

Abstract
Family trees have vast applications in multiple fields from genetics to anthropology and economics. However, the collection of extended family trees is tedious and usually relies on resources with limited geographical scope and complex data usage restrictions. Here, we collected 86 million profiles from publicly-available online data shared by genealogy enthusiasts. After extensive cleaning and validation, we obtained population-scale family trees, including a single pedigree of 13 million individuals. We leveraged the data to partition the genetic architecture of longevity by inspecting millions of relative pairs and to provide insights into the geographical dispersion of families. We also report a simple digital procedure to overlay other datasets with our resource in order to empower studies with population-scale genealogical data.
One Sentence Summary
Using massive crowd-sourced genealogy data, we created a population-scale family tree resource for scientific studies.
Family trees are mathematical graph structures that can capture mating and parenthood among humans. As such, the edges of the trees represent potential transmission lines for a wide variety of genetic, cultural, socio-demographic, and economic factors. Quantitative genetics is built on dissecting the interplay of these factors by overlaying data on family trees and analyzing the correlation of various classes of relatives (1–3). In addition, family trees can serve as a multiplier for genetic information through study designs that leverage genotype or phenotype data from relatives (4–7), analyzing parent-of-origin effects (8), refining heritability measures (9, 10), or improving individual risk assessment (11, 12). Beyond classical genetic applications, large-scale family trees have played an important role across disciplines, including human evolution (13, 14), anthropology (15), and economics (16).
Despite the range of applications, constructing population-scale family trees has been a labor-intensive process. Previous approaches mainly relied on local data repositories such as churches or vital record offices (14, 17, 18). But these approaches have limitations (19, 20): they require non-trivial resources to digitize the records and organize the data, the resulting trees are usually limited in geographical scope, and the data may be subject to strict usage protections. These challenges reduce demographic accessibility and complicates fusion with information such as genomic or health data.
Constructing and validating population scale family trees
Here, we leveraged genealogy-driven social media data to construct population-scale family trees. To this end, we focused on Geni.com, a crowd-sourcing website in the genealogy domain. Users can create individual profiles and upload family trees. The website automatically scans profiles to detect similarities and offers the option to merge the profiles when a match is detected. By merging, larger family trees are created that can be collaboratively co-managed to improve their accuracy. After obtaining relevant permissions, we downloaded over 86 million publicly available profiles (21). The input data consists of millions of individual profiles, each of which describes a person and any putative connections to other individuals in the dataset, along with any auxiliary data about the creator of the profile. Similar to other crowdsourcing projects (22), a small group of participants contributed the majority of genealogy profiles (fig. S1).
We organized the profiles into graph topologies that preserve the genealogical relationships between individuals (Fig. 1A). Biology dictates that a family tree should form a directed acyclic graph (DAG) where each individual has an in-degree that is less than or equal to two. However, 0.3% of the profiles resided in invalid biological topologies that included cycles (e.g. a person that is both the parent and child of another person) or an individual with more than two parents. We developed an automated pipeline to resolve local conflicts and prune invalid topologies (fig. S2) and benchmarked the performance of the pipeline against human genealogists (21). This resulted in >90% concordance between the pipeline and human decisions to resolve conflicts, generating 5.3 million disjoint family trees.
Fig. 1. Overview of the collected data.
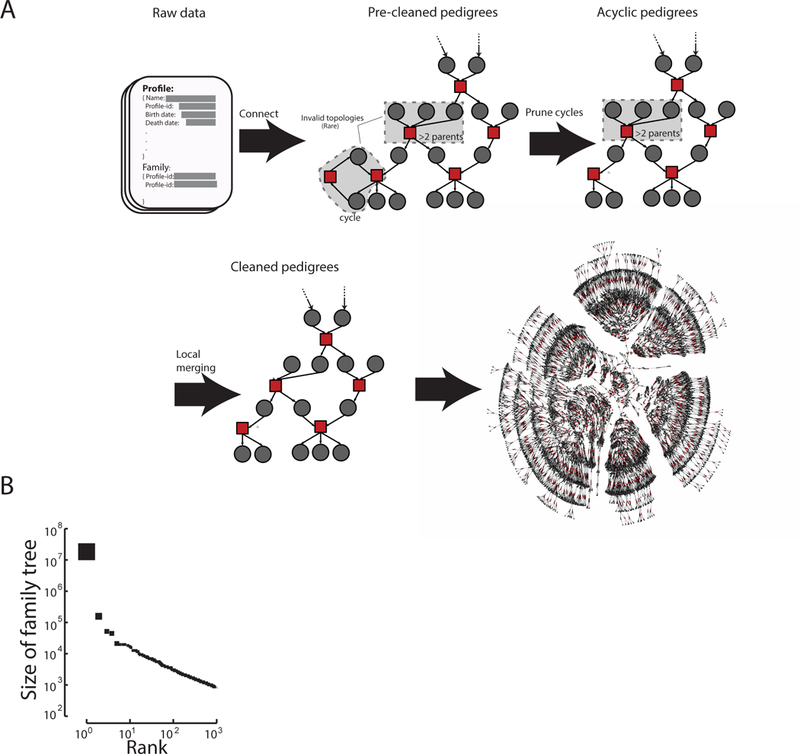
(A) The basic algorithmic steps to form valid pedigree structures from the input data available via the Geni API. Gray: profiles; Red: marriages (See fig. S2 for a comprehensive overview). The last step shows an example of a real pedigree from the website with ~6,000 individuals spanning about 7 generations (B) The size distribution of the largest 1,000 family trees after data cleaning sorted by size.
