

Summary
Melanoma frequently harbors oncogenic mutations in the BRAF gene, which drives melanoma growth. Therefore, BRAF kinase inhibitors (BRAFi) are developed and approved for treating BRAF-mutant melanoma. However, the efficacy of BRAFi is limited due to acquired resistance, and in over 40% of melanoma, the causes of BRAFi resistance remain unknown. Here, using a human phospho-receptor tyrosine kinase array we identified Anaplastic Lymphoma Kinase (ALK) as a driver of acquired BRAFi resistance in melanoma. We found that ALK ligand FAM150A was necessary for ALK activation and ALK via the PI3K/AKT pathway was sufficient to confer resistance to BRAFi. ALK inhibitor (ALKi) ceritinib inhibited BRAFi-resistant melanoma in cell culture and mice. Residual BRAFi and ALKi dual resistant melanoma cells from ceritinib-treated mice were sensitive to a broad-spectrum anti-apoptotic protein inhibitor, AT101. Collectively, our results provide a framework for treating BRAF-mutant melanoma that sequentially uses different targeted therapies based on post-treatment tumor evolution.
Subject Areas: Biological Sciences, Cell Biology, Functional Aspects of Cell Biology, Cancer
Graphical Abstract
Highlights
-
•
ALK confers resistance to BRAF inhibitors in melanoma via the PI3K/AKT pathway
-
•
Pharmacological inhibition of ALK inhibits BRAF inhibitor-resistant melanoma
-
•
An ALK ligand, FAM150A, activates ALK in BRAF inhibitor-resistant melanoma
-
•
BRAF inhibitor and ALK inhibitor dual resistant melanoma are sensitive to AT101
Biological Sciences; Cell Biology; Functional Aspects of Cell Biology; Cancer
Introduction
Melanoma is an aggressive cancer that frequently metastasizes to various vital organs (Miller and Mihm, 2006, Zbytek et al., 2008). Although treatment of melanoma at early stages is generally effective, the median survival of patients with metastatic melanoma is only 4.5–12.5 months (Miller and Mihm, 2006, Schadendorf et al., 2015). Genomic sequencing of melanoma has identified oncogenic mutations in the BRAF gene in over 50% of tumors (Cancer Genome Atlas Network, 2015, Davies et al., 2002). Acquiring oncogenic mutations in the BRAF gene cause constitutive activation of the BRAF→MEK→ERK pathway and is necessary for melanoma growth and progression (Davies et al., 2002, Wellbrock et al., 2004). These findings have encouraged the development and approval of several BRAF and MEK kinase inhibitors by the US Food and Drug Administration for treating unresectable metastatic melanoma (Chapman et al., 2011, Flaherty et al., 2012). However, although patients with melanoma initially respond swiftly and robustly to BRAF kinase-targeted therapy, they show acquired resistance within a few months, resulting in disease progression. Owing to the high impact of this problem, intensive efforts have focused on identifying the causes of resistance to BRAF and MEK kinase inhibitors and several mechanisms have been identified (Nazarian et al., 2010, Wong and Ribas, 2016). These mechanisms can be broadly categorized as either dependent or independent of the mitogen-activated protein kinase (MAPK) pathway (Johnson et al., 2015, Rizos et al., 2014).
Anaplastic Lymphoma Kinase (ALK) was initially identified as a chimeric protein fused with nucleophosmin, a protein driving the development and progression of anaplastic large cell lymphoma (Morris et al., 1994). Since its identification, various genetic alterations in ALK gene have also been identified. The most prevalent of these are ALK gene fusions (e.g., EML4-ALK) and amplifications or activating mutations (e.g., F1174L/V, F1245C/L/V) found in multiple cancers, including lung cancer, neuroblastomas, lymphomas, or renal cell carcinoma (George et al., 2008, Lamant et al., 1999, Roskoski, 2013, Soda et al., 2007).
Here, using a human phospho-receptor tyrosine kinase (RTK) array, we found that ALK activation caused resistance to BRAF kinase inhibitor (BRAFi). We show that activated ALK-stimulated the phosphatidylinositol 3-kinase (PI3K)/AKT pathway in BRAFi-resistant melanoma cells and ALK inhibition inhibited the growth of ALK-activated BRAFi-resistant melanoma. Furthermore, melanoma cells that were resistant to both BRAFi and ALK inhibitor (ALKi) were still sensitive to AT-101, a broad-spectrum inhibitor of anti-apoptotic proteins, providing an alternative targeted therapeutic approach for their treatment.
Results
Human RTK Array Identifies Alterations in RTK Phosphorylation in Vemurafenib-Resistant Melanoma Cells
Dysregulation of RTKs such as epidermal growth factor receptor (EGFR), insulin growth factor-1 receptor (IGF-1R), or hepatocyte growth factor receptor (HGFR)/MET can cause BRAFi resistance (Cross et al., 2014, Krepler et al., 2016, Villanueva et al., 2010), indicating that RTKs are important mediators of resistance to BRAFi-based therapies. To identify other RTKs that regulate the response of BRAF-mutant melanoma to BRAFi, we first generated and characterized BRAFi vemurafenib-resistant cells by treating BRAF-mutant cell line A375 with vemurafenib for 6 weeks (Figures 1A–1C). We then analyzed both parental A375 cells (A375-P) and vemurafenib-resistant A375 cells (A375-R) using a human phospho-RTK array that can detect changes in the phosphorylation of 49 different human RTKs (Figure 1D). We found increased phosphorylation of several RTKs in A375-R cells compared with A375-P cells (Figures 1E and 1F). Specifically, increased phosphorylation occurred for EGFR, HGFR/MET, RYK, ROR2, and EPHA2, all of which were previously implicated in causing BRAFi resistance (Anastas et al., 2014, Cross et al., 2014, Krepler et al., 2016, Miao et al., 2015, O'Connell et al., 2013, Vergani et al., 2011, Wang et al., 2015). We also detected increased phosphorylation in VEGFR2, ALK, and DDR1 (Figures 1E and 1F). Collectively, these results indicate that RTK activation has an important role in acquired BRAFi resistance.
Figure 1.

Human Phospho-RTK Arrays Identified Changes in RTK Phosphorylation in BRAF Inhibitor Vemurafenib-Resistant Cells
(A) Schematic representation of the generation of vemurafenib-resistant A375 melanoma cells.
(B) A375 parental (A375-P) and A375 vemurafenib-resistant (A375-R) melanoma cells were treated with DMSO or the indicated concentrations of vemurafenib for 72 h and analyzed for survival using the MTT assay. Relative survival (%) for A375-P and A375-R cells relative to DMSO treated cells (0) is shown.
(C) Indicated proteins were analyzed in A375-P and A375-R cells after 6 h treatment with DMSO or 1 μM vemurafenib. ACTINB was used as the loading control.
(D) Schematic representation of the steps for analyzing A375-P and A375-R protein lysates using Proteome Profiler Human Phospho-RTK Array.
(E) Proteome Profiler Human Phospho-RTK Array membranes showing relative RTK phosphorylation in A375-R cells and A375-P cells.
(F) Spot intensity fold changes are plotted for individual RTK phosphorylation changes normalized to control spot in A375-R cells relative to A375-P cells as bar chart (left) or as numbers in the table on right.
Data are presented as mean ± SD. ****p < 0.0001.
ALK Causes Acquired Resistance to Vemurafenib in BRAF-Mutant Melanoma Cells
To determine if increased phosphorylation of RTKs in the BRAF-mutant melanoma cell line was sufficient to confer vemurafenib resistance, we individually expressed ALK, VEGFR2, or DDR1 in BRAF-mutant A375 melanoma cell line and analyzed if their expression caused vemurafenib resistance. Stable ALK expression in A375 cells led to vemurafenib resistance (Figure 2A). In contrast, stable expression of VEGFR2 or DDR1 did not cause resistance (Figure S1). Similarly, ectopic expression of ALK in two other BRAF-mutant melanoma cell lines, M229 and SKMEL-28, also resulted in vemurafenib resistance (Figure 2A).
Figure 2.

Ectopic Expression of Anaplastic Lymphoma Kinase (ALK) in Melanoma Conferred Resistance to BRAF Inhibitor
(A) BRAF-mutant melanoma cells A375, M229, and SKMEL-28 ectopically expressing an empty vector (EV) or ALK expression construct were treated with either DMSO or the indicated concentrations of vemurafenib. Relative cell survival (%) for each cell line in reference to DMSO-treated cells is shown.
(B) BRAF-mutant melanoma cells A375, M229, and SKMEL-28 ectopically expressing an empty vector (EV) or ALK were treated with either DMSO or the indicated concentrations of dabrafenib. Relative cell survival (%) for each cell line in reference to DMSO-treated cells is shown.
