Piergentili, A. , Pigini, M. , Quaglia, W. , … Carrieri, A. (2004). α2‐Adrenoreceptors profile modulation. 2. Biphenyline analogues as tools for selective activation of the α2C‐subtype. Journal of Medicinal Chemistry, 47(25), 6160–6173. 10.1021/jm0408215 [DOI] [PubMed] [Google Scholar]
- Gilsbach, R. , & Hein, L. (2012). Are the pharmacology and physiology of α2‐adrenoceptors determined by α2‐heteroreceptors and autoreceptors respectively? British Journal of Pharmacology, 165(1), 90–102. 10.1111/j.1476-5381.2011.01533.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilsbach, R. , Röser, C. , Beetz, N. , Brede, M. , Hadamek, K. , Haubold, M. , … Hein, L. (2009). Genetic dissection of α2‐adrenoceptor functions in adrenergic versus non‐adrenergic cells. Molecular Pharmacology, 75, 1160–1170. 10.1124/mol.109.054544 [DOI] [PubMed] [Google Scholar]
- Giovannitti, J. A. Jr. , Thoms, S. M. , & Crawford, J. J. (2015). α‐2 Adrenergic receptor agonists: A review of current clinical applications. Anesthesia Progress, 62(1), 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guneli, E. , Yavasoglu, N. U. K. , Apaydin, S. , Uyar, M. , & Uyar, M. (2007). Analysis of the antinociceptive effect of systemic administration of tramadol and dexmedetomidine combination on rat models of acute and neuropathic pain. Pharmacology Biochemistry and Behavior, 88(1), 9–17. [DOI] [PubMed] [Google Scholar]
- Gyires, K. , Zádori, Z. S. , Török, T. , & Mátyus, P. (2009). α2‐Adrenoceptor subtypes‐mediated physiological, pharmacological actions. Neurochemistry International, 55(7), 447–453. [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida, K. , & Eisenach, J. C. (2011). A tropomyosine receptor kinase inhibitor blocks spinal neuroplasticity essential for the anti‐hypersensitivity effects of gabapentin and clonidine in rats with peripheral nerve injury. The Journal of Pain: Official Journal of the American Pain Society, 12(1), 94–100. 10.1016/j.jpain.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida, K. I. , Clayton, B. A. , Johnson, J. E. , & Eisenach, J. C. (2008). Brain derived nerve growth factor induces spinal noradrenergic fiber sprouting and enhances clonidine analgesia following nerve injury in rats. Pain, 136(3), 348–355. 10.1016/j.pain.2007.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida, K. I. , & Eisenach, J. C. (2010). Spinal α2‐adrenoceptor‐mediated analgesia in neuropathic pain reflects brain‐derived nerve growth factor and changes in spinal cholinergic neuronal function. Anesthesiology: The Journal of the American Society of Anesthesiologists, 113(2), 406–412. 10.1097/ALN.0b013e3181de6d2c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida, K. I. , Peters, C. M. , Gutierrez, S. , & Eisenach, J. C. (2012). Depletion of endogenous noradrenaline does not prevent spinal cord plasticity following peripheral nerve injury. The Journal of Pain, 13(1), 49–57. 10.1016/j.jpain.2011.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidary, N. , Sahraei, H. , Afarinesh, M. R. , Bahari, Z. , & Meftahi, G. H. (2018). Investigating the inhibition of NMDA glutamate receptors in the basolateral nucleus of the amygdala on the pain and inflammation induced by formalin in male Wistar rats. Frontiers in Biology, 13(2), 149–155. 10.1007/s11515-018-1491-5 [DOI] [Google Scholar]
- Holbech, J. V. , Jung, A. , Jonsson, T. , Wanning, M. , Bredahl, C. , & Bach, F. W. (2017). Combination treatment of neuropathic pain: Danish expert recommendations based on a Delphi process. Journal of Pain Research, 10, 1467–1475. 10.2147/JPR.S138099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino, H. , Obata, H. , & Saito, S. (2015). Antihyperalgesic effect of duloxetine and amitriptyline in rats after peripheral nerve injury: Influence of descending noradrenergic plasticity. Neuroscience Letters, 602, 62–67. 10.1016/j.neulet.2015.06.041 [DOI] [PubMed] [Google Scholar]