The largest family tree in the processed data spanned 13 million individuals who were connected by shared ancestry and marriage (Fig. 1B). On average, the tree spanned 11 generations between each terminal descendant and their founders (fig. S3). The size of this pedigree fits what is expected as familial genealogies coalesce at a logarithmic rate compared to the size of the population (23).
We evaluated the structure of the tree by inspecting the genetic segregation of unilineal markers. We obtained mitochondria (mtDNA) and Y-STR haplotypes to compare multiple pairs of relatives in our graph (21). The mtDNA data was available for 211 lineages and spanned a total of 1768 transmission events (i.e. graph edges), whereas the Y-STR data was available for 27 lineages that spanned 324 total transmission events. Using a prior of no more than a single non-paternity event per lineage, we estimated a non-maternity rate of 0.3% per meiosis and non-paternity rate of 1.9% per meiosis. This rate of non-paternity matched previous rates of Y-chromosome studies (24, 25) and the non-maternity rate was close to historical rates of adoption of an unrelated member in the US (26). Taken together, these results demonstrate that millions of genealogists can collaborate in order to produce high quality population-scale family trees.
Extracting demographic data
We found that lifespan in the Geni.com profiles was largely concordant with reports generated by traditional demographic approaches. First, we extracted demographic information from the collected profiles with exact birth and death dates, which show higher quality compared to profiles with only year resolution for these events (fig. S4). The data reflected historical events and trends such as elevated death rates at military age during the American Civil War, WWI, and WWII, and a reduction in child mortality during the 20th century (Fig. 2A). We compared the average lifespan in our collection to a worldwide historical analysis covering the years 1840–2000 (27). We found an R2=0.95 between the expected lifespan from historical data and the Geni dataset (Fig. 2B) and a 98% concordance with historical distributions reported by the Human Mortality Database (HMD) (Figs. 2C; fig. S5).
Fig. 2. Analysis and validation of demographic data.
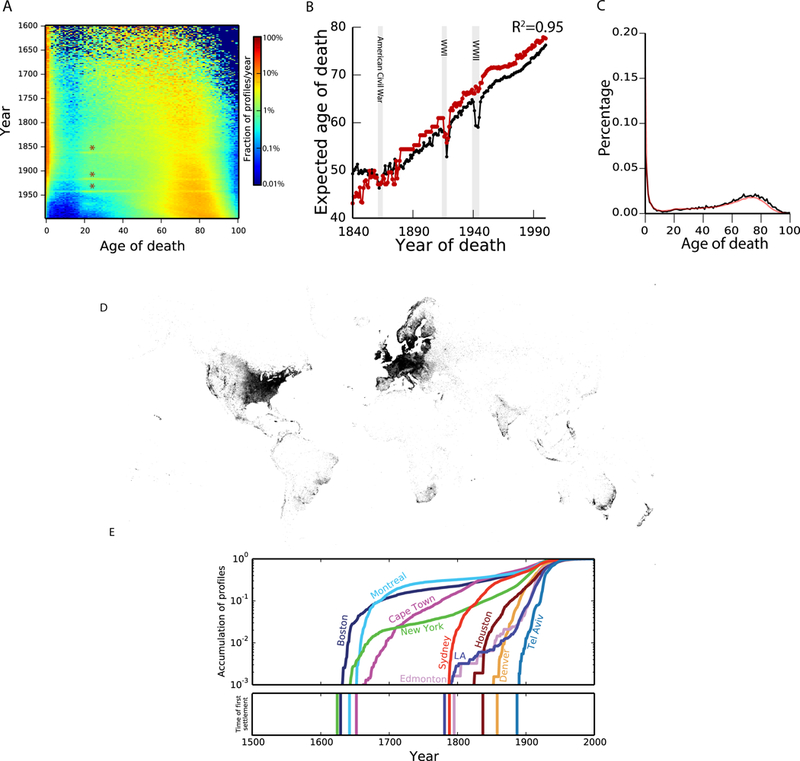
(A) Distribution of life expectancy per year. The colors correspond to the frequency of profiles of individuals who died at a certain age for each year. Stars indicate deaths during military ages in the civil war, WWI, and WWII (B) The expected lifespan in Geni (black) and the Oeppen & Vaupel study (red, ref: 30) as a function of year of death (C) Comparing the lifespan distributions versus Geni (black) and HMD (red) (Also see fig. S5A) (D) The geographic distribution of the annotated place of birth information. Every pixel corresponds to a profile in the dataset (E) Validation of geographical assignment by historical trends. Top: the cumulative distribution of profiles since 1500 for each city on a logarithmic scale as a function of time. Bottom: year of first settlement in the city.