(C) A375 cells ectopically expressing empty vector (EV) or ALK expression construct were treated with 2 μM vemurafenib for 4 weeks. Images of representative plates with surviving colonies are shown. Scale bar, 200 μM.
Data are presented as mean ± SD. **p < 0.01, ***p < 0.001. See also Figure S1.
Next, we tested if the resistance due to ALK expression was specific to vemurafenib, or if it was a general mechanism of BRAFi resistance. To this end, we tested if ectopic ALK expression in BRAF-mutant melanoma cells (A375, M229, SKMEL-28) caused resistance to another BRAFi, dabrafenib. Our results showed that ALK-expressing melanoma cells were also resistant to dabrafenib compared with the empty vectors alone (Figure 2B).
Finally, to determine the applicability of our results to a long-term treatment scenario, we performed clonogenic assays using BRAF-mutant A375 cells. We treated A375 cells expressing an empty vector or ALK with vemurafenib and monitored the emergence of colonies over 4 weeks. Similar to our short-term survival assays, we found that in clonogenic assays also ALK-expressing A375 cells had a significantly higher number of drug-resistant colonies than empty vector-expressing A375 cells (Figure 2C). Collectively, these results show that ectopic expression of ALK causes BRAFi resistance.
BRAF Kinase Inhibitor-Resistant Cells Have Increased ALK Phosphorylation and Activated Downstream Signaling
Next, we asked if ALK was activated in vemurafenib-resistant melanoma cell lines. To this end, we analyzed three pairs of parental and vemurafenib-resistant melanoma cell lines, including A375 parental (A375-P) and resistant (A375-R), SKMEL-239 parental (SKMEL-239-P) and resistant (SKMEL-239-R), and M229 parental (M229-P) and resistant (M229-R) melanoma cell lines. We measured phosphorylation of tyrosine 1278 within the ALK activation loop (Tartari et al., 2008) and found increased ALK phosphorylation in A375-R and M229-R cell lines compared with parental cell lines (A375-P and M229-P), but did not observe more phosphorylation in SKMEL-239-R cells (Figure 3A). Consistent with ALK-stimulated downstream signaling, increased ALK phosphorylation correlated with increased p-ERK1/2 and pAKT levels in these cell lines (Figure 3A). However, vemurafenib-resistant SKMEL-239 cells did not have increased ALK phosphorylation and had lower pAKT levels (Figure 3A). However, interestingly ectopic expression of ALK in SKMEL-239 cells also caused resistance to vemurafenib (Figures S2A and S2B). However, we believe for reasons not fully clear to us that the SKMEL-239 cells acquire resistance to BRAFi via mechanisms independent of activating ALK, as evidenced by lack of increased ALK phosphorylation in SKMEL-239-R cells (Figure 3A). In particular, a previous study has identified that the SKMEL-239-R cells become resistance to BRAFi due to dimerization of aberrantly spliced BRAF (V600E) (Poulikakos and Rosen, 2011). Similarly, ectopic ALK expression in BRAF-mutant melanoma cells (A375, M229, SKMEL-28) activated the PI3K/AKT pathway, as shown by increased pAKT levels (Figure 3B). The increased pAKT was dependent upon ALK activity in BRAFi-resistant A375 cells, because treatment of these cells with ALK inhibitor (ALKi) ceritinib reduced the pAKT level (Figure 3C). Similarly, treating ALK-expressing A375 cells with ceritinib inhibited the pAKT level (Figure S3A).
Figure 3.

Ectopic Expression of ALK Stimulated the PI3K/AKT Pathway and BRAFi-Resistant Melanoma Showed Genetic Signatures Consistent with ALK Activation
(A) Parental and BRAF inhibitor (BRAFi)-resistant A375, SKMEL-239, and M229 cells were analyzed for the indicated protein by immunoblotting. ACTINB was used as the loading control.
(B) A375, M229, and SKMEL-28 cells ectopically expressing an empty vector or ALK were analyzed by western blot for the indicated proteins. ACTINB was used as the loading control.
(C) A375 parental (A375-P) cells, A375 BRAFi-resistant cells (A375-R), and A375-R cells treated with ceritinib (1 μM) for 24 h were analyzed by immunoblotting for the indicated proteins. ACTINB was used as the loading control.
(D) mRNA expression for the indicated ALK-activated genes was measured in A375-P, A375-R, and A375-R cells treated with ceritinib (1 μM) for 24 h. mRNA expression for indicated genes relative to A375-P cells is shown. ACTINB mRNA expression was used for normalization.
Data are presented as mean ± SD. ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, respectively. See also Figures S2 and S3.
Next, we analyzed the expression of ALK-activated genes (Piva et al., 2006) in parental and BRAFi-resistant A375 cells. We found that expression of several ALK-activated genes was higher in BRAFi-resistant A375 and M229 cells than in parental cells (Figures 3D and S3B and Table S1). We also found that this expression was dependent on ALK activity, because treatment of BRAFi-resistant A375 and M229 cells with ceritinib inhibited the expression of ALK-activated genes (Figures 3D and S3B). Similarly, ectopic expression of ALK in A375 cells resulted in increased expression of ALK-stimulated genes compared with A375 cells expressing an empty vector (Figure S3C). Collectively, these results demonstrate that the ALK-driven transcriptional program is upregulated in BRAFi-resistant melanoma.
PI3K/AKT Pathway Activation Downstream of ALK Is Necessary to Cause BRAFi Resistance
Based on our finding that pAKT levels were higher in cells ectopically expressing ALK in some BRAFi-resistant cell lines, we asked if the PI3K pathway downstream of ALK-mediated the ALK-associated drug resistance. To test this, we expressed constitutively active PI3K (PI3KCA) in parental A375 cells and measured their sensitivity to vemurafenib with two assays. Our results showed that ectopic PI3KCA expression in parental A375 cells resulted in vemurafenib resistance in the Methylthiazolyldiphenyl-tetrazolium bromide (MTT) cell survival assay (Figures 4A and 4B) and the soft-agar assay (Figures 4C and 4D). In addition, we tested if ectopic PI3KCA expression in BRAFi-resistant A375 cells reduced sensitivity to ALK inhibition by ceritinib. We found that ectopic PI3KCA expression conferred ceritinib resistance in the MTT cell survival assay (Figures 4E and 4F) and the soft-agar assay (Figures 4G and 4H). In addition, we analyzed how PI3K inhibition affected the expression of ALK-activated genes (Piva et al., 2006). We found that treating BRAFi-resistant A375 or M229 cells with a PI3K inhibitor (PI3Ki) pictilisib downregulated expression of several ALK-activated genes (Figures 4I, 4J, and S3D). Collectively, these results show that the PI3K/AKT pathway downstream of ALK activates the majority of ALK-activated genes.
Figure 4.

ALK-Mediated BRAFi-Resistance by Activating the PI3K/AKT Pathway
(A) A375 cells ectopically expressing either an empty vector or constitutively active PI3K (PI3KCA) were analyzed for the indicated proteins by immunoblotting. ACTINB was used as a loading control.
(B) A375 cells ectopically expressing either an empty vector or constitutively active PI3K (PI3KCA) were treated with DMSO or with the indicated concentrations of vemurafenib and analyzed for survival using the MTT assay and for anchorage-independent growth using the soft-agar assay. Relative cell survival (%) in reference to DMSO-treated cells is shown.
(C) A375 cells ectopically expressing either an empty vector (EV) or constitutively active PI3K (PI3KCA) were treated with DMSO or vemurafenib (1 μM). Representative images for soft-agar colonies under the indicated conditions are shown. Scale bar, 500 μm.
(D) Relative soft-agar colony size (%) for experiments is presented in panel (C).
(E) A375 cells ectopically expressing either an empty vector or constitutively active PI3K (PI3KCA) were analyzed for indicated proteins using immunoblotting. ACTINB was used as a loading control.
(F) A375 cells ectopically expressing either an empty vector or constitutively active PI3K (PI3KCA) were treated with DMSO or with the indicated concentrations of ceritinib and analyzed for survival using the MTT assay and for anchorage-independent growth using the soft-agar assay. Relative cell survival (%) in reference to DMSO-treated cells is shown.
(G) A375 cells ectopically expressing either an empty vector (EV) or constitutively active PI3K (PI3KCA) were treated with DMSO or ceritinib (1 μM). Representative images for soft-agar colonies under the indicated conditions are shown. Scale bar, 500 μm.
(H) Relative soft-agar colony size in experiments is presented in panel (G).
(I) mRNA expression for the indicated ALK-activated genes was measured in A375 parental (A375-P), A375 vemurafenib-resistant (A375-R), and A375-R cells treated PI3K inhibitor pictilisib (1 μM) for 24 h. mRNA expression is shown relative to A375-P cells. ACTINB mRNA expression was used for normalization.