- Iida, H. , Ohata, H. , Iida, M. , Watanabe, Y. , & Dohi, S. (1999). Direct effects of α1‐and α2‐adrenergicagonists on spinal and cerebral pial vessels in dogs. Anesthesiology: The Journal of the American Society of Anesthesiologists, 91(2), 479–485. [DOI] [PubMed] [Google Scholar]
- Iyengar, S. , Webster, A. A. , Hemrick‐Luecke, S. K. , Xu, J. Y. , & Simmons, R. M. A. (2004). Efficacy of duloxetine, a potent and balanced serotonin‐norepinephrine reuptake inhibitor in persistent pain models in rats. Journal of Pharmacology and Experimental Therapeutics, 311(2), 576–584. [DOI] [PubMed] [Google Scholar]
- Jackson, K. C. (2006). Pharmacotherapy for neuropathic pain. Pain Practice, 6(1), 27–33. 10.1111/j.1533-2500.2006.00055.x [DOI] [PubMed] [Google Scholar]
- Jacobsen, L. M. , Eriksen, G. S. , Pedersen, L. M. , & Gjerstad, J. (2010). Catechol‐O‐methyltransferase (COMT) inhibition reduces spinal nociceptive activity. Neuroscience Letters, 473(3), 212–215. 10.1016/j.neulet.2010.02.049 [DOI] [PubMed] [Google Scholar]
- Jasmin, L. , Tien, D. , Weinshenker, D. , Palmiter, R. D. , Green, P. G. , Janni, G. , & Ohara, P. T. (2002). The NK1 receptor mediates both the hyperalgesia and the resistance to morphine in mice lacking noradrenaline. Proceedings of the National Academy of Sciences, 99(2), 1029–1034. 10.1073/pnas.012598599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalapesi, F. B. , Coroneo, M. T. , & Hill, M. A. (2005). Human ganglion cells express the α‐2 adrenergic receptor: Relevance to neuroprotection. The British Journal of Ophthalmology, 89(6), 758–763. 10.1136/bjo.2004.053025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalipatnapu, S. , & Chattopadhyay, A. (2007). Membrane organization and function of the serotonin 1A receptor. Cellular and Molecular Neurobiology, 27(8), 1097–1116. 10.1007/s10571-007-9189-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki, Y. , Kumamoto, E. , Furue, H. , & Yoshimura, M. (2003). α2Adrenoceptor‐mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology: The Journal of the American Society of Anesthesiologists, 98(3), 682–689. 10.1097/00000542-200303000-00016 [DOI] [PubMed] [Google Scholar]
- Kiani, J. , Sajedi, F. , Nasrollahi, S. A. , & Esna‐Ashari, F. (2015). A randomized clinical trial of efficacy and safety of the topical clonidine and capsaicin in the treatment of painful diabetic neuropathy. Journal of Research in Medical Sciences, 20(4), 359–363. [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. K. , Park, J. Y. , & Hwang, J. H. (2009). Comparison of antiallodynic effect of intrathecal morphine, brimonidine and rilmenidine between neuritis and ligation injury induced neuropathic pain. Korean Journal of Anesthesiology, 56(4), 425–432. 10.4097/kjae.2009.56.4.425 [DOI] [PubMed] [Google Scholar]
- Kimura, M. , Saito, S. , & Obata, H. (2012). Dexmedetomidine decreases hyperalgesia in neuropathic pain by increasing acetylcholine in the spinal cord. Neuroscience Letters, 529(1), 70–74. 10.1016/j.neulet.2012.08.008 [DOI] [PubMed] [Google Scholar]
- Kronschläger, M. T. , Drdla‐Schutting, R. , Gassner, M. , Honsek, S. D. , Teuchmann, H. L. , & Sandkühler, J. (2016). Gliogenic LTP spreads widely in nociceptive pathways. Science, 354(6316), 1144–1148. 10.1126/science.aah5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, G. , Park, H. , Park, H. S. , & Lee, J. G. (2012). Modulation of α 1 adrenergic receptors on urinary bladder in rat spinal cord injury model. International Neurourology Journal, 16(2), 62–68. 10.5213/inj.2012.16.2.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , & Eisenach, J. C. (2001). α2A‐Adrenoceptor stimulation reduces capsaicin‐induced glutamate release from spinal cord synaptosomes. Journal of Pharmacology and Experimental Therapeutics, 299(3), 939–944. [PubMed] [Google Scholar]