Next, we extracted the geographic locations of life events using a combination of an automated geo-parsing pipeline and structured text manually curated and approved by genealogists (21) (fig. S6A). Overall, we were able to place about 16 million profiles into longitude/latitude coordinates, typically at fine-scale geographic resolution, without major differences in quality between the automated geo-parsing and manual curations for subsequent analyses (fig. S6B) (21). The profiles were distributed across a wide range of locations in the Western World (Fig. 2D; fig. S7) with 55% from Europe and 30% from North America. We analyzed profiles in ten cities across the globe and found that the first appearance of profiles was only after the known first settlement date for nearly all of the cities, suggesting good spatiotemporal assignment of profiles (Fig. 2E). Movie S1 presents the place of birth of individuals in the Geni dataset in 5 year intervals from 1400 to 1900 along with known migration events.
We were concerned that the Geni.com profiles might suffer from certain socio-economic ascertainment biases and therefore would not reflect the local population. To evaluate this concern, we collected ~80,000 publicly available death certificates from the Vermont Department of Health for every death in this state between 1985 and 2010. These records have extensive information for each individual, including education level, place of birth, and a cause of death in an ICD-9 code. Approximately one thousand individuals in Geni overlapped this death certificate collection. We compared the education level, birth state, and ICD-9 code between these ~1000 Geni profiles and the entire Vermont collection. For all three parameters, we found >98% concordance between the distribution of these key sociodemographic attributes in the Geni profiles in Vermont and the entire state of Vermont (Table S1–S3). Overall, this high level of consistency argues against severe socioeconomic ascertainment. Table S4 reports key demographic and genetic attributes for various familial relationships from parent-child via great-great-grandparents to fourth cousins.
Characterizing the genetic architecture of longevity
We leveraged the Geni dataset to characterize the genetic architecture of human longevity, which exhibits complex genetics likely to involve a range of physiological and behavioral endophenotypes (28, 29). Narrow-sense heritability (h2) of longevity has been estimated to be around 15%−30% (Table S5) (30–35). Genome-wide association studies have had limited success in identifying genetic variants associated with longevity (36–38). This relatively large proportion of missing heritability can be explained by the following: (A) longevity has non-additive components that create upward bias in estimates of heritability (39), (B) estimators of heritability are biased due to unaccounted environmental effects (10), (C) the trait is highly polygenic and requires larger cohorts to identify the underlying variants (40). We thus sought to harness our resource and build a model for the sources of genetic variance in longevity that jointly evaluates additivity, dominance, epistasis, shared household effects, spatiotemporal trends, and random noise.
We adjusted longevity to be the difference between the age of death from the expected lifespan using a model that we trained with 3 million individuals. Our model includes spatiotemporal and sex effects and was the best among 10 different models that adjusted various spatio-temporal attributes (fig. S8). We also validated this model by estimating the narrow-sense heritability of longevity (h2) according to the mid-parent design (41) with nearly 130,000 parent-child trios. This process yielded h2mid-parent = 12.2% (s.e.=0.4%) (Fig. 3A), which is on the lower end but in the range of previous heritability estimates (Table S5). Consistent with previous studies, we did not observe any temporal trend in mid-parent heritability (Fig. 3B).
Fig. 3. The genetic architecture of longevity.