(J) A375-P, A375-R, and A375-R cells treated with pictilisib (1 μM) for 24 h were analyzed by immunoblotting for the indicated proteins. ACTINB was used as the loading control.
(K) A375-P cells were treated with the indicated concentrations of vemurafenib, pictilisib, or both for 3 days and analyzed for survival using the MTT assay. Relative cell survival (%) relative to DMSO-treated cells is shown.
(L) A375-R cells were treated with the indicated concentrations of vemurafenib, pictilisib, or both for 3 days and analyzed for survival using the MTT assay. Relative cell survival (%) relative to DMSO (0)-treated cells is shown.
Data are presented as mean ± SD. ns, not significant p value; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, respectively. See also Figure S3.
Finally, we treated BRAFi-sensitive parental or resistant A375 cells with vemurafenib, pictilisib, or both. We found that although parental cells were not sensitive to pictisilib, BRAFi-resistant A375 cells were (Figures 4K, 4L, and S3E). Similarly, ALK-expressing A375 cells although resistant to vemurafenib were sensitive to pictilisib (Figure S3F). These results further strengthen the idea that the PI3K pathway downstream of ALK mediates BRAFi resistance. Moreover, this result is consistent with other findings for the role of PI3K in causing resistance to BRAF inhibitors (Deuker et al., 2015, Lassen et al., 2014, Villanueva et al., 2010).
ALK Ligand FAM150A Expression Activates ALK Phosphorylation and Confers Vemurafenib Resistance
As ALK is an RTK, we hypothesized that increased phosphorylation in BRAFi-resistant cells could be due to increased ligand expression. To test this possibility, we collected conditioned medium from either parental or BRAFi-resistant A375 cells and determined if this conditioned medium conferred resistance to vemurafenib. Our results showed that that conditioned medium from BRAFi-resistant cells conferred vemurafenib resistance in A375 parental cells (Figure 5A). Based on these results, we next analyzed the expression of two known ALK-activating ligands, FAM150A and FAM150B, in parental and BRAFi-resistant melanoma cells (Guan et al., 2015, Reshetnyak et al., 2015). Therefore, we asked if the mRNA expression of either FAM150A or FAM150B was higher in BRAFi-resistant melanoma cells than in parental cells (A375-P/R, SKMEL-239P/R, M229-P/R). We found upregulated FAM150A expression in BRAFi-resistant A375 and M229 cells, but not in SKMEL-239 cells (Figure 5B), which was consistent with higher p-ALK levels in A375-R and M229-R cells. In contrast, FAM150B expression did not correlate with ALK phosphorylation in parental and vemurafenib-resistant cell lines (Figure S4). Treating BRAF-mutant melanoma cells with BRAFi causes them to secrete a number of proteins, which are collectively referred to as the therapy-induced secretome (TIS); the TIS stimulates the growth and survival of surrounding cells and confers drug resistance (Obenauf et al., 2015). Therefore, we analyzed FAM150A expression in A375 cells after vemurafenib treatment, finding that vemurafenib treatment in parental A375 increased FAM150A expression (Figure 5C). To confirm that FAM150A was responsible for increased ALK phosphorylation in vemurafenib-resistant A375 cells, we overexpressed FAM150A in A375-P cells and analyzed their sensitivity to vemurafenib and ALK-mediated signaling. Consistent with FAM150A being an ALK ligand, FAM150A overexpression increased ALK phosphorylation and stimulated downstream signaling, as measured by increased pAKT (Figure 5D). Furthermore, similar to ALK expression, ectopic FAM150A expression conferred vemurafenib resistance (Figure 5E). Collectively, these results show that FAM150A plays an important role in increasing ALK-mediating signaling in melanoma.
Figure 5.

FAM150A-Activated ALK Phosphorylation in Melanoma Cells and Stimulated BRAFi Resistance
(A) A375 parental cells were cultured with conditioned medium harvested from A375 parental (A375-P CM) or vemurafenib-resistant A375 cells (A375-R CM), then treated with DMSO or the indicated concentrations of vemurafenib for 3 days, and analyzed for survival using the MTT assay. Relative cell survival (%) relative to DMSO-treated cells is shown.
(B) mRNA expression for FAM150A was measured for the indicated pairs of parental and BRAFi-resistant cells (A375, SKMEL-239, M229). mRNA expression is shown relative to respective parental melanoma cells. ACTINB mRNA expression was used for normalization.
(C) mRNA expression for FAM150A was measured in A375 cells treated with DMSO or vemurafenib (1 μM) for 24 h FAM150A mRNA expression relative to DMSO-treated A375 cells is shown. ACTINB mRNA expression was used for normalization.
(D) A375 cells overexpressing empty vector (−) or FAM150A (+) were analyzed for indicated proteins by immunoblotting. ACTINB was used as a loading control.
(E) A375 cells ectopically expressing an empty vector (EV) or FAM150A were treated with DMSO or the indicated concentrations of vemurafenib for 3 days and analyzed for survival by the MTT assay. Relative cell survival (%) relative to DMSO-treated cells is shown.
Data are presented as mean ± SD. *p < 0.05, ***p < 0.001, ****p < 0.0001. See also Figure S4.
Pharmacological Inhibition of ALK Blocks Melanoma Growth in Cell Culture and in Mice
Because we found upregulated ALK-mediated signaling in BRAFi-resistant melanoma cells, we asked whether ALK inhibition would inhibit cell proliferation and soft-agar growth of melanoma cells. Therefore, we treated the parental and BRAFi-resistant melanoma cell lines (A375-P/R, M229-P/R, SKMEL-239-P/R) with vemurafenib or ceritinib. As expected, BRAFi-sensitive melanoma cells showed reduced growth with vemurafenib treatment, whereas BRAFi-resistant cells were not inhibited (Figures 6A and S5). Consistent with the level of ALK phosphorylation, ceritinib treatment inhibited the growth of BRAFi-resistant A375 and M229 cells (Figure 6A). Furthermore, ceritinib treatment inhibited the growth of BRAFi-resistant A375 cells in soft agar (Figures 6B and 6C). Similar results were obtained using short hairpin RNA targeting ALK (Figures S6A and S6B). Notably, ceritinib treatment of BRAFi-resistant SKMEL-239 cells, in which the level of ALK phosphorylation was not higher than that in the parental cells, only partially inhibited proliferation (Figure S5). Finally, we performed animal experiments to test if ALK inhibition would sensitize BRAFi-resistant melanoma cells to the ALK inhibitor ceritinib. To this end, we injected BRAFi-resistant A375 cells into the flank of athymic nude mice and then treated them with vehicle, vemurafenib, or ceritinib. We found that ceritinib treatment of BRAFi-resistant melanoma cells inhibited tumor growth in mice (Figures 6D and 6E). Collectively, these results demonstrate that ALK inhibition is a potential strategy to treat BRAFi-resistant melanoma with increased ALK pathway activity.
Figure 6.

Inhibition of ALK Impaired BRAFi-Resistant Melanoma in Cell Culture and in Mice
(A) Parental A375 (A375-P) and BRAFi-resistant A375 cells (A375-R) were treated with DMSO or the indicated concentrations of vemurafenib or ceritinib for 3 days and analyzed for survival using the MTT assay. Relative cell survival (%) in reference to DMSO-treated cells is shown.
(B) A375-P and BRAFi-resistant A375-R cells were treated with vemurafenib (1 μM) or ceritinib (1 μM) and analyzed for anchorage-independent growth by the soft-agar assay. Representative images for soft-agar colonies for the indicated melanoma cell lines under the indicated treatment conditions are shown. Scale bar, 500 μm.
(C) Relative colony size for the indicated melanoma cell lines at the indicated treatment conditions is shown.
(D) Vemurafenib-resistant A375 cells (A375-R) were subcutaneously injected into the flanks of athymic nude mice (n = 5) and treated with vehicle, vemurafenib (30 mg/kg), or ceritinib (25 mg/kg). Average tumor volumes (n = 5) at the indicated weeks are plotted.
(E) Representative tumor images for the indicated conditions for the experiment in panel (D).
Data are presented as mean ± SD. ns, not significant p value. *p < 0.05, ***p < 0.001, and ****p < 0.0001. See also Figures S5 and S6.
