

Abstract

The design, synthesis, and conformational analysis of an oligobenzanilide helix mimetic scaffold capable of simultaneous mimicry of two faces of an α-helix is reported. The synthetic methodology provides access to diverse monomer building blocks amenable to solid-phase assembly in just four synthetic steps. The conformational flexibility of model dimers was investigated using a combination of solid and solution state methodologies supplemented with DFT calculations. The lack of noncovalent constraints allows for significant conformational plasticity in the scaffold, thus permitting it to successfully mimic residues i, i+2, i+4, i+6, i+7, and i+9 of a canonical α-helix.
Protein–protein interactions (PPIs) are central to all biological processes and offer a vast array of potential therapeutic targets.1−3 However, selective targeting of protein–protein interfaces remains an enormous challenge. In comparison to the small, well-defined pocket of an enzyme–substrate interaction (300–500 Å2), the interface of a typical PPI is large (1000–2000 Å2) and topographically featureless.4,5 The most common molecular recognition scaffold in multiprotein complexes is the α-helix, and as such, numerous strategies to target this class of PPI using rationally designed molecules have been reported, including both peptidic and nonpeptidic approaches, a concept known as peptidomimetics.6,7
Typically, helix mimetics possess two common features: a rigid scaffold to mimic the rod-like morphology of the helical backbone and side chains to recapitulate the spatial orientation of hotspot residues on the native helix.8 The majority of scaffolds reported have focused on mimicry of key recognition residues located on a single face of an α-helix (i, i+3/4, and i+7).
However, given that roughly one-third of PPIs interact through more than one helical face, there is an obvious requirement for helix mimetics that simultaneously mimic more than one helical face.9 Embellishment of previously reported scaffolds has resulted in multifacial helix mimetics such as bis- and tris-benzamides, and benzoylureas and novel scaffolds such as diphenylacetylenes and azaoctanes have also been developed (Figure 1).10−15
Figure 1.
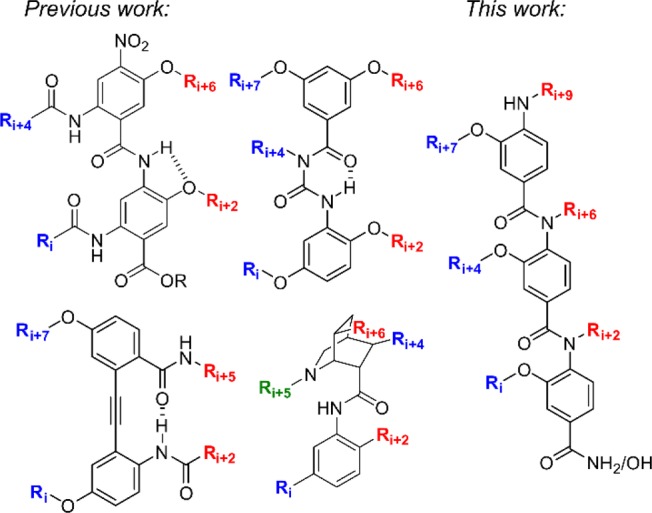
Previously published multifacial α-helix mimetics: bis-benzamides, benzoylureas, diphenylacetylenes, azaoctanes, and our novel oligobenzanilide.
Common to all two-faced helix mimetics published are lengthy, complex syntheses that limit their potential as generic tools for PPI inhibition. As part of our interest in developing methods to inhibit complex protein–protein interactions, we sought to design a novel synthetically accessible two-faced α-helix mimetic scaffold. Given the prevalence of oligobenzamide scaffolds, we chose to develop a two-faced scaffold based upon this linkage. Herein, we describe the design, synthesis, and conformational analysis of a novel 3-O-alkyated, 4-N-dialkylated helix mimetic that is capable of simultaneously mimicking residues i, i+2, i+4, i+6, i+7, and i+9 of a canonical α-helix.