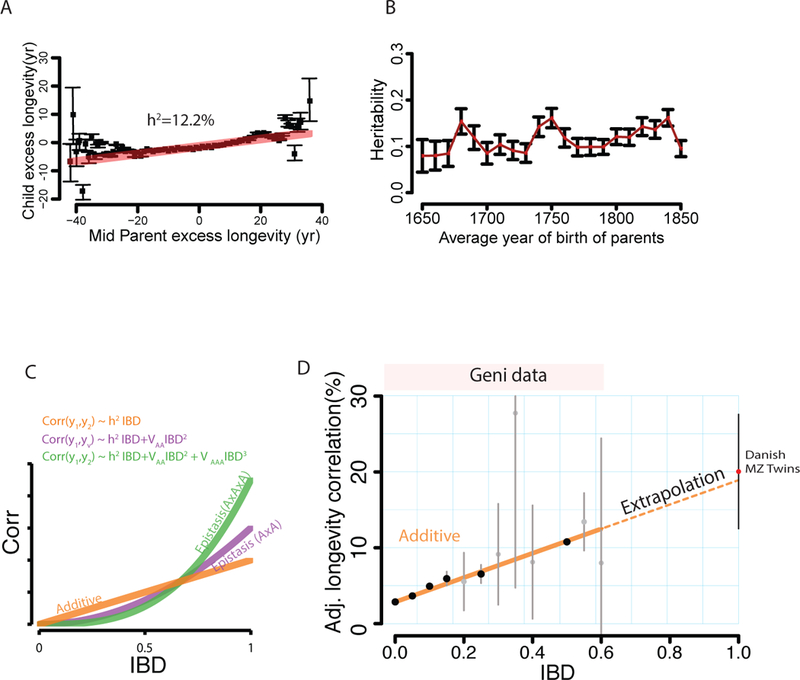
(A) The regression (red) of child longevity on its mid-parent longevity (defined as difference of age of death from the expected lifespan). Black: the average longevity of children binned by the mid-parent value. Gray: estimated 95% confidence intervals (B) The estimated narrow-sense heritability (red squares) with 95% confidence intervals (black bars) obtained by the mid-parent design stratified by the average decade of birth of the parents (C) The correlation of a trait as a function of IBD under strict additive (h2, orange), squared (VAA, purple), and cubic (VAAA, green) epistasis architectures after dormancy adjustments (D) The average longevity correlation as a function of IBD (black circles) grouped in 5% increments (gray: 95% CI) after adjusting for dominancy. Dotted line: the extrapolation of the models towards MZ twins from the Danish Twin Registry (red circle).
We partitioned the source of genetic variance of longevity using more than three million pairs of relatives from full sibling to 4th cousin (21). We measured the variance explained by an additive component, pairwise epistatic model, 3-way epistasis, and dominancy (Fig. 3C). To mitigate correlations due to non-genetic factors, these three million pairs were all sex-concordant to address residual sex differences not accounted for by our longevity adjustments (fig. S9) and do not include relatives who are likely to have died due to environmental catastrophes or in major wars (fig. S10). We also refined the genetic correlation of the relatives by considering multiple genealogical paths (fig. S11–S13).
The analysis of longevity in these 3 million of pairs of relatives showed a robust additive genetic component, a small impact of dominance, and no detectable epistasis (Fig. 3D; Table S6) (21). Additivity was highly significant (padditive<10−318) with an estimated h2sex-concordant/relatives = 16.1% (s.e.=0.4%), similar to the heritability estimated from sex-concordant parent-child pairs h2concordant/parent-child = 15.0% (s.e.=0.4%). The maximum-likelihood estimate for dominance was around 4% but the epistatic terms converged to zero despite the substantial amount of data. Other model selection procedures such as Mean Squared Error (MSE) analysis and Bayesian Information Criterion (BIC) argued against pervasive epistatic contribution to longevity variance in the population (21).
We tested the ability of our model to predict the longevity correlation of an orthogonal dataset of 810 monozygotic (MZ) twin pairs collected by the Danish Twin Registry (Fig. 3D) (42). Our inferred model for longevity accurately predicted the observed correlation of this twin cohort with 1% difference, well within the sampling error for the mean twin correlation (s.e. = 3.2%). We also evaluated an extensive array of additional analyses that included various adjustments for environmental components and other confounders (fig. S14–15) (21). In all cases, additivity explained 15.8%−16.9% of the longevity estimates, dominance explained 2%−4% and no evidence for epistatic interactions could be detected using our procedure.