Ceritinib-Resistant Residual Tumor Cells Are Sensitive to a Broad-Spectrum Inhibitor of Anti-apoptotic Proteins
Owing to genetic and epigenetic alterations, cancer cells become resistant to a wide variety of anti-cancer therapies (Gottesman et al., 2016, Salgia and Kulkarni, 2018, Shaffer et al., 2017). Thus, almost all therapies leave behind residual drug-resistant cells, which are an active focus of research (Rambow et al., 2018, van Dongen et al., 2015). Targeting these cells is important to prevent recurrence and necessary to fully eradicate the disease. Therefore, we isolated melanoma tumors from control and ceritinib-treated mice and analyzed the residual cells (Figure 7A). For each condition, we generated two replicate cell lines from tumors isolated from mice that were treated with either vehicle (control group) or ceritinib (treatment group). First, we tested if residual tumor cells from the ceritinib-treated group were resistant to ceritinib. To this end, we performed an MTT-based survival assay and measured their growth in soft agar after treatment with DMSO or ceritinib. We found that tumor cells from ceritinib-treated mice were significantly more ceritinib resistant than melanoma cells from vehicle-treated mice (Figure 7B). We obtained similar results in the soft-agar assay, in which residual tumor cells from ceritinib-treated mice grew better in soft-agar than the tumor cells from vehicle control mice (Figures 7C and 7D). Previous studies found that failure to undergo apoptosis after drug treatment can drive acquired drug resistance in cancer cells (Gottesman, 2002, Housman et al., 2014, Mohammad et al., 2015). Consistent with this, ceritinib-resistant melanoma cells were substantially more resistant to apoptosis induction after ceritinib treatment (Figure S7A). Furthermore, analysis of a series of anti and pro-apoptotic proteins in ceritinib-sensitive versus ceritinib-resistant cells resulted in the identification of Bcl-2, cIAP2, cIAP1, and survivin as potential mediators of resistance to ceritinib in these cells (Figure S7B). These findings indicated the possibility of targeting ceritinib-resistant cells with pharmacological inhibitors of anti-apoptotic proteins.
Figure 7.

Residual Tumor-Derived BRAF and ALK Inhibitor Dual Resistant Melanoma Cells Were Sensitive to a Broad-Spectrum Anti-apoptotic Protein inhibitor, AT101
(A) Schematic showing steps for generating cell lines from melanoma tumors isolated from mice treated with either vehicle or ceritinib.
(B) Tumor-derived BRAFi-resistant and ceritinib-sensitive cells (A357-R-Cer-S) and BRAFi and ceritinib dual resistant A375 cells (A375-R-Cer-R) cells were treated with indicated concentrations of ceritinib for 3 days and analyzed by MTT assay. Relative cell survival (%) in reference to DMSO-treated cells is shown.
(C) Tumor-derived A375-R-Cer-S and A375-R-Cer R cells were treated with DMSO or ceritinib (1 μM) and analyzed for anchorage-independent growth by soft-agar assay. Representative images for soft-agar colonies are shown. Scale bar, 500 μm.
(D) Relative colony size for indicated melanoma cell lines at the indicated treatment conditions is shown.
(E) Tumor-derived A375-R-Cer-S and A375-R-Cer-R cells were treated with the indicated concentrations of broad anti-apoptotic protein inhibitor AT101 (10 μM) for 72 h and analyzed for survival using the MTT assay. Relative cell survival (%) relative to DMSO-treated cells is shown.
(F) Mice tumor-derived A375-R-Cer-S and A375-R-Cer-R cells were treated with AT101 (10 μM) and analyzed for anchorage-independent growth by the soft-agar assay. Representative images for soft-agar colonies for the indicated conditions are shown. Scale bar, 500 μm.
(G) Relative colony size for the indicated melanoma cell lines at indicated treatment conditions is shown.
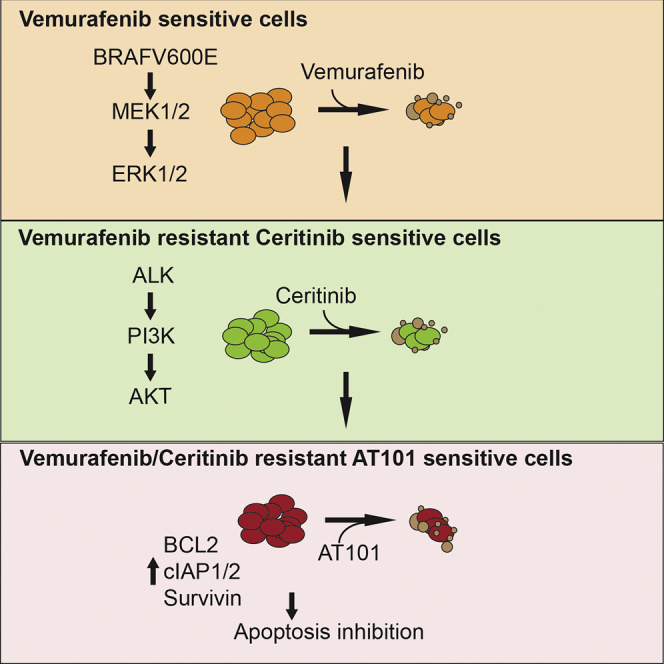
(H) Model summarizing the findings of our study.
Data are presented as mean ± SD. ns, not significant p value. *p < 0.05, **p < 0.01, and ****p < 0.0001. See also Figure S7.
Several anti-apoptotic proteins are important for cancer cell survival and mediate acquired resistance to targeted therapeutic agents (Adams and Cory, 2018, Dai et al., 2013, Srivastava et al., 1999). This knowledge has been used to develop high-quality inhibitors of anti-apoptotic proteins, which are clinically used for cancer therapy (Cory et al., 2016, Montero and Letai, 2018). With this in mind, we tested four small-molecule inhibitors: IAP inhibitor, AT406 (Cai et al., 2011); MCL1 inhibitor, S63845 (Kotschy et al., 2016); BCL2 inhibitor, venetoclax (Souers et al., 2013); and a broad-spectrum anti-apoptotic protein inhibitor, AT101 (Wang et al., 2006). AT101 inhibits multiple anti-apoptotic proteins, such as BCL2, BCL-XL, and MCL1. After treatment, we analyzed cell survival by MTT assay and found that AT406, S63845, and venetoclax did not significantly inhibit the survival of BRAFi and ALKi dual resistant melanoma cells (Figure S7C). However, AT101 treatment strongly inhibited the growth and survival of these dual resistant cells (Figures 7E–7G). Collectively, these results demonstrate that residual cells that escape ceritinib treatment become resistant to apoptosis. These findings provide targets for treating BRAFi and ALKi dual resistant melanoma cells.
Discussion
Acquired resistance to targeted therapeutic agents such as BRAFi makes it challenging to use personalized therapies to treat melanoma and other cancers. One way to overcome this limitation is to understand the mechanism that causes resistance and use this information to revise the therapeutic regimen. Here, we found that activation of ALK RTK is a mechanism for causing acquired resistance to BRAFi and show that ALK targeting can be used to treat BRAFi-resistant melanoma. We also show that BRAFi-resistant ALK-sensitive cells can evolve to become resistant to apoptosis, but are sensitive to broad-spectrum anti-apoptotic protein inhibitors. A summary of our results is presented in Figure 7H and is also described below.
ALK in Acquired Resistance to BRAF Kinase Inhibitors
BRAFi resistance can be due to a wide variety of mechanisms. For example, a multi-center trial analyzed clinical and genomic data using 132 tissue samples obtained from 100 patients who progressed on BRAFi therapy, from three large, previously published studies of BRAFi resistance (Haq et al., 2013, Johnson et al., 2015, Long et al., 2014, Villanueva et al., 2013). This study only identified resistance mechanisms in 58% patients; mechanisms included NRAS or KRAS mutations (20%), BRAF splice variants (16%), BRAF (V600E/K) amplifications (13%), MEK1/2 mutations (7%), and non-mitogen-activated protein kinase pathway alterations (11%). This study demonstrated that the mechanism of resistance remains unknown in over 40% of BRAFi-resistant melanomas.
ALK is an oncogenic driver in non-small cell lung cancer (NSCLC) and anaplastic large cell lymphomas. In these cancers, ALK alterations are chromosomal rearrangements that result in fusion oncogenes (Bolger and Sherman, 1991, Lamant et al., 1999, Morris et al., 1995). In addition, ALK gene amplification and activating ALK mutations have also been observed in other cancer types. ALK was initially considered to be an orphan receptor; however, FAM150A and FAM150B were recently identified as ALK-activating ligands that were capable of activating ALK-mediated signaling (Guan et al., 2015, Reshetnyak et al., 2015).
A previous study showed that ALK inhibitor can potentiate the effectiveness of the MEK inhibitor trametinib (Verduzco et al., 2018). Here, we found that ALK is activated in BRAFi-resistant melanoma via the action of FAM150A, and that ectopic expression of ALK conferred BRAFi resistance. We also showed that the ALK inhibitor ceritinib inhibited the growth of BRAFi-resistant melanoma both in cell culture and in mice. These results identify a non-genetic mechanism for ALK RTK activation and demonstrate that ALK targeting in BRAFi-resistant melanoma may be a viable therapeutic option for BRAFi-resistant melanoma therapy.