- Li, X. , Guo, Q. , Gao, J. , Yang, J. , Zhang, W. , Liang, Y. , … Zhang, Y. (2013). The adenylyl cyclase inhibitor MDL‐12,330 A potentiates insulin secretion via blockade of voltage‐dependent K+ channels in pancreatic beta cells. PLoS One, 8(10), e77934 10.1371/journal.pone.0077934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, T. F. , Yeh, Y. C. , Lin, F. S. , Wang, Y. P. , Lin, C. J. , Sun, W. Z. , & Fan, S. Z. (2008). Effect of combining dexmedetomidine and morphine for intravenous patient‐controlled analgesia. British Journal of Anaesthesia, 102(1), 117–122. 10.1093/bja/aen320 [DOI] [PubMed] [Google Scholar]
- Llorca‐Torralba, M. , Borges, G. , Neto, F. , Mico, J. A. , & Berrocoso, E. (2016). Noradrenergic locus coeruleus pathways in pain modulation. Neuroscience, 338, 93–113. 10.1016/j.neuroscience.2016.05.057 [DOI] [PubMed] [Google Scholar]
- Lopez‐Canul, M. , Palazzo, E. , Dominguez‐Lopez, S. , Luongo, L. , Lacoste, B. , Comai, S. , … Gobbi, G. (2015). Selective melatonin MT2 receptor ligands relieve neuropathic pain through modulation of brainstem descending antinociceptive pathways. Pain, 156(2), 305–317. 10.1097/01.j.pain.0000460311.71572.5f [DOI] [PubMed] [Google Scholar]
- Malmberg, A. B. , Hedley, L. R. , Jasper, J. R. , Hunter, J. C. , & Basbaum, A. I. (2001). Contribution of α2 receptor subtypes to nerve injury‐induced pain and its regulation by dexmedetomidine. British Journal of Pharmacology, 132(8), 1827–1836. 10.1038/sj.bjp.0704032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cesare Mannelli, L. , Ghelardini, C. , Micheli, L. , Del Bello, F. , Giannella, M. , Piergentili, A. , … Quaglia, W. (2017). Synergic stimulation of serotonin 5‐HT1A receptor and α2‐adrenoceptors for neuropathic pain relief: Preclinical effects of 2‐substituted imidazoline derivatives. European Journal of Pharmacology, 810, 128–133. 10.1016/j.ejphar.2017.06.023 [DOI] [PubMed] [Google Scholar]
- Matsushita, Y. , Manabe, M. , Kitamura, N. , & Shibuya, I. (2018). Adrenergic receptors inhibit TRPV1 activity in the dorsal root ganglion neurons of rats. PLoS One, 13(1), e0191032 10.1371/journal.pone.0191032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meske, D. S. , Xie, J. Y. , Oyarzo, J. , Badghisi, H. , Ossipov, M. H. , & Porreca, F. (2014). Opioid and noradrenergic contributions of tapentadol in experimental neuropathic pain. Neuroscience Letters, 562, 91–96. 10.1016/j.neulet.2013.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljanich, G. , Rauck, R. , & Saulino, M. (2013). Spinal mechanisms of pain and analgesia. Pain Practice, 13(2), 114–130. 10.1111/j.1533-2500.2012.00564.x [DOI] [PubMed] [Google Scholar]
- Millan, M. J. (2002). Descending control of pain. Progress in Neurobiology, 66(6), 355–474. 10.1016/S0301-0082(02)00009-6 [DOI] [PubMed] [Google Scholar]
- Morioka, N. , Tanabe, H. , Inoue, A. , Dohi, T. , & Nakata, Y. (2009). Noradrenaline reduces the ATP‐stimulated phosphorylation of p38 MAP kinase via β‐adrenergic receptors–cAMP–protein kinase A‐dependent mechanism in cultured rat spinal microglia. Neurochemistry International, 55(4), 226–234. 10.1016/j.neuint.2009.03.004 [DOI] [PubMed] [Google Scholar]
- Muthuraman, A. , Singh, N. , Singh Jaggi, A. , & Ramesh, M. (2014). Drug therapy of neuropathic pain: Current developments and future perspectives. Current Drug Targets, 15(2), 210–253. [PubMed] [Google Scholar]
- Naaz, S. , & Ozair, E. (2014). Dexmedetomidine in current anaesthesia practice—A review. Journal of Clinical and Diagnostic Research, 8(10), 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeson, R. , & Goodchild, C. S. (2002). Antinociceptive role of 5‐HT1A receptors in rat spinal cord. British Journal of Anaesthesia, 88(5), 679–684. 10.1093/bja/88.5.679 [DOI] [PubMed] [Google Scholar]