Our novel scaffold was designed by combining two previously published scaffolds onto a single helix mimetic: Wilson’s N-alkylated triaryl amide and Boger’s 3-O-alkylated triaryl amide were combined to give a 3-O-,4-N-dialkylated scaffold capable of simultaneous mimicry of two helical faces (Figure 1).16,17 The synthetic route to the individual monomers is shown in Scheme 1. Notable features of the synthetic route are (i) a common starting material to access most monomers, (ii) compatibility with the Fmoc/tBu protocol for solid-phase synthesis (SPS), and (iii) the wide variety of functionality, both natural and unnatural, that may be incorporated at both positions on the individual monomers.
Scheme 1. Synthesis of Fmoc-Protected Monomers.

3-Fluoro-4-nitrobenzoic acid was used as the starting material for monomer synthesis. The nitro group simultaneously activates the 3′-position for nucleophilic aromatic substitution and “protects” the aniline required for installation of the second side chain and subsequent coupling reactions. Using this SNAr approach, it is possible to incorporate functionality found across the majority of proteinogenic amino acids as well as the enormous chemical space that has not been sampled by nature. For most monomers, catalytic hydrogenation afforded the primary aniline in near quantitative yields. For monomers with functionality susceptible to hydrogenation, a tin(II) chloride-based reduction was used. The second side chain was introduced onto the primary aniline through a one-pot reductive amination using a picoline–borane complex (pic-BH3) as the stoichiometric reductant. All aldehydes corresponding to hydrophobic amino acids side chains are commercially available. Unoptimized yields for these hydrophobic aldehydes were typically >80%. Aldehydes for installation of polar side chains were typically obtained by oxidation of the hydroxycarbamate to the aldehyde with Dess–Martin periodinane.
To render the monomers compatible with Fmoc/tBu SPS, the secondary anilines were Fmoc protected to afford the tertiary amide in reasonable yields (40–70%). Using this four-step protocol, we generated a library of 13 diverse monomers containing charged, polar, and hydrophobic functionality in both positions (Table 1 and Figure S1).
Table 1. 3-O, 4-N-Dialkylated Monomers Synthesized.

In comparison to previously published helix mimetics, oligomers were assembled by loading the monomers directly onto a solid support (Scheme 2). HATU mediated coupling under microwave heating allowed the Fmoc-protected monomers to be loaded onto a rink amide resin, whereas monomers could be loaded on a 2-chlorotrityl resin at room temperature by stirring with DIPEA. To effect amide bond formation, the benzoic acid was activated in situ to the acyl chloride using Ghosez’s reagent in anhydrous N-methyl-2-pyrollidone (NMP) according to previously published methodology (Scheme 3).18 Products were isolated by mass-directed purifications. To exemplify the methodology, three dimers and two trimers were successfully produced bearing a range of hydrophobic and polar side chains (Figures 2 and S2 and Table 2).
Scheme 2. Monomer Resin Loading.

Scheme 3. Solid Phase Oligomer Synthesis.

Figure 2.
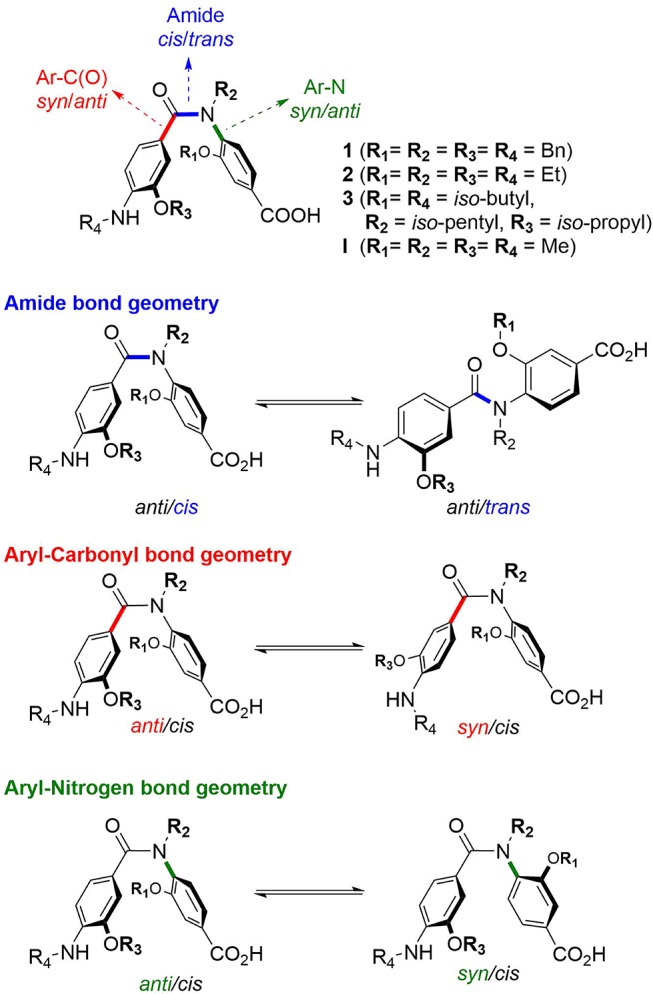
3-O, 4-N-Dialkylated benzanilide dimers synthesized and the conformational and stereochemical isomerization available to the helix mimetic scaffold.