However, our studies largely focused on BRAFi (vemurafenib and dabrafenib) and our results are also expected to be relevant in the context of BRAFi and MEKi combination therapies (e.g., dabrafenib and trametinib). This prediction is based on our results that ALK-mediated BRAFi resistance occurs due to the activation PI3K/AKT pathway rather than by activation of MAPK pathway downstream of BRAF and is supported by a previous study that has shown PI3K/AKT pathway activation causes resistance to BRAFi and MEKi combination therapy (Atefi et al., 2011). In this regard, previous studies have shown that SKMEL-239-R cells are resistant to BRAFi (Poulikakos and Rosen, 2011), whereas M229-R cells are resistant to both BRAFi and MEKi (Nazarian et al., 2010). Finally, our vemurafenib resistant A375 were significantly more resistant to MEK inhibition than parental A375 cells (Figure S7D).
Role of PI3K/AKT Downstream of ALK in Conferring Resistance in Melanoma
As mentioned above, a large majority of BRAFi resistance mechanisms rely on reactivating the MAPK pathway to confer BRAFi resistance. However, MAPK-independent mechanisms have also been identified. Among these, the second most predominant mechanism is activation of the PI3K pathway by various mechanisms, including activating mutations in PI3KCA, AKT1, AKT3, and inactivating mutation or deletion of PTEN.
Oncogenic forms of ALK are known to enhance cancer growth and development by stimulating signaling pathways, such as ERK or PI3K/AKT (Hallberg and Palmer, 2016). We found that melanoma cells with increased phosphorylation of ALK had heightened PI3K signaling, as observed by increased pAKT levels. We also showed that this activation was crucial for ALK to confer BRAFi resistance because constitutive activation of PI3K was sufficient to confer resistance to ALK inhibitor, similar to the effect of constitutive ALK activation. Furthermore, treatment of ALK-expressing cells and BRAFi-resistant cells showed that they were sensitive to PI3K inhibitor, and cells ectopically expressing ALK could be sensitized via treatment with PI3K inhibitor. These results show that PI3K downstream of ALK is necessary for conferring resistance to BRAFi in melanoma.
Sequential Targeted Therapy-Based Therapeutic Approach for Extending the Therapeutic Responses in Patients with BRAF-Mutant Melanoma
The overarching goal for studies of the mechanisms of resistance to targeted therapeutic agents is to develop new therapeutic approaches to treat drug-resistant tumors and extend the survival of patients with cancer. For example, the development of dual BRAF + MEK or BRAF + ERK targeting approaches, which have benefited patients with metastatic melanoma with BRAFi-resistant tumors, were based on the finding that the MAPK pathway via MEK activation or ERK activation causes most BRAFi resistance (Flaherty et al., 2012). Similar approaches, such as those based on the identification of secondary mutations in EGFR in NSCLC, BCR-ABL in chronic myelogenous leukemia, and EZH2 in non-Hodgkin lymphoma, have identified new therapeutic approaches to address treatment resistance and improve outcomes for patients with cancer (Bisserier and Wajapeyee, 2018, Cross et al., 2014, Gibaja et al., 2016, Pao et al., 2005, Shah et al., 2004). In our study, we present an approach that is based on tracking the evolution of drug resistance in cancer cells in real time. We show that BRAFi-resistant and potentially BRAFi/MEKi-dual resistant tumors with higher p-ALK levels can be treated with the ALK inhibitor ceritinib. Also, we show that residual tumor cells that become resistant to ceritinib-based therapy and are dual resistant to BRAFi + ALKi can be treated using a broadly acting anti-apoptotic protein inhibitor. Collectively, these results underpin the need to progressively identify and adjust therapeutic regimens for patients undergoing cancer treatment, with the ultimate effect of more effective cancer treatment and extended patient survival.
Limitations of the Study
This study relied upon in vitro-generated BRAFi-resistant cell lines, patient-derived BRAFi-resistant melanoma cell lines, and in vivo xenograft-based mouse models. However, we could not optimize any phospho-ALK antibody to reliably detect changes in paraffin-fixed patient-derived BRAFi-resistant melanoma samples that were available to us. Thus the clinical relevance of our findings remains to be determined.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We gratefully acknowledge grants from the National Institutes of Health: R01CA195077-01A1 (N.W.), R01CA200919-01 (N.W.), 1R01 CA218008-01A1 (N.W.), and R03CA221926. N.W. is also supported by Research Scholar Grant from American Cancer Society [128347-RSG-15-212-01-TBG and a grant from Department of Defence (Award number W81XWH-18-1-0069).
Author Contributions
R.J. and N.W. conceived and designed the experiments. R.J. performed most of the experiments with help from P.M. R.J. and N.W. analyzed and interpreted the data. R.J. and N.W. prepared the figures and co-wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: June 28, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.06.001.
Supplemental Information
References
- Adams J.M., Cory S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018;25:27–36. doi: 10.1038/cdd.2017.161. [DOI] [PMC free article] [PubMed] [Google Scholar]; Adams, J.M., and Cory, S.. (2018). The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 25, 27-36. [DOI] [PMC free article] [PubMed]
- Anastas J.N., Kulikauskas R.M., Tamir T., Rizos H., Long G.V., von Euw E.M., Yang P.T., Chen H.W., Haydu L., Toroni R.A. WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J. Clin. Invest. 2014;124:2877–2890. doi: 10.1172/JCI70156. [DOI] [PMC free article] [PubMed] [Google Scholar]; Anastas, J.N., Kulikauskas, R.M., Tamir, T., Rizos, H., Long, G.V., von Euw, E.M., Yang, P.T., Chen, H.W., Haydu, L., Toroni, R.A., et al. (2014). WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J. Clin. Invest. 124, 2877-2890. [DOI] [PMC free article] [PubMed]
- Atefi M., von Euw E., Attar N., Ng C., Chu C., Guo D., Nazarian R., Chmielowski B., Glaspy J.A., Comin-Anduix B. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One. 2011;6:e28973. doi: 10.1371/journal.pone.0028973. [DOI] [PMC free article] [PubMed] [Google Scholar]; Atefi, M., von Euw, E., Attar, N., Ng, C., Chu, C., Guo, D., Nazarian, R., Chmielowski, B., Glaspy, J.A., Comin-Anduix, B., et al. (2011). Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One 6, e28973. [DOI] [PMC free article] [PubMed]
- Bisserier M., Wajapeyee N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood. 2018;131:2125–2137. doi: 10.1182/blood-2017-08-804344. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bisserier, M., and Wajapeyee, N.. (2018). Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood 131, 2125-2137. [DOI] [PMC free article] [PubMed]
- Bolger M.B., Sherman M.A. Computer modeling of combining site structure of anti-hapten monoclonal antibodies. Methods Enzymol. 1991;203:21–45. doi: 10.1016/0076-6879(91)03004-z. [DOI] [PubMed] [Google Scholar]; Bolger, M.B., and Sherman, M.A.. (1991). Computer modeling of combining site structure of anti-hapten monoclonal antibodies. Methods Enzymol. 203, 21-45. [DOI] [PubMed]
- Cai Q., Sun H., Peng Y., Lu J., Nikolovska-Coleska Z., McEachern D., Liu L., Qiu S., Yang C.Y., Miller R. A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J. Med. Chem. 2011;54:2714–2726. doi: 10.1021/jm101505d. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cai, Q., Sun, H., Peng, Y., Lu, J., Nikolovska-Coleska, Z., McEachern, D., Liu, L., Qiu, S., Yang, C.Y., Miller, R., et al. (2011). A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J. Med. Chem. 54, 2714-2726. [DOI] [PMC free article] [PubMed]
- Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cancer Genome Atlas Network. (2015). Genomic classification of cutaneous melanoma. Cell 161, 1681-1696. [DOI] [PMC free article] [PubMed]