- Nakai, K. , Nakae, A. , Oba, S. , Hosokawa, K. , Mashimo, T. , & Ueda, K. (2015). Anti‐allodynic effects following intrathecal administration of α1‐and α2‐adrenergic receptor agonists in a rat model of trigeminal neuropathic pain. Medical Research Archives, 2(3). 10.18103/mra.v2i3.387 [DOI] [Google Scholar]
- Nakajima, K. , Obata, H. , Iriuchijima, N. , & Saito, S. (2012). An increase in spinal cord noradrenaline is a major contributor to the antihyperalgesic effect of antidepressants after peripheral nerve injury in the rat. Pain, 153(5), 990–997. 10.1016/j.pain.2012.01.029 [DOI] [PubMed] [Google Scholar]
- Nasirinezhad, F. , Hosseini, M. , & Salari, S. (2015). Anti‐allodynic efficacy of NMDA antagonist peptide and noradrenaline alone and in combination in rodent neuropathic pain model. The Korean Journal of Pain, 28(2), 96–104. 10.3344/kjp.2015.28.2.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Novellis, V. , Palazzo, E. , Rossi, F. , De Petrocellis, L. , Petrosino, S. , Guida, F. , … Endocannabinoid Research Group (2008). The analgesic effect of N‐arachidonoyl‐serotonin, a FAAH inhibitor and TRPV1 receptor antagonist, associated with changes in rostral ventromedial medulla and locus coeruleus cell activity in rats. Neuropharmacology, 55(7), 1105–1113. 10.1016/j.neuropharm.2008.06.023 [DOI] [PubMed] [Google Scholar]
- Obata, H. (2017). Analgesic mechanisms of antidepressants for neuropathic pain. International Journal of Molecular Sciences, 18(11), 2483 10.3390/ijms18112483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata, H. , Saito, S. , Koizuka, S. , Nishikawa, K. , & Goto, F. (2005). The monoamine‐mediated antiallodynic effects of intrathecally administered milnacipran, a serotonin noradrenaline reuptake inhibitor, in a rat model of neuropathic pain. Anesthesia & Analgesia, 100(5), 1406–1410. 10.1213/01.ANE.0000149546.97299.A2 [DOI] [PubMed] [Google Scholar]
- Ohnami, S. , Kato, A. , Ogawa, K. , Shinohara, S. , Ono, H. , & Tanabe, M. (2012). Effects of milnacipran, a 5‐HT and noradrenaline reuptake inhibitor, on C‐fibre‐evoked field potentials in spinal long‐term potentiation and neuropathic pain. British Journal of Pharmacology, 167(3), 537–547. 10.1111/j.1476-5381.2012.02007.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oladosu, F. A. , Maixner, W. , & Nackley, A. G. (2015). Alternative splicing of G‐protein coupled receptors: Relevance to pain management. Mayo Clinic Proceedings, 90(8), 1135–1151. 10.1016/j.mayocp.2015.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov, M. H. , Dussor, G. O. , & Porreca, F. (2010). Central modulation of pain. The Journal of Clinical Investigation, 120(11), 3779–3787. 10.1172/JCI43766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov, M. H. , Harris, S. , Lloyd, P. , & Messineo, E. (1990). An isobolographic analysis of the antinociceptive effect of systemically and intrathecally administered combinations of clonidine and opiates. Journal of Pharmacology and Experimental Therapeutics, 255(3), 1107–1116. [PubMed] [Google Scholar]
- Ossipov, M. H. , Morimura, K. , & Porreca, F. (2014). Descending pain modulation and chronification of pain. Current Opinion in Supportive and Palliative Care, 8(2), 143–151. 10.1097/SPC.0000000000000055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou‐Yang, H. D. , Zeng, W. A. , Li, Q. , He, W. X. , Wang, P. Z. , Lin, L. L. , … Liu, X. G. (2008). Effects of intrathecal ouabain and tizanidine injection for treatment of neuropathic pain in rats. Nan fang yi ke da xue xue bao= Journal of Southern Medical University, 28(10), 1760–1763. [PubMed] [Google Scholar]