We also estimated the additive and epistatic components using a method that allows rapid estimation of variance components of extremely large relationship matrices, called sparse Cholesky factorization linear mixed models (Sci-LMM) (43). This method takes into account a kinship coefficient matrix of 250 million pairs of related individuals in the Geni dataset and includes adjustment for population structure, sex, and year of birth. We observed an additivity of 17.8% (s.e=0.84%) and a pairwise epistatic component that was not significantly different from zero (21).
Taken together, our results across multiple study designs (fig. S16) indicated that the limited ability of GWA studies so far to associate variants with longevity cannot be attributed to statistical epistasis. Importantly, this does not rule out the existence of molecular interactions between genes contributing to this trait (44–47). Based on a large number of data points and study designs, we measured an additive component (h2 ≃16%) that is considerably smaller than the value generally cited in the literature of 25%. These results indicate that previous studies are likely to have overestimated the heritability of longevity. As such, we should lower our expectations about our ability to predict longevity from genomic data and presumably to identify causal genetic variants.
Assessment of theories of familial dispersion
Familial dispersion is a major driving force of various genetic, economical, and demographic processes (48). Previous work has primarily relied on vital records from a limited geographical scope (49, 50) or used indirect inference from genetic datasets that mainly illuminate distant historical events (51).
We harnessed our resource to evaluate patterns of human migration. First, we analyzed sex-specific migration patterns (21) to resolve conflicting results regarding sex bias in human migration (52). Our results indicate that females migrate more than males in Western societies but over shorter distances. The median mother-child distances were significantly larger (Wilcox, one-tailed, p<10−90) by a factor of 1.6x than father-child distances (Fig. 4A). This trend appeared throughout the 300 years of our analysis window, including in the most recent birth cohort, and was observed both in North American (Wilcox, one-tailed, p<10−23) and European duos (Wilcox, one-tailed, p<10−87). On the other hand, we found that the average mother-child distances (fig. S17) were significantly shorter than the father-child distances (t-test, p<10−90), suggesting that long-range migration events are biased towards males. Consistent with this pattern, fathers displayed a significantly (p<10−83) higher frequency than mothers to be born in a different country than their offspring (Fig. 4B). Again, this pattern was evident when restricting the data to North American or European duos. Taken together, males and females in Western societies show different migration distributions in which patrilocality occurs only in relatively local migration events and large-scale events that usually involve a change of country are more common in males than females.
Fig. 4. Analysis of familial dispersion.
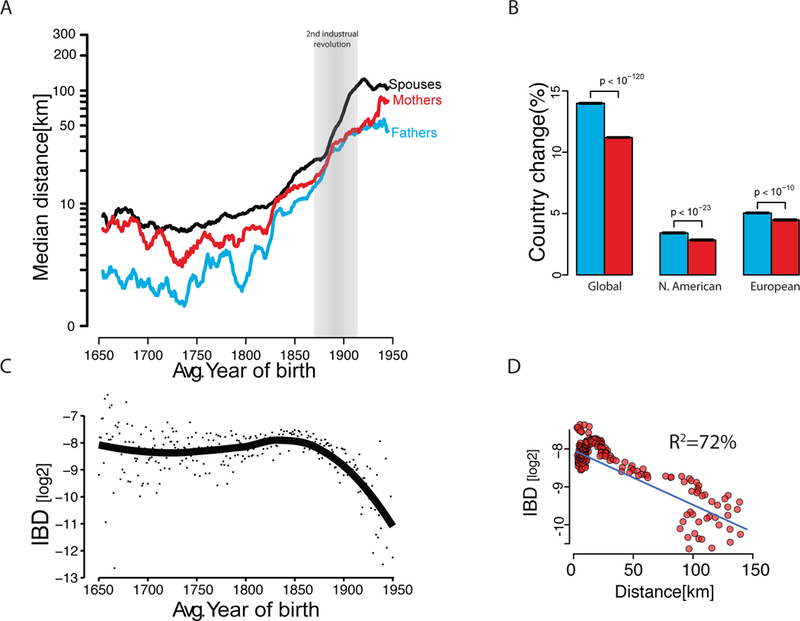
(A) The median distance [log10 x+1] of father-offspring places of birth (cyan), mother-offspring (red), and marital radius (black) as a function of time (average year of birth) (B) The rate of change in the country of birth for father-offspring (cyan) or mother-offspring (red) stratified by major geographic areas (C) The average IBD [log2] between couples as a function of average year of birth. Individual dots represent the measured average per year. Black line denotes the smooth trend using locally weighted regression (D) The IBD of couples as a function of marital radius. Blue line denotes best linear regression line in log-log space.