- Chapman P.B., Hauschild A., Robert C., Haanen J.B., Ascierto P., Larkin J., Dummer R., Garbe C., Testori A., Maio M. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chapman, P.B., Hauschild, A., Robert, C., Haanen, J.B., Ascierto, P., Larkin, J., Dummer, R., Garbe, C., Testori, A., Maio, M., et al. (2011). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507-2516. [DOI] [PMC free article] [PubMed]
- Cory S., Roberts A.W., Colman P.M., Adams J.M. Targeting BCL-2-like proteins to kill cancer cells. Trends Cancer. 2016;2:443–460. doi: 10.1016/j.trecan.2016.07.001. [DOI] [PubMed] [Google Scholar]; Cory, S., Roberts, A.W., Colman, P.M., and Adams, J.M.. (2016). Targeting BCL-2-like proteins to kill cancer cells. Trends Cancer 2, 443-460. [DOI] [PubMed]
- Cross D.A., Ashton S.E., Ghiorghiu S., Eberlein C., Nebhan C.A., Spitzler P.J., Orme J.P., Finlay M.R., Ward R.A., Mellor M.J. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cross, D.A., Ashton, S.E., Ghiorghiu, S., Eberlein, C., Nebhan, C.A., Spitzler, P.J., Orme, J.P., Finlay, M.R., Ward, R.A., Mellor, M.J., et al. (2014). AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4, 1046-1061. [DOI] [PMC free article] [PubMed]
- Dai H., Ding H., Meng X.W., Lee S.H., Schneider P.A., Kaufmann S.H. Contribution of Bcl-2 phosphorylation to Bak binding and drug resistance. Cancer Res. 2013;73:6998–7008. doi: 10.1158/0008-5472.CAN-13-0940. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dai, H., Ding, H., Meng, X.W., Lee, S.H., Schneider, P.A., and Kaufmann, S.H.. (2013). Contribution of Bcl-2 phosphorylation to Bak binding and drug resistance. Cancer Res. 73, 6998-7008. [DOI] [PMC free article] [PubMed]
- Davies H., Bignell G.R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M.J., Bottomley W. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]; Davies, H., Bignell, G.R., Cox, C., Stephens, P., Edkins, S., Clegg, S., Teague, J., Woffendin, H., Garnett, M.J., Bottomley, W., et al. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949-954. [DOI] [PubMed]
- Deuker M.M., Marsh Durban V., Phillips W.A., McMahon M. PI3'-kinase inhibition forestalls the onset of MEK1/2 inhibitor resistance in BRAF-mutated melanoma. Cancer Discov. 2015;5:143–153. doi: 10.1158/2159-8290.CD-14-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]; Deuker, M.M., Marsh Durban, V., Phillips, W.A., and McMahon, M.. (2015). PI3'-kinase inhibition forestalls the onset of MEK1/2 inhibitor resistance in BRAF-mutated melanoma. Cancer Discov. 5, 143-153. [DOI] [PMC free article] [PubMed]
- Flaherty K.T., Infante J.R., Daud A., Gonzalez R., Kefford R.F., Sosman J., Hamid O., Schuchter L., Cebon J., Ibrahim N. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]; Flaherty, K.T., Infante, J.R., Daud, A., Gonzalez, R., Kefford, R.F., Sosman, J., Hamid, O., Schuchter, L., Cebon, J., Ibrahim, N., et al. (2012). Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 367, 1694-1703. [DOI] [PMC free article] [PubMed]
- George R.E., Sanda T., Hanna M., Frohling S., Luther W., 2nd, Zhang J., Ahn Y., Zhou W., London W.B., McGrady P. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]; George, R.E., Sanda, T., Hanna, M., Frohling, S., Luther, W., 2nd, Zhang, J., Ahn, Y., Zhou, W., London, W.B., McGrady, P., et al. (2008). Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455, 975-978. [DOI] [PMC free article] [PubMed]
- Gibaja V., Shen F., Harari J., Korn J., Ruddy D., Saenz-Vash V., Zhai H., Rejtar T., Paris C.G., Yu Z. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene. 2016;35:558–566. doi: 10.1038/onc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gibaja, V., Shen, F., Harari, J., Korn, J., Ruddy, D., Saenz-Vash, V., Zhai, H., Rejtar, T., Paris, C.G., Yu, Z., et al. (2016). Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene 35, 558-566. [DOI] [PMC free article] [PubMed]
- Gottesman M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]; Gottesman, M.M.. (2002). Mechanisms of cancer drug resistance. Annu. Rev. Med. 53, 615-627. [DOI] [PubMed]
- Gottesman M.M., Lavi O., Hall M.D., Gillet J.P. Toward a better understanding of the complexity of cancer drug resistance. Annu. Rev. Pharmacol. Toxicol. 2016;56:85–102. doi: 10.1146/annurev-pharmtox-010715-103111. [DOI] [PubMed] [Google Scholar]; Gottesman, M.M., Lavi, O., Hall, M.D., and Gillet, J.P.. (2016). Toward a better understanding of the complexity of cancer drug resistance. Annu. Rev. Pharmacol. Toxicol. 56, 85-102. [DOI] [PubMed]
- Guan J., Umapathy G., Yamazaki Y., Wolfstetter G., Mendoza P., Pfeifer K., Mohammed A., Hugosson F., Zhang H., Hsu A.W. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife. 2015;4:e09811. doi: 10.7554/eLife.09811. [DOI] [PMC free article] [PubMed] [Google Scholar]; Guan, J., Umapathy, G., Yamazaki, Y., Wolfstetter, G., Mendoza, P., Pfeifer, K., Mohammed, A., Hugosson, F., Zhang, H., Hsu, A.W., et al. (2015). FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife 4, e09811. [DOI] [PMC free article] [PubMed]
- Hallberg B., Palmer R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016;27(Suppl 3):iii4–iii15. doi: 10.1093/annonc/mdw301. [DOI] [PubMed] [Google Scholar]; Hallberg, B., and Palmer, R.H.. (2016). The role of the ALK receptor in cancer biology. Ann. Oncol. 27 Suppl 3, iii4-iii15. [DOI] [PubMed]
- Haq R., Yokoyama S., Hawryluk E.B., Jonsson G.B., Frederick D.T., McHenry K., Porter D., Tran T.N., Love K.T., Langer R. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc. Natl. Acad. Sci. U S A. 2013;110:4321–4326. doi: 10.1073/pnas.1205575110. [DOI] [PMC free article] [PubMed] [Google Scholar]; Haq, R., Yokoyama, S., Hawryluk, E.B., Jonsson, G.B., Frederick, D.T., McHenry, K., Porter, D., Tran, T.N., Love, K.T., Langer, R., et al. (2013). BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc. Natl. Acad. Sci. U S A 110, 4321-4326. [DOI] [PMC free article] [PubMed]
- Housman G., Byler S., Heerboth S., Lapinska K., Longacre M., Snyder N., Sarkar S. Drug resistance in cancer: an overview. Cancers (Basel) 2014;6:1769–1792. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]; Housman, G., Byler, S., Heerboth, S., Lapinska, K., Longacre, M., Snyder, N., and Sarkar, S.. (2014). Drug resistance in cancer: an overview. Cancers (Basel) 6, 1769-1792. [DOI] [PMC free article] [PubMed]
- Johnson D.B., Menzies A.M., Zimmer L., Eroglu Z., Ye F., Zhao S., Rizos H., Sucker A., Scolyer R.A., Gutzmer R. Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer. 2015;51:2792–2799. doi: 10.1016/j.ejca.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]; Johnson, D.B., Menzies, A.M., Zimmer, L., Eroglu, Z., Ye, F., Zhao, S., Rizos, H., Sucker, A., Scolyer, R.A., Gutzmer, R., et al. (2015). Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 51, 2792-2799. [DOI] [PMC free article] [PubMed]
- Kotschy A., Szlavik Z., Murray J., Davidson J., Maragno A.L., Le Toumelin-Braizat G., Chanrion M., Kelly G.L., Gong J.N., Moujalled D.M. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–482. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]; Kotschy, A., Szlavik, Z., Murray, J., Davidson, J., Maragno, A.L., Le Toumelin-Braizat, G., Chanrion, M., Kelly, G.L., Gong, J.N., Moujalled, D.M., et al. (2016). The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477-482. [DOI] [PubMed]
- Krepler C., Xiao M., Sproesser K., Brafford P.A., Shannan B., Beqiri M., Liu Q., Xu W., Garman B., Nathanson K.L. Personalized preclinical trials in BRAF inhibitor-resistant patient-derived xenograft models identify second-line combination therapies. Clin. Cancer Res. 2016;22:1592–1602. doi: 10.1158/1078-0432.CCR-15-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]; Krepler, C., Xiao, M., Sproesser, K., Brafford, P.A., Shannan, B., Beqiri, M., Liu, Q., Xu, W., Garman, B., Nathanson, K.L., et al. (2016). Personalized preclinical trials in BRAF inhibitor-resistant patient-derived xenograft models identify second-line combination therapies. Clin. Cancer Res. 22, 1592-1602. [DOI] [PMC free article] [PubMed]