- Özdoǧan, Ü. K. , Lähdesmäki, J. , Hakala, K. , & Scheinin, M. (2004). The involvement of α2A‐adrenoceptors in morphine analgesia, tolerance and withdrawal in mice. European Journal of Pharmacology, 497(2), 161–171. 10.1016/j.ejphar.2004.06.051 [DOI] [PubMed] [Google Scholar]
- Panczyk, K. , Golda, S. , Waszkielewicz, A. , Zelaszczyk, D. , Gunia‐Krzyzak, A. , & Marona, H. (2015). Serotonergic system and its role in epilepsy and neuropathic pain treatment: A review based on receptor ligands. Current Pharmaceutical Design, 21(13), 1723–1740. 10.2174/1381612821666141121114917 [DOI] [PubMed] [Google Scholar]
- Paqueron, X. , Conklin, D. , & Eisenach, J. C. (2003). Plasticity in action of intrathecal clonidine to mechanical but not thermal nociception after peripheral nerve injury. Anesthesiology: The Journal of the American Society of Anesthesiologists, 99(1), 199–204. 10.1097/00000542-200307000-00030 [DOI] [PubMed] [Google Scholar]
- Parent, A. J. , Tetreault, P. , Roux, M. , Belleville, K. , Longpré, J. M. , Beaudet, N. , … Sarret, P. (2016). Descending nociceptive inhibition is modulated in a time‐dependent manner in a double‐hit model of chronic/tonic pain. Neuroscience, 315, 70–78. 10.1016/j.neuroscience.2015.11.065 [DOI] [PubMed] [Google Scholar]
- Park, H. J. , Kim, Y. H. , Koh, H. J. , Park, C. S. , Kang, S. H. , Choi, J. H. , & Moon, D. E. (2012). Analgesic effects of dexmedetomidine in vincristine‐evoked painful neuropathic rats. Journal of Korean Medical Science, 27(11), 1411–1417. 10.3346/jkms.2012.27.11.1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, R. , Qu, C. , Xie, J. Y. , Porreca, F. , & Dickenson, A. H. (2018). Selective deficiencies in descending inhibitory modulation in neuropathic rats: Implications for enhancing noradrenergic tone. Pain, 159(9), 1887–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, K. , Jin, X. H. , Liu, S. L. , & Ji, F. H. (2015). Effect of intraoperative dexmedetomidine on post‐craniotomy pain. Clinical Therapeutics, 37(5), 1114–1121. 10.1016/j.clinthera.2015.02.011 [DOI] [PubMed] [Google Scholar]
- Pertovaara, A. (2004). Antinociceptive properties of fadolmidine (MPV‐2426), a novel α2‐adrenoceptor agonist. CNS Drug Reviews, 10(2), 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertovaara, A. (2006). Noradrenergic pain modulation. Progress in Neurobiology, 80(2), 53–83. 10.1016/j.pneurobio.2006.08.001 [DOI] [PubMed] [Google Scholar]
- Philipp, M. , Brede, M. , & Hein, L. (2002). Physiological significance of α2‐adrenergic receptor subtype diversity: One receptor is not enough. American Journal of Physiology‐Regulatory, Integrative and Comparative Physiology, 283(2), 287–295. [DOI] [PubMed] [Google Scholar]
- Rahman, W. , D'Mello, R. , & Dickenson, A. H. (2008). Peripheral nerve injury–induced changes in spinal α2‐adrenoceptor–mediated modulation of mechanically evoked dorsal horn neuronal responses. The Journal of Pain, 9(4), 350–359. 10.1016/j.jpain.2007.11.010 [DOI] [PubMed] [Google Scholar]
- Rahman, W. , & Dickenson, A. H. (2011). Recent developments in neuropathic pain mechanisms: Implications for treatment. Reviews in Pain, 5(2), 21–25. 10.1177/204946371100500204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, F. , Zhang, H. , Qi, C. , Gao, M. , Wang, H. , & Li, X. (2015). Blockade of transient receptor potential cation channel subfamily V member 1 promotes regeneration after sciatic nerve injury. Neural Regeneration Research, 10(8), 1324–1331. 10.4103/1673-5374.162770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl, M. S. , Schnell, S. A. , Overland, A. C. , Chabot‐Doré, A. J. , Taylor, A. M. , Ribeiro‐Da‐Silva, A. , … Stone, L. S. (2009). Coexpression of α2A‐adrenergic and δ‐opioid receptors in substance P‐containing terminals in rat dorsal horn. Journal of Comparative Neurology, 513(4), 385–398. 10.1002/cne.21982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh, D. H. , Kim, H. W. , Yoon, S. Y. , Seo, H. S. , Kwon, Y. B. , Han, H. J. , … Lee, J. H. (2008). Intrathecal clonidine suppresses phosphorylation of the N‐methyl‐D‐aspartate receptor NR1 subunit in spinal dorsal horn neurons of rats with neuropathic pain. Anesthesia & Analgesia, 107(2), 693–700. 10.1213/ane.0b013e31817e7319 [DOI] [PubMed] [Google Scholar]