Table 2. 3-O, 4-N-Dialkylated Benzanilide Dimers and Trimers Synthesized.

It is well established that N-alkylated benzanilides preferentially adopt a “folded” conformation about the amide bond in which the aryl rings are cis (formally E/s-trans).19−21
Clearly, a permanent cis relationship about the amide bond is not representative of the elongated structure of the α-helix and as such is unlikely to lead to effective perturbation of α-helix-mediated PPIs. However, the N-alkylated benzanilide scaffolds have been shown to be as potent as both (i) the native peptide and (ii) benzamide scaffolds with preorganization endowed through intramolecular hydrogen bonding networks.16
To study the conformational space of our scaffold, we selected a triage of ab initio, solution-, and solid-state techniques. For ease of analysis, representative dimers 14–16 were chosen as model compounds for experimental studies. These dimers (Figure 2) contain all the features common to more extended oligomers: (i) a central amide linkage and thus the potential for cis–trans isomers, (ii) the potential for syn/anti conformers with regards to the O-alkoxy substituent, and (iii) the potential for restricted rotation about the aryl-nitrogen (Ar–N) bond.
Single crystals suitable for X-ray diffraction of compound 16 were generated by slow evaporation of a solution of hexane:ethyl acetate (1:1). The unit cell contained two chemically identical but crystallographically independent structures (Figure 3). As expected, both structures clearly demonstrate the cis-isomer about the amide bond with the two 3-O-substituents on opposing faces, presumably minimizing unfavorable steric clashes. A1,3 strain between phenyl rings results in the aromatic rings lying roughly perpendicular to the plane of the amide bond with the aromatic rings partially facing each other. This perpendicular cis-anti conformer is consistent with previous solid-state structures of tertiary benzanilides.22 Interestingly, the amide bond displays a significant distortion from planarity (12.1° for 16-A, 16.3° for 16-B, Table S1) placing the latter within the top 5% of the acyclic amides in which the amide bond is not constrained with a ring (nonlactam).
Figure 3.
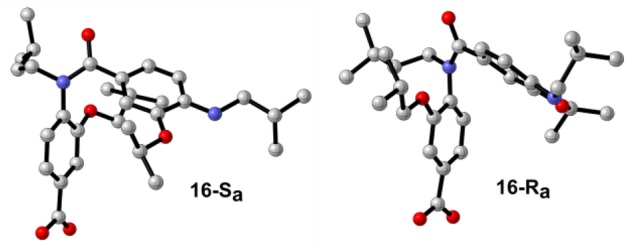
X-ray structures of 16 (16-Sa and 16-Ra).
An important feature highlighted by the solid-state structure is the presence of axial chirality. Restricted rotation about the Ar–N axis of benzanilides is long known, and many ortho-substituted anilides displaying axial chirality have been documented.22−25 This bond rotation in helix mimetics is significant in allowing scope for side chains to recapitulate native interactions through an induced-fit binding mechanism. In multifacial helix mimetics, rotation about the Ar–N bond corresponds to interconversion between helical faces (Figure 2).