Next, we inspected the marital radius (the distance between mates’ places of birth) and its effect on the genetic relatedness of couples (21). The isolation by distance theory of Malécot predicts that increases in the marital radius should exponentially decrease the genetic relatedness of individuals (53). But the magnitude of these forces is also a function of factors such as taboos against cousin marriages (54).
We started by analysing temporal changes in the birth locations of couples in our cohort. Prior to the Industrial Revolution (<1750), most marriages occurred between people born only 10km from each other (Fig. 4A [black line]). Similar patterns were found when analysing European-born individuals (fig. S18) or North American-born individuals (fig. S19). After the beginning of the second Industrial Revolution (1870), the marital radius rapidly increased and reached ~100km for most marriages in the birth cohort in 1950. Next, we analysed the genetic relatedness (IBD) of couples as measured by tracing their genealogical ties (Fig. 4C). Between 1650 and 1850, the average IBD of couples was relatively stable and on the order of ~4th cousins, whereas IBD exhibited a rapid decrease post-1850. Overall, the median marital radius for each year showed a strong correlation (R2=72%) with the expected IBD between couples. Every 70km increase in the marital radius correlated with a decrease in the genetic relatedness of couples by one meiosis event (Fig. 4D). This correlation matches previous isolation by distance forces in continental regions (55). However, this trend was not consistent over time and exhibits three phases. For the pre-1800 birth cohorts, the correlation between marital distance and IBD was insignificant (p>0.2) and weak (R2=0.7%) (fig. S20A). Couples born around 1800–1850 showed a two-fold increase in their marital distance from 8km in 1800 to 19km in 1850. Marriages are usually about 20–25 years after birth and around this time (1820–1875) rapid transportation changes took place, such as the advent of railroad travel in most of Europe and the United States. However, the increase in marital distance was significantly (p<10−13) coupled with an increase in genetic relatedness, contrary to the isolation by distance theory (fig. S20B). Only for the cohorts born after 1850, did the data match (R2=80%) the theoretical model of isolation by distance (fig. S20C).
Taken together, the data shows a 50-year lag between the advent of increased familial dispersion and the decline of genetic relatedness between couples. During this time, individuals continued to marry relatives despite the increased distance. From these results, we hypothesize that changes in 19th century transportation were not the primary cause for decreased consanguinity. Rather, our results suggest that shifting cultural factors played a more important role in the recent reduction of genetic relatedness of couples in Western societies.
Discussion
In this work, we leveraged genealogy-driven media to build a dataset of human pedigrees of massive scale that covers nearly every country in the Western world. Multiple validation procedures indicated that it is possible to obtain a dataset that has similar quality to traditionally collected studies, but at much greater scale and lower cost.
We envision that this and similar large datasets can address quantitative aspects of human families, including genetics, anthropology, public health, and economics. Our tree and demographic data are available in a de-identified format, enabling static analysis of the Geni dataset. We also offer a dynamic method that enables fusing other datasets with our data based on digital consent of participants using the Geni API (fig. S21) (21). We have been using this one-click mechanism to overlay thousands of genomes with family trees on DNA.Land (56). Other projects can use a similar strategy to add large pedigrees to their existing data collection.
More generally, similar to previous studies (57, 58), our work demonstrates the synergistic power of a collaboration between basic research and consumer genetic genealogy datasets. With ever-growing digitization of humanity and the rise of consumer genetics (59), we believe that such collaborative efforts can be a valuable path to reach the dramatic scale of information needed to address fundamental questions in biomedical research.
Supplementary Material
Acknowledgments:
We thank D. Zielinski, G. Japhet, and J. Novembre for valuable comments and the Erlich lab members for constant support in pursuing this project. The authors also thank the Vermont Health Department for providing all death certificates. This study was supported by a generous gift from Andria and Paul Heafy (Y.E), the Burroughs Wellcome Fund Career Awards at the Scientific Interface (Y.E.), the Broad Institute’s SPARC: Catalytic Funding for Novel Collaborative Projects award (Y.E. and D.M.), by NIH grants R01 MH101244 and R03 HG006731 (A.L.P.), and Israeli Science Foundation grant 1678/12 (D.G). A.G., and Y.E. conducted the downloading, indexing, and organizing the data. J.K., A.G., M.W., B.M., M.G., M.S., and Y.E. developed the procedures to cleaning the family trees and extract demographic information. J.K., T.S., O.W., D.G., G.B., D.G.M, A.L.P, and Y.E. were involved in analysing the genetic architecture of longevity. J.P., M.W. and Y.E. conducted the analysis of human migration. J.K., T.S., O.W., D.G.M, A.L.P, and Y.E. wrote the manuscript. T.S and Y.E became employees of MyHeritage.com, the parent company of Geni.com, during the course of this study. The other authors did not declare relevant competing interests. The Geni dataset without names is available from Yaniv Erlich under the terms described on FamiLinx.org. The code for the API integration is available on https://github.com/TeamErlich/geni-integration-example, the code for Sci-LMM is available on: https://github.com/TalShor/SciLMM, and the code to download Geni profiles is available on: https://github.com/erlichya/geni-download. The Human Mortality Database (HMD) is available on http://www.mortality.org/. The Danish Twin Registry (DTR) data is available upon request from the University of Southern Denmark (https://www.sdu.dk/en/om_sdu/institutter_centre/ist_sundhedstjenesteforsk/centre/dtr). The findings, opinions and recommendations expressed therein are those of the authors and are not necessarily those of the DTR. The Vermont Death Certificate collection were obtained upon request from the Chief of Public Health Statistics of the Vermont Department of Health (http://www.healthvermont.gov/stats).