- Roh, D. H. , Seo, H. S. , Yoon, S. Y. , Song, S. , Han, H. J. , Beitz, A. J. , & Lee, J. H. (2010). Activation of spinal α2 adrenoceptors, but not μ‐opioid receptors, reduces the intrathecal N‐Methyl‐d‐Aspartate‐induced increase in spinal NR1 subunit phosphorylation and nociceptive behaviors in the rat. Anesthesia & Analgesia, 110(2), 622–629. 10.1213/ANE.0b013e3181c8afc1 [DOI] [PubMed] [Google Scholar]
- Ruscheweyh, R. , Wilder‐Smith, O. , Drdla, R. , Liu, X. G. , & Sandkühler, J. (2011). Long‐term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Molecular Pain, 7(1), 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallinen, J. , Höglund, I. , Engström, M. , Lehtimäki, J. , Virtanen, R. , Sirviö, J. , … Haapalinna, A. (2007). Pharmacological characterization and CNS effects of a novel highly selective α2C‐adrenoceptor antagonist JP‐1302. British Journal of Pharmacology, 150(4), 391–402. 10.1038/sj.bjp.0707005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkühler, J. , & Gruber‐Schoffnegger, D. (2012). Hyperalgesia by synaptic long‐term potentiation (LTP): An update. Current Opinion in Pharmacology, 12(1), 18–27. 10.1016/j.coph.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel, A. , Meyer‐Friessem, C. H. , Reichl, S. U. , Zahn, P. K. , & Pogatzki‐Zahn, E. M. (2013). Is intraoperative dexmedetomidine a new option for postoperative pain treatment? A meta‐analysis of randomized controlled trials. Pain, 154, 1140–1149. 10.1016/j.pain.2013.03.029 [DOI] [PubMed] [Google Scholar]
- Schnabel, A. , Reichl, S. U. , Poepping, D. M. , Kranke, P. , Pogatzki‐Zahn, E. M. , & Zahn, P. K. (2013). Efficacy and safety of intraoperative dexmedetomidine for acute postoperative pain in children: A meta‐analysis of randomized controlled trials. Pediatric Anesthesia, 23(2), 170–179. 10.1111/pan.12030 [DOI] [PubMed] [Google Scholar]
- Schwartz, S. , Etropolski, M. S. , Shapiro, D. Y. , Rauschkolb, C. , Vinik, A. I. , Lange, B. , … Haeussler, J. (2015). A pooled analysis evaluating the efficacy and tolerability of tapentadol extended release for chronic, painful diabetic peripheral neuropathy. Clinical Drug Investigation, 35(2), 95–108. 10.1007/s40261-014-0249-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenchuk, M. R. , & Sherman, S. (2000). Effectiveness of tizanidine in neuropathic pain: An open‐label study. The Journal of Pain, 1(4), 285–292. 10.1054/jpai.2000.9435 [DOI] [PubMed] [Google Scholar]
- Solanki, S. L. , Bharti, N. A. , Batra, Y. K. , Jain, A. , Kumar, P. , & Nikhar, S. (2013). The analgesic effect of intrathecal dexmedetomidine or clonidine, with bupivacaine, in trauma patients undergoing lower limb surgery: A randomised, double‐blind study. Anaesthesia and Intensive Care, 41(1), 51–56. 10.1177/0310057X1304100110 [DOI] [PubMed] [Google Scholar]
- Stone, L. S. , German, J. P. , Kitto, K. F. , Fairbanks, C. A. , & Wilcox, G. L. (2014). Morphine and clonidine combination therapy improves therapeutic window in mice: Synergy in antinociceptive but not in sedative or cardiovascular effects. PLoS One, 9(10), e109903 10.1371/journal.pone.0109903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone, L. S. , & Wilcox, G. L. (2004). α‐2‐Adrenergic and opioid receptor additivity in rat locuscoeruleus neurons. Neuroscience Letters, 361(1–3), 265–268. 10.1016/j.neulet.2003.12.065 [DOI] [PubMed] [Google Scholar]