To calculate the barrier to rotation about the Ar–N bond, variable temperature (VT) NMR spectroscopy was used. Given rotation about the Ar–N bond of unsymmetrically substituted anilides interconverts not only enantiomeric conformers but diastereotopic protons, it is possible to measure the rate of, and therefore barrier to, rotation. The 1H NMR spectra of 14–16 at 298 K in DMSO-d6 all displayed a single set of broadened resonances indicative of an exchange process but with no distinction between the putative diastereotopic protons (Figure S3). This is indicative of “fast” exchange in solution at room temperature.
To investigate the nature of the exchange, low temperature NMR spectra were taken at the slow-exchange limit. As expected, equal intensity signal doubling due to restricted rotation about the Ar–N axis was observed. For dimers 14, 15, and 16, anisochronicity was observed for all methylene protons on the 3-O-alkoxy and amide substituents (spin-systems: 14 = AB; 15 = ABX3; 16 = varies). In addition, dimer 3 demonstrated diastereotopic methyl doublets (spin-system = AX3X3′). For all dimers studied, no magnetic inequivalence was observed for protons on the secondary aniline substituent due to the increased distance from the chiral Ar–N axis.
Modeling the coalescence of the methylene protons in dimer 14 gave a barrier to rotation of 14.9 kcal mol–1 ± 0.59. For dimer 16, modeling the coalescence of the methyl doublets gave a barrier to rotation of 13.9 kcal mol–1 ± 0.41 (Figures 4, S4, and S5). Both values correspond to an estimated millisecond half-life for racemization. Thus, although interconversion between enantiomers is slow on the NMR time scale, the helix mimetic scaffold is not atropisomeric at room temperature (Oki’s definition >1000 s at 25 °C26), and as such, the “helical” faces mimicked by the scaffold are interconverting rapidly at 298 K and thus sample a vast array of conformational space. Unfortunately, spectral crowding prevented us from obtaining accurate energies for Ar–N bond rotation for compound 15.
Figure 4.
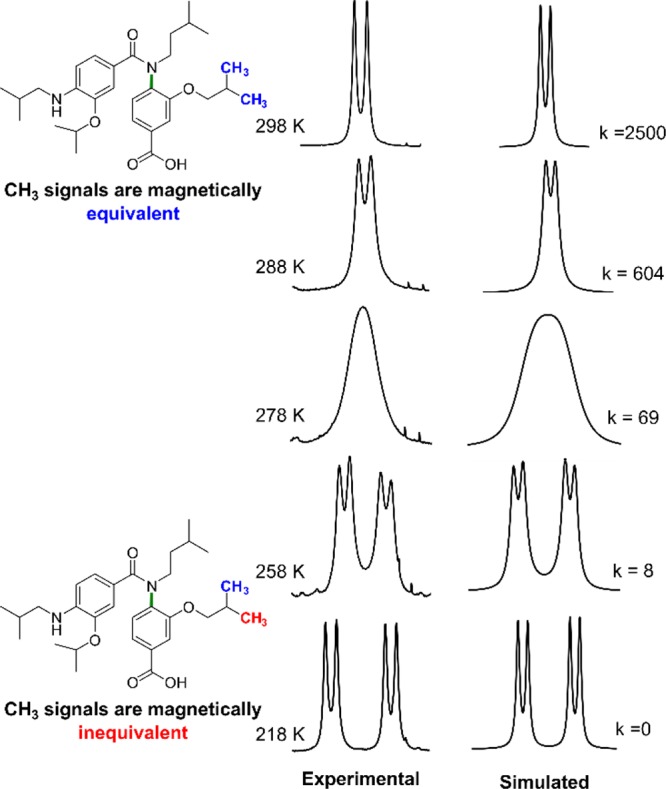
Experimental and simulated NMR spectrum of 16 at the slow-exchange limit.
