

Abstract
Objective
Multiple single‐nucleotide polymorphisms (SNPs) conferring susceptibility to osteoarthritis (OA) mark imbalanced expression of positional genes in articular cartilage, reflected by unequally expressed alleles among heterozygotes (allelic imbalance [AI]). We undertook this study to explore the articular cartilage transcriptome from OA patients for AI events to identify putative disease‐driving genetic variation.
Methods
AI was assessed in 42 preserved and 5 lesioned OA cartilage samples (from the Research Arthritis and Articular Cartilage study) for which RNA sequencing data were available. The count fraction of the alternative alleles among the alternative and reference alleles together (φ) was determined for heterozygous individuals. A meta‐analysis was performed to generate a meta‐φ and P value for each SNP with a false discovery rate (FDR) correction for multiple comparisons. To further validate AI events, we explored them as a function of multiple additional OA features.
Results
We observed a total of 2,070 SNPs that consistently marked AI of 1,031 unique genes in articular cartilage. Of these genes, 49 were found to be significantly differentially expressed (fold change <0.5 or >2, FDR <0.05) between preserved and paired lesioned cartilage, and 18 had previously been reported to confer susceptibility to OA and/or related phenotypes. Moreover, we identified notable highly significant AI SNPs in the CRLF1,WWP2, and RPS3 genes that were related to multiple OA features.
Conclusion
We present a framework and resulting data set for researchers in the OA research field to probe for disease‐relevant genetic variation that affects gene expression in pivotal disease‐affected tissue. This likely includes putative novel compelling OA risk genes such as CRLF1,WWP2, and RPS3.
Introduction
Due to the increased proportion of elderly persons in the human population, osteoarthritis (OA) has become one of the major musculoskeletal diseases 1. While all joint tissues have been implicated in OA pathology, the disease is characterized primarily by progressive degradation and calcification of articular cartilage 2. Both gene‐targeted research 3, 4, 5 and genome‐wide research 6, 7, 8, 9 showed that a multitude of genes are involved in the currently irreversible destruction of articular cartilage that precedes total joint replacement surgery, which is at present the only effective treatment for end‐stage OA. In this regard, numerous studies have shown altered regulation of gene expression that reflects, attenuates, and/or stimulates OA‐mediated cartilage degradation 10, 11, 12, 13. Moreover, multiple OA risk alleles of single‐nucleotide polymorphisms (SNPs) were shown to consistently modulate OA pathology by altering transcription of the respective genes in articular cartilage, commonly referred to as an allelic imbalance (AI) 14, 15, 16, 17, 18, 19. Notable recent examples are the genes ALDH1A2 18 and MGP 20. Hence, it is clear that in‐cis genetic regulation of transcription plays a substantial role in cartilage homeostasis and, therefore, in OA pathophysiology.
Despite the evidence for in‐cis genetic regulation of transcription in OA susceptibility, genome‐wide association studies (GWAS) have thus far failed to explain the larger part of the hereditary component of OA 21. In this regard, a large number of the tested SNPs in GWAS likely bear no biologic function in relation to the addressed phenotype or disease‐relevant tissues 22, resulting in massive inflation of possibly biologically irrelevant statistical tests and thus the multiple testing correction penalty. Consequently, large numbers of SNPs that do bear biologic functionality in the context of OA are missed. Furthermore, SNPs that reside within linkage disequilibrium (LD) blocks are hard to interpret, as association analysis is inherently unable to distinguish disease‐relevant alleles from merely statistically associated alleles.
In previous studies, we and others have used targeted approaches to address AI events of putative as well as established OA susceptibility genes 14, 16, 17, 18, 23, 24. Given the successful identification of the transcriptional consequences of multiple OA‐associated SNPs, we have aimed to characterize, on a transcriptome‐wide scale, novel SNPs that tag AI of genes expressed in articular cartilage, and we have subsequently identified those that appear to confer susceptibility to OA. Finally, further using the RNA sequencing data set, we ran analyses to identify AI genes whose expression was additionally modified with severity of OA pathophysiology as reflected by differential expression between preserved and lesioned OA cartilage.
Materials and Methods
Cohorts
Ethical approval for the Research Arthritis and Articular Cartilage (RAAK) study was obtained from the medical ethics committee of the Leiden University Medical Center (P08.239), and informed consent was obtained from all patients included. For the current study, RNA sequencing data were available from preserved and lesioned cartilage from 21 patients (6 with hip OA and 15 with knee OA), complemented by an additional 21 preserved samples (from 14 patients with hip OA and 7 patients with knee OA) and 5 lesioned samples (from 2 patients with hip OA and 3 patients with knee OA) (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract). For cartilage sampling details, see refs. 19 and 25.