- Sugiyama, Y. , Kataoka, T. , Tasaki, Y. , Kondo, Y. , Sato, N. , Naiki, T. , … Kimura, K. (2018). Efficacy of tapentadol for first‐line opioid‐resistant neuropathic pain in Japan. Japanese Journal of Clinical Oncology, 48(4), 362–366. 10.1093/jjco/hyy023 [DOI] [PubMed] [Google Scholar]
- Suzuki, R. , Rygh, L. J. , & Dickenson, A. H. (2004). Bad news from the brain: Descending 5‐HT pathways that control spinal pain processing. Trends in Pharmacological Sciences, 25(12), 613–617. 10.1016/j.tips.2004.10.002 [DOI] [PubMed] [Google Scholar]
- Taylor, B. K. , & Westlund, K. N. (2017). The noradrenergic locus coeruleus as a chronic pain generator. Journal of Neuroscience Research, 95(6), 1336–1346. 10.1002/jnr.23956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang, T. , Beggs, S. , Wan, X. , & Salter, M. W. (2009). P2X4‐receptor‐mediated synthesis and release of brain‐derived neurotrophic factor in microglia is dependent on calcium and p38‐mitogen‐activated protein kinase activation. Journal of Neuroscience, 29(11), 3518–3528. 10.1523/JNEUROSCI.5714-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschentke, T. M. , Christoph, T. , Kögel, B. , Schiene, K. , Hennies, H. H. , Englberger, W. , … De Vry, J. (2007). (−)‐(1R, 2R)‐3‐(3‐dimethylamino‐1‐ethyl‐2‐methyl‐propyl)‐phenol hydrochloride (tapentadol HCl): A novel μ‐opioid receptor agonist/norepinephrine reuptake inhibitor with broad‐spectrum analgesic properties. Journal of Pharmacology and Experimental Therapeutics, 323(1), 265–276. 10.1124/jpet.107.126052 [DOI] [PubMed] [Google Scholar]
- Tzschentke, T. M. , De Vry, J. , Terlinden, R. , Hennies, H. H. , Lange, C. , Strassburger, W. , … Friderichs, E. (2006). Tapentadol hydrochloride. Drugs of the Future, 31(12), 1053–1061. 10.1358/dof.2006.031.12.1047744 [DOI] [Google Scholar]
- Unterberger, U. , Moskvina, E. , Scholze, T. , Freissmuth, M. , & Boehm, S. (2002). Inhibition of adenylyl cyclase by neuronal P2Y receptors. British Journal of Pharmacology, 135(3), 673–684. 10.1038/sj.bjp.0704514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uslusoy, F. , Nazıroğlu, M. , & Çiğ, B. (2017). Inhibition of the TRPM2 and TRPV1 channels through Hypericum perforatum in sciatic nerve injury‐induced rats demonstrates their key role in apoptosis and mitochondrial oxidative stress of sciatic nerve and dorsal root ganglion. Frontiers in Physiology, 8, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachhani, A. , Barvaliya, M. , Naik, V. , & Tripathi, C. B. (2014). Cardiovascular abnormalities with single dose of tapentadol. Journal of Postgraduate Medicine, 60(2), 189–191. 10.4103/0022-3859.132341 [DOI] [PubMed] [Google Scholar]
- Vadivelu, N. , Kai, A. , Maslin, B. , Kodumudi, G. , Legler, A. , & Berger, J. M. (2015). Tapentadol extended release in the management of peripheral diabetic neuropathic pain. Therapeutics and Clinical Risk Management, 11, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaegter, H. B. , & Graven‐Nielsen, T. (2016). Pain modulatory phenotypes differentiate subgroups with different clinical and experimental pain sensitivity. Pain, 157(7), 1480–1488. 10.1097/j.pain.0000000000000543 [DOI] [PubMed] [Google Scholar]
- Vergelli, C. , Ciciani, G. , Cilibrizzi, A. , Crocetti, L. , Di Cesare Mannelli, L. , Ghelardini, C. , … Giovannoni, M. P. (2015). Synthesis of five and six‐membered heterocycles bearing an arylpiperazinylalkyl side chain as orally active antinociceptive agents. Bioorganic & Medicinal Chemistry, 23(19), 6237–6245. 10.1016/j.bmc.2015.08.043 [DOI] [PubMed] [Google Scholar]