2D NOESY and selNOE experiments at 298 K revealed through-space correlations characteristic of a cis conformation about the central amide bond (Figure S6), in line with a fast exchange at room temperature and existence of cis conformations. However, no characteristic correlation showing existence of a trans conformation was observed by 2D NMR spectroscopy, presumably due to the large distance (>5 Å) between the two aromatic moieties of interest in the trans conformation.
To further explore the barriers to rotation about the Ar–N and amide bonds, DFT calculations were carried out, and energy barriers of the rotations about the amide, Ar–C(O) and Ar–N bonds were calculated based on methods previously used by Miller27 (details in Supporting Information). A simplified model dimer was used in which all alkyl groups were replaced by methyl groups (I, Figure 2). Rotations solely about the amide bond were found to have energy barriers as low as ΔG298 = 14.1 kcal/mol (I-TS2, Figures 5, S12, and S15) and only led to cis/trans conformational exchanges (i.e., no anti/syn conformational exchange, Figures S12 and S15). Interestingly, as previously reported by others,27−29 concerted amide and Ar–N bond rotations occurred with slightly lower energy barriers of ΔG298 = 12.1 kcal/mol (I-TS1, Figures 5, S10, and S19) and led to concerted cis/anti and syn/trans conformational exchanges (i.e. anti/cis ↔ syn/trans or syn/cis ↔ anti/trans, Figures S10 and S19). These concerted rotations are a consequence of the amide carbonyl oxygen and the ortho-O-alkoxy group of the benzamide “pushing” each other during the rotations (Figure 5). Rotations about the Ar–C(O) bond were found to occur with low energy barriers ΔG298 < 10 kcal/mol for both trans and cis conformations about the amide bond (Figures S21 and S22). Finally, rotations about the Ar–N bond in a cis conformation were found to have the highest energy barrier ΔG298 = 16.7 kcal/mol due to a steric clash between the ortho-O-alkoxy group of the benzamide and the N-Me group of the amide moiety (III-TS5, Figure 5).
Figure 5.
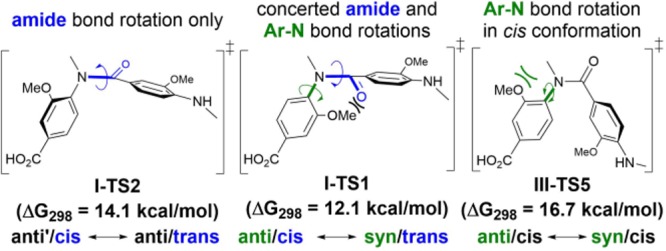
Examples of energy barriers ΔG298 for selected bond rotations (see details in the Supporting Information)
Numerous pathways are possible for the exchange between the two crystallographically observed enantiomers. However, exchange between the two enantiomers via constant cis conformations is likely to be the least favorable pathway due to the high energy barrier of the Ar–N bond rotation (Figure 5). Alternatively, pathways via extended conformations are more favorable, involving bond rotations with lower energy barriers with a maximum of 14.1 kcal/mol (Figure S30), in good agreement with the experimental values of 14.9 and 13.9 kcal/mol.
The Boltzmann population of both conformers was calculated based on the energy required for rotation about the amide bond or Ar–N bond or a concerted rotation (Table S15). Given the energy difference between the cis and trans conformations, it is not surprising to find that at equilibrium, >99% of the scaffold exists in the folded conformation at 298 K. However, this does not preclude the molecule from adopting a trans conformation. The low energy barriers associated with these bond rotations strongly suggests that our scaffold can sample an extended “helix mimetic” conformation at room temperature.