- Lamant L., Dastugue N., Pulford K., Delsol G., Mariame B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood. 1999;93:3088–3095. [PubMed] [Google Scholar]; Lamant, L., Dastugue, N., Pulford, K., Delsol, G., and Mariame, B.. (1999). A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 93, 3088-3095. [PubMed]
- Lassen A., Atefi M., Robert L., Wong D.J., Cerniglia M., Comin-Anduix B., Ribas A. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol. Cancer. 2014;13:83. doi: 10.1186/1476-4598-13-83. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lassen, A., Atefi, M., Robert, L., Wong, D.J., Cerniglia, M., Comin-Anduix, B., and Ribas, A.. (2014). Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol. Cancer 13, 83. [DOI] [PMC free article] [PubMed]
- Long G.V., Fung C., Menzies A.M., Pupo G.M., Carlino M.S., Hyman J., Shahheydari H., Tembe V., Thompson J.F., Saw R.P. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]; Long, G.V., Fung, C., Menzies, A.M., Pupo, G.M., Carlino, M.S., Hyman, J., Shahheydari, H., Tembe, V., Thompson, J.F., Saw, R.P., et al. (2014). Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 5, 5694. [DOI] [PubMed]
- Miao B., Ji Z., Tan L., Taylor M., Zhang J., Choi H.G., Frederick D.T., Kumar R., Wargo J.A., Flaherty K.T. EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 2015;5:274–287. doi: 10.1158/2159-8290.CD-14-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]; Miao, B., Ji, Z., Tan, L., Taylor, M., Zhang, J., Choi, H.G., Frederick, D.T., Kumar, R., Wargo, J.A., Flaherty, K.T., et al. (2015). EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 5, 274-287. [DOI] [PMC free article] [PubMed]
- Miller A.J., Mihm M.C., Jr. Melanoma. N. Engl. J. Med. 2006;355:51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]; Miller, A.J., and Mihm, M.C., Jr. (2006). Melanoma. N. Engl. J. Med. 355, 51-65. [DOI] [PubMed]
- Mohammad R.M., Muqbil I., Lowe L., Yedjou C., Hsu H.Y., Lin L.T., Siegelin M.D., Fimognari C., Kumar N.B., Dou Q.P. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015;35(Suppl):S78–S103. doi: 10.1016/j.semcancer.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mohammad, R.M., Muqbil, I., Lowe, L., Yedjou, C., Hsu, H.Y., Lin, L.T., Siegelin, M.D., Fimognari, C., Kumar, N.B., Dou, Q.P., et al. (2015). Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 35 Suppl, S78-S103. [DOI] [PMC free article] [PubMed]
- Montero J., Letai A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018;25:56–64. doi: 10.1038/cdd.2017.183. [DOI] [PMC free article] [PubMed] [Google Scholar]; Montero, J., and Letai, A.. (2018). Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 25, 56-64. [DOI] [PMC free article] [PubMed]
- Morris S.W., Kirstein M.N., Valentine M.B., Dittmer K., Shapiro D.N., Look A.T., Saltman D.L. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1995;267:316–317. doi: 10.1126/science.267.5196.316-b. [DOI] [PubMed] [Google Scholar]; Morris, S.W., Kirstein, M.N., Valentine, M.B., Dittmer, K., Shapiro, D.N., Look, A.T., and Saltman, D.L.. (1995). Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 267, 316-317. [DOI] [PubMed]
- Morris S.W., Kirstein M.N., Valentine M.B., Dittmer K.G., Shapiro D.N., Saltman D.L., Look A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]; Morris, S.W., Kirstein, M.N., Valentine, M.B., Dittmer, K.G., Shapiro, D.N., Saltman, D.L., and Look, A.T.. (1994). Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 263, 1281-1284. [DOI] [PubMed]
- Nazarian R., Shi H., Wang Q., Kong X., Koya R.C., Lee H., Chen Z., Lee M.K., Attar N., Sazegar H. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nazarian, R., Shi, H., Wang, Q., Kong, X., Koya, R.C., Lee, H., Chen, Z., Lee, M.K., Attar, N., Sazegar, H., et al. (2010). Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973-977. [DOI] [PMC free article] [PubMed]
- O'Connell M.P., Marchbank K., Webster M.R., Valiga A.A., Kaur A., Vultur A., Li L., Herlyn M., Villanueva J., Liu Q. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. 2013;3:1378–1393. doi: 10.1158/2159-8290.CD-13-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]; O'Connell, M.P., Marchbank, K., Webster, M.R., Valiga, A.A., Kaur, A., Vultur, A., Li, L., Herlyn, M., Villanueva, J., Liu, Q., et al. (2013). Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. 3, 1378-1393. [DOI] [PMC free article] [PubMed]
- Obenauf A.C., Zou Y., Ji A.L., Vanharanta S., Shu W., Shi H., Kong X., Bosenberg M.C., Wiesner T., Rosen N. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368–372. doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]; Obenauf, A.C., Zou, Y., Ji, A.L., Vanharanta, S., Shu, W., Shi, H., Kong, X., Bosenberg, M.C., Wiesner, T., Rosen, N., et al. (2015). Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 520, 368-372. [DOI] [PMC free article] [PubMed]
- Pao W., Miller V.A., Politi K.A., Riely G.J., Somwar R., Zakowski M.F., Kris M.G., Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pao, W., Miller, V.A., Politi, K.A., Riely, G.J., Somwar, R., Zakowski, M.F., Kris, M.G., and Varmus, H.. (2005). Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2, e73. [DOI] [PMC free article] [PubMed]
- Piva R., Pellegrino E., Mattioli M., Agnelli L., Lombardi L., Boccalatte F., Costa G., Ruggeri B.A., Cheng M., Chiarle R. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J. Clin. Invest. 2006;116:3171–3182. doi: 10.1172/JCI29401. [DOI] [PMC free article] [PubMed] [Google Scholar]; Piva, R., Pellegrino, E., Mattioli, M., Agnelli, L., Lombardi, L., Boccalatte, F., Costa, G., Ruggeri, B.A., Cheng, M., Chiarle, R., et al. (2006). Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J. Clin. Invest. 116, 3171-3182. [DOI] [PMC free article] [PubMed]
- Poulikakos P.I., Rosen N. Mutant BRAF melanomas–dependence and resistance. Cancer Cell. 2011;19:11–15. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]; Poulikakos, P.I., and Rosen, N.. (2011). Mutant BRAF melanomas-dependence and resistance. Cancer Cell 19, 11-15. [DOI] [PubMed]
- Rambow F., Rogiers A., Marin-Bejar O., Aibar S., Femel J., Dewaele M., Karras P., Brown D., Chang Y.H., Debiec-Rychter M. Toward minimal residual disease-directed therapy in melanoma. Cell. 2018;174:843–855.e19. doi: 10.1016/j.cell.2018.06.025. [DOI] [PubMed] [Google Scholar]; Rambow, F., Rogiers, A., Marin-Bejar, O., Aibar, S., Femel, J., Dewaele, M., Karras, P., Brown, D., Chang, Y.H., Debiec-Rychter, M., et al. (2018). Toward minimal residual disease-directed therapy in melanoma. Cell 174, 843-855.e19. [DOI] [PubMed]
- Reshetnyak A.V., Murray P.B., Shi X., Mo E.S., Mohanty J., Tome F., Bai H., Gunel M., Lax I., Schlessinger J. Augmentor alpha and beta (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. U S A. 2015;112:15862–15867. doi: 10.1073/pnas.1520099112. [DOI] [PMC free article] [PubMed] [Google Scholar]; Reshetnyak, A.V., Murray, P.B., Shi, X., Mo, E.S., Mohanty, J., Tome, F., Bai, H., Gunel, M., Lax, I., and Schlessinger, J.. (2015). Augmentor alpha and beta (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. U S A 112, 15862-15867. [DOI] [PMC free article] [PubMed]
- Rizos H., Menzies A.M., Pupo G.M., Carlino M.S., Fung C., Hyman J., Haydu L.E., Mijatov B., Becker T.M., Boyd S.C. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin. Cancer Res. 2014;20:1965–1977. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]; Rizos, H., Menzies, A.M., Pupo, G.M., Carlino, M.S., Fung, C., Hyman, J., Haydu, L.E., Mijatov, B., Becker, T.M., Boyd, S.C., et al. (2014). BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin. Cancer Res. 20, 1965-1977. [DOI] [PubMed]
- Roskoski R., Jr. Anaplastic lymphoma kinase (ALK): structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 2013;68:68–94. doi: 10.1016/j.phrs.2012.11.007. [DOI] [PubMed] [Google Scholar]; Roskoski, R., Jr. (2013). Anaplastic lymphoma kinase (ALK): structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 68, 68-94. [DOI] [PubMed]