- Vorobeychik, Y. , Gordin, V. , Mao, J. , & Chen, L. (2011). Combination therapy for neuropathic pain. CNS Drugs, 25(12), 1023–1034. 10.2165/11596280-000000000-00000 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Zhang, H. , Feng, Y. P. , Meng, H. , Wu, L. P. , Wang, W. , … Li, Y.‐Q. (2014). Morphological evidence for a neurotensinergic periaqueductal gray‐rostral ventromedial medulla‐spinal dorsal horn descending pathway in rat. Frontiers in Neuroanatomy, 8, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, H. , & Pertovaara, A. (2006). Spinal and pontine α2‐adrenoceptors have opposite effects on pain‐related behavior in the neuropathic rat. European Journal of Pharmacology, 551(1–3), 41–49. 10.1016/j.ejphar.2006.08.064 [DOI] [PubMed] [Google Scholar]
- Willoughby, D. , Wachten, S. , Masada, N. , & Cooper, D. M. (2010). Direct demonstration of discrete Ca2+ microdomains associated with different isoforms of adenylyl cyclase. Journal of Cell Science, 123(1), 107–117. 10.1242/jcs.062067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrzosek, A. , Woron, J. , Dobrogowski, J. , Jakowicka‐Wordliczek, J. , & Wordliczek, J. (2015). Topical clonidine for neuropathic pain. Cochrane Database of Systematic Reviews, 8, CD010967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav, G. , Jain, G. , Samprathi, A. , Baghel, A. , & Singh, D. K. (2016). Role of preemptive tapentadol in reduction of postoperative analgesic requirements after laparoscopic cholecystectomy. Journal of Anaesthesiology, Clinical Pharmacology, 32(4), 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ydemann, M. , Nielsen, B. N. , Henneberg, S. , Jakobsen, J. C. , Wetterslev, J. , Lauritsen, T. , … Reinhardt, S. (2018). Intraoperative clonidine for prevention of postoperative agitation in children anaesthetised with sevoflurane (PREVENT AGITATION): A randomised, placebo‐controlled, double‐blind trial. The Lancet Child & Adolescent Health, 2(1), 15–24. 10.1016/S2352-4642(17)30127-X [DOI] [PubMed] [Google Scholar]
- Yekkirala, A. S. , Roberson, D. P. , Bean, B. P. , & Woolf, C. J. (2017). Breaking barriers to novel analgesic drug development. Nature Reviews Drug Discovery, 16(8), 545–564. 10.1038/nrd.2017.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo, J. H. , Yoon, S. Y. , Kim, S. J. , Oh, S. B. , Lee, J. H. , Beitz, A. J. , & Roh, D. H. (2016). Clonidine, an α‐2 adrenoceptor agonist relieves mechanical allodynia in oxaliplatin‐induced neuropathic mice; potentiation by spinal p38 MAPK inhibition without motor dysfunction and hypotension. International Journal of Cancer, 138(10), 2466–2476. 10.1002/ijc.29980 [DOI] [PubMed] [Google Scholar]
- Yoon, S. Y. , Kang, S. Y. , Kim, H. W. , Kim, H. C. , & Roh, D. H. (2015). Clonidine reduces nociceptive responses in mouse orofacial formalin model: Potentiation by σ‐1 receptor antagonist BD1047 without impaired motor coordination. Biological and Pharmaceutical Bulletin, 38(9), 1320–1327. 10.1248/bpb.b15-00183 [DOI] [PubMed] [Google Scholar]
- Zakir, H. M. , Mostafeezur, R. M. , Suzuki, A. , Hitomi, S. , Suzuki, I. , Maeda, T. , … Kitagawa, J. (2012). Expression of TRPV1 channels after nerve injury provides an essential delivery tool for neuropathic pain attenuation. PLoS One, 7(9), e44023 10.1371/journal.pone.0044023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, T. T. , Wu, J. R. , Chen, Z. Y. , Liu, Z. X. , & Miao, B. (2014). Effects of dexmedetomidine on P2X4Rs, p38‐MAPK and BDNF in spinal microglia in rats with spared nerve injury. Brain Research, 1568, 21–30. 10.1016/j.brainres.2014.04.025 [DOI] [PubMed] [Google Scholar]
- Zhuo, M. (2007). Neuronal mechanism for neuropathic pain. Molecular Pain, 3(1), 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur, E. (2014). Topical treatment of neuropathic pain using compounded medications. The Clinical Journal of Pain, 30(1), 73–91. 10.1097/AJP.0b013e318285d1ba [DOI] [PubMed] [Google Scholar]