We described the design, synthesis, and conformational analysis of a novel oligo 3-O, 4-N-dialkylated benzanilide helix mimetic scaffold capable of simultaneous mimicry of side chains i, i+2, i+4, i+6, i+7, and i+9 of a canonical α-helix. The synthetic route is the shortest and most straightforward described to date for multifacial helix mimetics. A combination of solid-state, solution-phase, and in silico analyses show that the helix mimetic scaffold rapidly samples a wide variety of conformations in solution and can successfully mimic the relevant helical pharmacophore. The increased conformational plasticity in comparison with previous helix mimetic design is a shift for the proteomimetic field. To this date, the analysis of helix mimetics has focused on reporting RMSD values obtained from energy-minimized or solid-state structures. As noted by Burgess, it is evident that this is not representative of the equilibrating conformations present in solution.30−32 This work goes part way to demonstrating that a more sophisticated approach to characterization of helix mimetics is required if the values are to be considered a determinant in the quality of a scaffold. Furthermore, a degree of conformational plasticity has been noted to be advantageous for protein–surface recognition through an induced-fit-type mechanism. This is even more pertinent given that most secondary structure elements at protein interfaces are perturbed from geometric ideality. Future work on this scaffold will focus initially on inhibition of complex helix–protein interactions that are mediated by the binding of more than one helical face. For example, malaria parasite invasion of host red blood cells is critically dependent on the fully buried interaction which exists between myosin tail interacting protein (MTIP) and myosin A (MyoA),33 and in oncology, the Wnt signaling pathway is centered around β-catenin, which forms multifacial PPIs with T-cell factor 4 (Tcf-4)34 and B cell lymphoma 9 (BCL9).35
Acknowledgments
The authors thank Dr. Lisa Haigh, Imperial College London for mass spectrometry and Prof. Ed Tate, Imperial College London for support and advice. A.B. and C.R. thank Imperial College London for their Junior Research Fellowships. A.B. would like to thank the Wellcome Trust for funding for her JRF (Grant 105603/Z/14/Z).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.9b01115.
Supplementary figures, experimental procedures, compound characterization, copies of 1H and 13C NMR spectra, computational figures, and experimental procedures (PDF)
Accession Codes
CCDC 1902316 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033. Complementary FAIR data36 are available from the Imperial College Data Repository at DOI: 10.14469/hpc/5108.
The authors declare no competing financial interest.
Supplementary Material
References
- Arkin M. M. R.; Wells J. a. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing towards the Dream. Nat. Rev. Drug Discovery 2004, 3 (4), 301–317. 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Tang Y.; Wells J. A. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 2014, 21 (9), 1102–1114. 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf M. P. H.; Thorne T.; de Silva E.; Stewart R.; An H. J.; Lappe M.; Wiuf C. Estimating the Size of the Human Interactome. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (19), 6959–6964. 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller J. C.; Burgoyne N. J.; Jackson R. M. Predicting Druggable Binding Sites at the Protein-Protein Interface. Drug Discovery Today 2009, 14 (3–4), 155–161. 10.1016/j.drudis.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Hwang H.; Vreven T.; Janin J.; Weng Z. Protein-Protein Docking Benchmark Version 4.0. Proteins: Struct., Funct., Genet. 