References and Notes
- 1.Fisher RA, Trans. R. Soc. Edinb. 52, 399–433 (1919). [Google Scholar]
- 2.Wright S, J. Agric. Res. 20, 557–585 (1921). [Google Scholar]
- 3.Tenesa A, Haley CS, Nat. Rev. Genet. 14, 139–149 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Kong A et al. , Nat. Genet. 40, 1068–1075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowe JK et al. , PLoS Genet. 5, e1000365 (2009). [Google Scholar]
- 6.Gudbjartsson DF et al. , Nat. Genet. 47, 435–444 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Liu JZ, Erlich Y, Pickrell JK, Nat. Genet. 49, 325–331 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Kong A et al. , Nature. 462, 868–874 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ober C, Abney M, McPeek MS, Am. J. Hum. Genet. 69, 1068–1079 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaitlen N et al. , PLoS Genet. 9, e1003520 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ, Annu. Rev. Public Health. 31, 69–87 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Do CB, Hinds DA, Francke U, Eriksson N, PLoS Genet. 8, e1002973 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lahdenperä M, Lummaa V, Helle S, Tremblay M, Russell AF, Nature. 428, 178–181 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Moreau C et al. , Science. 334, 1148–1150 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Helgason A, Pálsson S, Guðbjartsson DF, Stefánsson K, Science. 319, 813–816 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Modalsli J, “Multigenerational persistence: Evidence from 146 years of administrative data” (Discussion papers by Statistics Norway, Research Department, 2016; https://EconPapers.repec.org/RePEc:ssb:dispap:850). [Google Scholar]
- 17.Gulcher JR, Stefansson K, in Encyclopedia of Life Sciences (Wiley, New York, 2001; 10.1002/9780470015902.a0006270). [DOI] [Google Scholar]
- 18.Cannon Albright LA, Hum. Hered. 65, 209–220 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Albright LAC, in AMIA Annual Symposium Proceedings (American Medical Informatics Association, 2006), vol. 2006, p. 1161. [Google Scholar]
- 20.Stefansdottir V et al. , J. Community Genet. 4, 1–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.See supplementary materials on Science Online.
- 22.Kittur A, Chi E, Pendleton BA, Suh B, Mytkowicz T, World Wide Web. 1, 19 (2007). [Google Scholar]
- 23.Chang JT, Adv. Appl. Probab, 1002–1026 (1999). [Google Scholar]
- 24.Anderson K, Curr. Anthropol. 47, 513–520 (2006). [Google Scholar]
- 25.King TE, Jobling MA, Mol. Biol. Evol. 26, 1093–1102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maza P, Child Welf. Res. Notes. 9, 1–11 (1984). [Google Scholar]
- 27.Oeppen J, Vaupel JW, Science. 296, 1029–1031 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Sebastiani P, Perls TT, Front. Genet. 3, 277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marioni RE et al. , Proc. Natl. Acad. Sci. 113, 13366–13371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Philippe P, Opitz JM, Am. J. Med. Genet. 2, 121–129 (1978). [DOI] [PubMed] [Google Scholar]
- 31.Mayer PJ, Am. J. Hum. Biol. 3, 49–58 (1991). [DOI] [PubMed] [Google Scholar]
- 32.Ljungquist B, Berg S, Lanke J, McClearn GE, Pedersen NL, Gerontol J. A. Biol. Sci. Med. Sci. 53, M441–M446 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Herskind AM et al. , Hum. Genet. 97, 319–323 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Mitchell BD et al. , Am. J. Med. Genet. 102, 346–352 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Kerber RA, O’Brien E, Smith KR, Cawthon RM, Gerontol J. A. Biol. Sci. Med. Sci. 56, B130–B139 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Sebastiani P et al. , PloS One. 7, e29848 (2012). [Google Scholar]