- Salgia R., Kulkarni P. The genetic/non-genetic duality of drug 'resistance' in cancer. Trends Cancer. 2018;4:110–118. doi: 10.1016/j.trecan.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; Salgia, R., and Kulkarni, P.. (2018). The genetic/non-genetic duality of drug 'resistance' in cancer. Trends Cancer 4, 110-118. [DOI] [PMC free article] [PubMed]
- Schadendorf D., Fisher D.E., Garbe C., Gershenwald J.E., Grob J.J., Halpern A., Herlyn M., Marchetti M.A., McArthur G., Ribas A. Melanoma. Nat. Rev. Dis. Primers. 2015;1:15003. doi: 10.1038/nrdp.2015.3. [DOI] [PubMed] [Google Scholar]; Schadendorf, D., Fisher, D.E., Garbe, C., Gershenwald, J.E., Grob, J.J., Halpern, A., Herlyn, M., Marchetti, M.A., McArthur, G., Ribas, A., et al. (2015). Melanoma. Nat. Rev. Dis. Primers 1, 15003. [DOI] [PubMed]
- Shaffer S.M., Dunagin M.C., Torborg S.R., Torre E.A., Emert B., Krepler C., Beqiri M., Sproesser K., Brafford P.A., Xiao M. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431–435. doi: 10.1038/nature22794. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shaffer, S.M., Dunagin, M.C., Torborg, S.R., Torre, E.A., Emert, B., Krepler, C., Beqiri, M., Sproesser, K., Brafford, P.A., Xiao, M., et al. (2017). Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431-435. [DOI] [PMC free article] [PubMed]
- Shah N.P., Tran C., Lee F.Y., Chen P., Norris D., Sawyers C.L. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]; Shah, N.P., Tran, C., Lee, F.Y., Chen, P., Norris, D., and Sawyers, C.L.. (2004). Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 305, 399-401. [DOI] [PubMed]
- Soda M., Choi Y.L., Enomoto M., Takada S., Yamashita Y., Ishikawa S., Fujiwara S., Watanabe H., Kurashina K., Hatanaka H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]; Soda, M., Choi, Y.L., Enomoto, M., Takada, S., Yamashita, Y., Ishikawa, S., Fujiwara, S., Watanabe, H., Kurashina, K., Hatanaka, H., et al. (2007). Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561-566. [DOI] [PubMed]
- Souers A.J., Leverson J.D., Boghaert E.R., Ackler S.L., Catron N.D., Chen J., Dayton B.D., Ding H., Enschede S.H., Fairbrother W.J. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]; Souers, A.J., Leverson, J.D., Boghaert, E.R., Ackler, S.L., Catron, N.D., Chen, J., Dayton, B.D., Ding, H., Enschede, S.H., Fairbrother, W.J., et al. (2013). ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202-208. [DOI] [PubMed]
- Srivastava R.K., Sasaki C.Y., Hardwick J.M., Longo D.L. Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription. J. Exp. Med. 1999;190:253–265. doi: 10.1084/jem.190.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]; Srivastava, R.K., Sasaki, C.Y., Hardwick, J.M., and Longo, D.L.. (1999). Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription. J. Exp. Med. 190, 253-265. [DOI] [PMC free article] [PubMed]
- Tartari C.J., Gunby R.H., Coluccia A.M., Sottocornola R., Cimbro B., Scapozza L., Donella-Deana A., Pinna L.A., Gambacorti-Passerini C. Characterization of some molecular mechanisms governing autoactivation of the catalytic domain of the anaplastic lymphoma kinase. J. Biol. Chem. 2008;283:3743–3750. doi: 10.1074/jbc.M706067200. [DOI] [PubMed] [Google Scholar]; Tartari, C.J., Gunby, R.H., Coluccia, A.M., Sottocornola, R., Cimbro, B., Scapozza, L., Donella-Deana, A., Pinna, L.A., and Gambacorti-Passerini, C.. (2008). Characterization of some molecular mechanisms governing autoactivation of the catalytic domain of the anaplastic lymphoma kinase. J. Biol. Chem. 283, 3743-3750. [DOI] [PubMed]
- van Dongen J.J., van der Velden V.H., Bruggemann M., Orfao A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. 2015;125:3996–4009. doi: 10.1182/blood-2015-03-580027. [DOI] [PMC free article] [PubMed] [Google Scholar]; van Dongen, J.J., van der Velden, V.H., Bruggemann, M., and Orfao, A.. (2015). Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood 125, 3996-4009. [DOI] [PMC free article] [PubMed]
- Verduzco D., Kuenzi B.M., Kinose F., Sondak V.K., Eroglu Z., Rix U., Smalley K.S.M. Ceritinib enhances the efficacy of trametinib in BRAF/NRAS-Wild-Type melanoma cell lines. Mol. Cancer Ther. 2018;17:73–83. doi: 10.1158/1535-7163.MCT-17-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]; Verduzco, D., Kuenzi, B.M., Kinose, F., Sondak, V.K., Eroglu, Z., Rix, U., and Smalley, K.S.M.. (2018). Ceritinib enhances the efficacy of trametinib in BRAF/NRAS-Wild-Type melanoma cell lines. Mol. Cancer Ther. 17, 73-83. [DOI] [PMC free article] [PubMed]
- Vergani E., Vallacchi V., Frigerio S., Deho P., Mondellini P., Perego P., Cassinelli G., Lanzi C., Testi M.A., Rivoltini L. Identification of MET and SRC activation in melanoma cell lines showing primary resistance to PLX4032. Neoplasia. 2011;13:1132–1142. doi: 10.1593/neo.111102. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vergani, E., Vallacchi, V., Frigerio, S., Deho, P., Mondellini, P., Perego, P., Cassinelli, G., Lanzi, C., Testi, M.A., Rivoltini, L., et al. (2011). Identification of MET and SRC activation in melanoma cell lines showing primary resistance to PLX4032. Neoplasia 13, 1132-1142. [DOI] [PMC free article] [PubMed]
- Villanueva J., Infante J.R., Krepler C., Reyes-Uribe P., Samanta M., Chen H.Y., Li B., Swoboda R.K., Wilson M., Vultur A. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4:1090–1099. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; Villanueva, J., Infante, J.R., Krepler, C., Reyes-Uribe, P., Samanta, M., Chen, H.Y., Li, B., Swoboda, R.K., Wilson, M., Vultur, A., et al. (2013). Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 4, 1090-1099. [DOI] [PMC free article] [PubMed]
- Villanueva J., Vultur A., Lee J.T., Somasundaram R., Fukunaga-Kalabis M., Cipolla A.K., Wubbenhorst B., Xu X., Gimotty P.A., Kee D. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; Villanueva, J., Vultur, A., Lee, J.T., Somasundaram, R., Fukunaga-Kalabis, M., Cipolla, A.K., Wubbenhorst, B., Xu, X., Gimotty, P.A., Kee, D., et al. (2010). Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18, 683-695. [DOI] [PMC free article] [PubMed]
- Wang G., Nikolovska-Coleska Z., Yang C.Y., Wang R., Tang G., Guo J., Shangary S., Qiu S., Gao W., Yang D. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J. Med. Chem. 2006;49:6139–6142. doi: 10.1021/jm060460o. [DOI] [PubMed] [Google Scholar]; Wang, G., Nikolovska-Coleska, Z., Yang, C.Y., Wang, R., Tang, G., Guo, J., Shangary, S., Qiu, S., Gao, W., Yang, D., et al. (2006). Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J. Med. Chem. 49, 6139-6142. [DOI] [PubMed]
- Wang J., Huang S.K., Marzese D.M., Hsu S.C., Kawas N.P., Chong K.K., Long G.V., Menzies A.M., Scolyer R.A., Izraely S. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J. Invest. Dermatol. 2015;135:532–541. doi: 10.1038/jid.2014.418. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang, J., Huang, S.K., Marzese, D.M., Hsu, S.C., Kawas, N.P., Chong, K.K., Long, G.V., Menzies, A.M., Scolyer, R.A., Izraely, S., et al. (2015). Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J. Invest. Dermatol. 135, 532-541. [DOI] [PMC free article] [PubMed]
- Wellbrock C., Karasarides M., Marais R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]; Wellbrock, C., Karasarides, M., and Marais, R.. (2004). The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 5, 875-885. [DOI] [PubMed]
- Wong D.J., Ribas A. Targeted therapy for melanoma. Cancer Treat Res. 2016;167:251–262. doi: 10.1007/978-3-319-22539-5_10. [DOI] [PubMed] [Google Scholar]; Wong, D.J., and Ribas, A.. (2016). Targeted therapy for melanoma. Cancer Treat Res. 167, 251-262. [DOI] [PubMed]
- Zbytek B., Carlson J.A., Granese J., Ross J., Mihm M.C., Jr., Slominski A. Current concepts of metastasis in melanoma. Expert Rev. Dermatol. 2008;3:569–585. doi: 10.1586/17469872.3.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zbytek, B., Carlson, J.A., Granese, J., Ross, J., Mihm, M.C., Jr., and Slominski, A.. (2008). Current concepts of metastasis in melanoma. Expert Rev. Dermatol. 3, 569-585. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.