2010, 78 (15), 3111–3114. 10.1002/prot.22830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock B. N.; Jochim A. L.; Arora P. S. Assessing Helical Protein Interfaces for Inhibitor Design. J. Am. Chem. Soc. 2011, 133 (36), 14220–14223. 10.1021/ja206074j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. Inhibition of Αlpha-Helix-Mediated Protein-Protein Interactions Using Designed Molecules. Nat. Chem. 2013, 5 (3), 161–173. 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- Clackson T.; Wells J. A. A Hot Spot of Binding Energy in a Hormone-Receptor Interface. Science 1995, 267 (5196), 383–386. 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- Jochim A. L.; Arora P. S. Assessment of Helical Interfaces in Protein-Protein Interactions. Mol. BioSyst. 2009, 5 (9), 924–926. 10.1039/b903202a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayatunga M. K. P.; Thompson S.; Hamilton A. D. Alpha-Helix Mimetics: Outwards and upwards. Bioorg. Med. Chem. Lett. 2014, 24 (3), 717–734. 10.1016/j.bmcl.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Marimganti S.; Cheemala M. N.; Ahn J. M. Novel Amphiphilic α-Helix Mimetics Based on a Bis-Benzamide Scaffold. Org. Lett. 2009, 11 (19), 4418–4421. 10.1021/ol901785v. [DOI] [PubMed] [Google Scholar]
- Thompson S.; Vallinayagam R.; Adler M. J.; Scott R. T. W.; Hamilton A. D. Double-Sided α-Helix Mimetics. Tetrahedron 2012, 68 (23), 4501–4505. 10.1016/j.tet.2011.11.010. [DOI] [Google Scholar]
- Thompson S.; Hamilton A. D. Amphiphilic α-Helix Mimetics Based on a Benzoylurea Scaffold. Org. Biomol. Chem. 2012, 10 (30), 5780–5782. 10.1039/c2ob25273b. [DOI] [PubMed] [Google Scholar]
- Jung K. Y.; Vanommeslaeghe K.; Lanning M. E.; Yap J. L.; Gordon C.; Wilder P. T.; Mackerell A. D.; Fletcher S. Amphipathic α-Helix Mimetics Based on a 1,2-Diphenylacetylene Scaffold. Org. Lett. 2013, 15 (13), 3234–3237. 10.1021/ol401197n. [DOI] [PubMed] [Google Scholar]
- Bayly A. R.; White A. J. P.; Spivey A. C. Design and Synthesis of a Prototype Scaffold for Five-Residue α-Helix Mimetics. Eur. J. Org. Chem. 2013, 2013 (25), 5566–5569. 10.1002/ejoc.201300478. [DOI] [Google Scholar]
- Campbell F.; Plante J. P.; Edwards T. A.; Warriner S. L.; Wilson A. J. N-Alkylated Oligoamide α-Helical Proteomimetics. Org. Biomol. Chem. 2010, 8 (10), 2344–2351. 10.1039/c001164a. [DOI] [PubMed] [Google Scholar]
- Shaginian A.; Whitby L. R.; Hong S.; Hwang I.; Farooqi B.; Searcey M.; Chen J.; Vogt P. K.; Boger D. L. Design, Synthesis, and Evaluation of an a-Helix Mimetic Library Targeting Protein-Protein Interactions. J. Am. Chem. Soc. 2009, 131 (15), 5564–5572. 10.1021/ja810025g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy N. S.; Prabhakaran P.; Azzarito V.; Plante J. P.; Hardie M. J.; Kilner C. A.; Warriner S. L.; Wilson A. J. Solid-Phase Methodology for Synthesis of O-Alkylated Aromatic Oligoamide Inhibitors of α-Helix-Mediated Protein-Protein Interactions. Chem. - Eur. J. 2013, 19 (18), 5546–5550. 10.1002/chem.201204098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itai A.; Toriumi Y.; Tomioka N.; Kagechika H.; Azumaya I.; Shudo K. Stereochemistry of N-Methylbenzanilide and Benzanilide. Tetrahedron Lett. 1989, 30 (45), 6177–6180. 10.1016/S0040-4039(01)93335-0. [DOI] [Google Scholar]
- Campbell F.; Plante J.; Carruthers C.; Hardie M. J.; Prior T. J.; Wilson A. J. Macrocyclic Scaffolds Derived from P-Aminobenzoic Acid. Chem. Commun. 2007, (22), 2240–2242. 10.1039/b704366j. [DOI] [PubMed] [Google Scholar]
- Saito S.; Toriumi Y.; Tomioka N.; Itai A. Theoretical Studies on Cis-Amide Preference in N-Methylanilides. J. Org. Chem. 1995, 60 (15), 4715–4720. 10.1021/jo00120a011. [DOI] [Google Scholar]