- 37.Deelen J et al. , Hum. Mol. Genet. 23, 4420–4432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erikson GA et al. , Cell. 165, 1002–1011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zuk O, Hechter E, Sunyaev SR, Lander ES, Proc. Natl. Acad. Sci. 109, 1193–1198 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyle EA, Li YI, Pritchard JK, Cell. 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Visscher PM, Hill WG, Wray NR, Nat. Rev. Genet. 9, 255–266 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Skytthe A, Kyvik KO, Holm NV, Christensen K, Scand. J. Public Health. 39, 75–78 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shor T, Geiger D, Erlich Y, Weissbrod O, bioRxiv (2018), doi: 10.1101/256396 [DOI] [Google Scholar]
- 44.Li W, Reich J, Hum. Hered. 50, 334–349 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Phillips PC, Nat. Rev. Genet. 9, 855–867 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cordell HJ, Nat. Rev. Genet. 10, 392–404 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei W-H, Hemani G, Haley CS, Nat. Rev. Genet. 15, 722–733 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Cavalli-Sforza LL, Menozzi P, Piazza A, The History and Geography of Human Genes (Princeton university press, 1994). [Google Scholar]
- 49.Wijsman EM, Cavalli-Sforza LL, Annu. Rev. Ecol. Syst. 15, 279–301 (1984). [Google Scholar]
- 50.Labouriau R, Amorim A, Genetics. 178, 601–603 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veeramah KR, Hammer MF, Nat. Rev. Genet. 15, 149–162 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Lawson Handley LJ, Perrin N, Mol. Ecol. 16, 1559–1578 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Malécot G, The Mathematics of Heredity (Freeman, 1970). [Google Scholar]
- 54.Cavalli-Sforza LL, Moroni A, Zei G, Consanguinity, Inbreeding, and Genetic Drift in Italy (Princeton University Press, 2004), vol. 39. [Google Scholar]
- 55.Relethford JH, Brennan ER, Hum. Biol, 315–327 (1982). [PubMed] [Google Scholar]
- 56.Yuan J et al. , Nat. Genet. (2018), doi: 10.1038/s41588-017-0021-8 [DOI] [PubMed] [Google Scholar]
- 57.Pickrell JK et al. , Nat. Genet. 48, 709 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han E et al. , Nat. Commun. 8, 14238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khan R, Mittelman D, Genome Biol. 14, 1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tarjan R, SIAM J Comput. 1, 146–160 (1972). [Google Scholar]
- 61.Gymrek M, McGuire AL, Golan D, Halperin E, Erlich Y, Science. 339, 321–324 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Walsh B, Genetics. 158, 897–912 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abney M, Bioinformatics. 25, 1561–1563 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abney M, McPeek MS, Ober C, Am J Hum Genet. 66, 629–650 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rao D, MacLean C, Morton N, Yee S, Am. J. Hum. Genet. 27, 509 (1975). [PMC free article] [PubMed] [Google Scholar]
- 66.Kempthorne O, Proc. R. Soc. Lond. B Biol. Sci. 143, 103–113 (1954). [PubMed] [Google Scholar]
- 67.Elwert F, Christakis NA, Am. J. Public Health. 98, 2092–2098 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rostila M, Saarela J, Kawachi I, Am. J. Epidemiol. 176, 338–346 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Chen G-B, Front. Genet. 5, 107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Golan D, Rosset S, arXiv:1305.5363 (2013). [Google Scholar]
- 71.Speed D, Balding DJ, Nat. Rev. Genet. 16, 33–44 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Henderson CR, Biometrics, 69–83 (1976). [Google Scholar]
- 73.Mrode RA, Linear Models for the Prediction of Animal Breeding Values (Cabi, 2014). [Google Scholar]
- 74.Meuwissen T, Luo Z, Genet. Sel. Evol. 24, 305 (1992). [Google Scholar]
- 75.Chen Y, Davis TA, Hager WW, Rajamanickam S, ACM Trans. Math. Softw. TOMS. 35, 22 (2008). [Google Scholar]
- 76.Loh P-R et al. , Nat Genet. 47, 1385–1392 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matilainen K, Mäntysaari EA, Lidauer MH, Strandén I, Thompson R, PloS One. 8, e80821 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kang HM et al. , Genetics. 178, 1709–1723 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lippert C et al. , Nat Methods. 8, 833–5 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.