- Adler T.; Bonjoch J.; Clayden J.; Font-Bardía M.; Pickworth M.; Solans X.; Solé D.; Vallverdú L. Slow Interconversion of Enantiomeric Conformers or Atropisomers of Anilide and Urea Derivatives of 2-Substituted Anilines. Org. Biomol. Chem. 2005, 3 (17), 3173–3183. 10.1039/b507202f. [DOI] [PubMed] [Google Scholar]
- Siddall T. H.; Prohaska C. A. Geminal Proton Nonequivalences and Related Phenomena in Some N-Substituted Amides. J. Am. Chem. Soc. 1966, 88 (6), 1172–1176. 10.1021/ja00958a018. [DOI] [Google Scholar]
- Shvo Y.; Taylor E. C.; Mislow K.; Raban M. Chemical Shift Nonequivalence of Diastereotopic Protons Due to Restricted Rotation Around Aryl-Nitrogen Bonds in Substituted Amides. J. Am. Chem. Soc. 1967, 89 (19), 4910–4917. 10.1021/ja00995a015. [DOI] [Google Scholar]
- Chabaud L.; Clayden J.; Helliwell M.; Page A.; Raftery J.; Vallverdú L. Conformational Studies of Tertiary Oligo-m-Benzanilides and Oligo-p-Benzanilides in Solution. Tetrahedron 2010, 66 (34), 6936–6957. 10.1016/j.tet.2010.06.037. [DOI] [Google Scholar]
- O̅ki M. Recent Advances in Atropisomerism. Top. Stereochem. 1983, 1–81. 10.1002/9780470147238.ch1. [DOI] [Google Scholar]
- Barrett K. T.; Metrano A. J.; Rablen P. R.; Miller S. J. Spontaneous Transfer of Chirality in an Atropisomerically Enriched Two-Axis System. Nature 2014, 509 (7498), 71–75. 10.1038/nature13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayden J.; Pink J. H. Concerted Rotation in a Tertiary Aromatic Amide: Towards a Simple Molecular Gear. Angew. Chem., Int. Ed. 1998, 37 (13–14), 1937–1939. . [DOI] [Google Scholar]
- Ahmed A.; Bragg R. A.; Clayden J.; Lai L. W.; McCarthy C.; Pink J. H.; Westlund N.; Yasin S. A. Barriers to Rotation about the Chiral Axis of Tertiary Aromatic Amides. Tetrahedron 1998, 54 (43), 13277–13294. 10.1016/S0040-4020(98)00814-X. [DOI] [Google Scholar]
- Ko E.; Liu J.; Perez L. M.; Lu G.; Schaefer A.; Burgess K. Universal Peptidomimetics. J. Am. Chem. Soc. 2011, 133 (3), 462–477. 10.1021/ja1071916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko E.; Raghuraman A.; Perez L. M.; Ioerger T. R.; Burgess K. Exploring Key Orientations at Protein-Protein Interfaces with Small Molecule Probes. J. Am. Chem. Soc. 2013, 135 (1), 167–173. 10.1021/ja3067258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin D.; Ko E.; Perez L. M.; Ioerger T. R.; Burgess K. Evaluating Minimalist Mimics by Exploring Key Orientations on Secondary Structures (EKOS). Org. Biomol. Chem. 2013, 11 (44), 7789–7801. 10.1039/c3ob41848k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douse C. H.; Maas S. J.; Thomas J. C.; Garnett J. A.; Cota E.; Tate E. W. Crystal Structures of Stapled and Hydrogen Bond Surrogate Peptides Targeting a Fully Buried Protein-Protein Interaction. ACS Chem. Biol. 2014, 9 (10), 2204–2209. 10.1021/cb500271c. [DOI] [PubMed] [Google Scholar]
- Grossmann T. N.; Yeh J. T.-H.; Bowman B. R.; Chu Q.; Moellering R. E.; Verdine G. L. Inhibition of oncogenic Wnt signaling through direct targeting of β-catenin. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (44), 17942–17947. 10.1073/pnas.1208396109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K.; et al. Targeted Disruption of the BCL9/β-catenin Complex Inhibits Oncogenic Wnt Signaling. Sci. Transl. Med. 2012, 4 (148), 148ra117. 10.1126/scitranslmed.3003808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barba A.; Dominguez S.; Cobas C.; Martinsen D. P.; Romain C.; Rzepa H. S.; Seoane F. Workflows Allowing Creation of Journal Article Supporting Information and Findable, Accessible, Interoperable and Reusable (FAIR)-Enabled Publication of Spectroscopic Data. ACS Omega 2019, 4 (2), 3280–3286. 10.1021/acsomega.8b03005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.