RNA sequencing data
After RNA isolation (RNeasy Mini Kit, RNA integrity number >7; Qiagen), paired‐end 100‐bp RNA library sequencing (Illumina TruSeq RNA Library Prep Kit, Illumina HiSeq 2000) resulted in an average of 10 million clusters. Reads were aligned using GSNAP (an R package within Bioconductor; https://rdrr.io/bioc/gmapR/) against the human (hg19) reference genome, while known Dutch SNPs (Genome of the Netherlands) were masked to aid in preventing potential reference alignment bias. AI events were assessed on SNPs called using SNVMix2 with default settings 26 with minimum coverage of 25 and at least 10 reads (R) per allele. AI is reported as the average fraction (φ) of the alternative allele reads (R alternative) among the total number of reads (R total = R alternative + R reference) at the position of the respective genetic variation per sample (i):
To detect SNPs that robustly mark imbalance, 2 binomial tests were performed per heterozygote and per SNP under the null hypothesis that the amount of imbalance is either greater or smaller than 0.477. A meta‐analysis (meta; http://www.r-project.org/) per SNP among heterozygous individuals (null hypothesis median φ = 0.49) was performed to generate a meta‐φ and P value per SNP with a false discovery rate (FDR) correction for multiple comparisons. To allow independent samples in the meta‐analysis only, the preserved cartilage of each sample pair (n = 21) was used complemented with the individual preserved (n = 21) and lesioned (n = 5) OA samples.
Unfortunately, SNVMix2 discards strand specificity. While in general this does not pose an issue when annotating the AI direction to an effector allele, it does so for A>T, T>A, G>C, and C>G SNPs. Therefore, for these SNPs (n = 119) we supplied (see Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract) minor allele frequencies (MAFs) and SNPs in strong LD for GWAS look‐ups. Using the edgeR package, fragments per gene were used to assess the dispersion by quantile‐adjusted conditional maximum likelihood 27. Subsequently, differential gene expression analysis was performed pairwise between preserved and lesioned samples for which we had RNA of both (n = 21) (Table 1) followed by FDR correction. Gene Ontology (GO) term enrichment analysis was performed using the tool DAVID available online 28.
Table 1.
Sample characteristics of preserved and lesioned OA articular cartilage in the Research Arthritis and Articular Cartilage studya
| Tissue type | No. of samples | Age, mean ± SD years | No. of men | No. of women | OA articular cartilage | ||
|---|---|---|---|---|---|---|---|
| Preserved | Lesioned | Preserved–lesioned pairs | |||||
| Knee | 25 | 69 ± 9 | 4 | 21 | 22 | 18 | 15 |
| Hip | 22 | 66 ± 9 | 5 | 17 | 20 | 8 | 6 |
| All | 47 | 68 ± 9 | 9 | 38 | 42 | 26 | 21 |
OA = osteoarthritic.
Genotype data
Using Illumina HumanOmniExpressExome chips, genome‐wide genotyping data were constructed for 216 samples from the RAAK study. SNPs with <95% call rate, Hardy‐Weinberg equilibrium <10−4, MAF <0.01, or located on the sex chromosomes were removed prior to imputation against the 1000 GenomesV3 March 2012 reference panel 29. We removed SNPs for which the imputation quality of 0.4 was not met 12, 30.
TaqMan assay
Conventional TaqMan genotyping was performed on both genomic DNA and articular cartilage complementary DNA (cDNA) 31 from 6 patients (2 females and 4 males) who underwent total joint replacement surgery of the knee due to primary OA. An allele‐specific custom TaqMan assay for rs7256319 (ThermoFisher Scientific) was used to quantify the allele ratio in cDNA samples. AI of cDNA was normalized against the genomic DNA ratio (with an inherent 1:1 allele ratio) as a reference.
Results
Transcriptome‐wide discovery of articular cartilage AI events
To understand how genetic variation contributes in‐cis to transcriptional regulation in articular cartilage on a transcriptome‐wide scale, we first called heterozygous SNPs (dbSNP144) using RNA sequencing data from articular cartilage derived from patients who underwent total replacement surgery of either the hip joint (n = 22) or knee joint (n = 25) due to primary OA (Table 1; also see Supplementary Table 1, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract). After filtering by the number of read counts per position (R reference ≥10, R alternative ≥10, and R total ≥25), selecting for heterozygous SNPs present in at least 2 individuals, removing SNPs present in multiple or no distinct transcripts, and discarding the HLA locus, we defined φ for 13,853 SNPs as the measure of imbalance (Figure 1A), which denotes the fraction of R alternative among R total. Possibly due to reference bias, a considerable number of SNPs were marked as allelic imbalanced by φ <0.1 or φ >0.9 (n = 418) and were subsequently removed prior to further analyses. As such, in Supplementary Table 2 (http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract) we show AI, defined as φ across all individuals heterozygous for the 13,435 SNPs.
Figure 1.
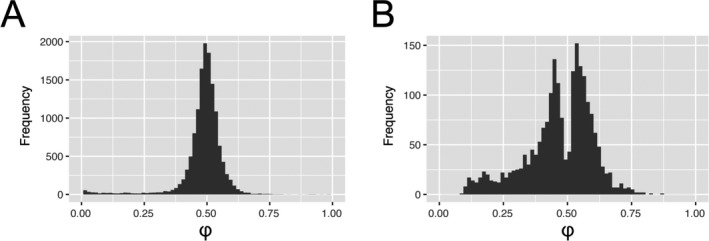
Distribution of allelic imbalance (AI) events in articular cartilage. AI is reported as the average fraction (φ) of the alternative allele reads among the total number of reads. A, AI was defined for 13,853 called variants after selecting for at least 2 heterozygotes, selecting single‐nucleotide polymorphisms (SNPs) present in only single genes and removing low counts. B, After filtering by allelic fraction (0.1<φ<0.9) and meta false discovery rate <0.05, 2,070 SNPs remained that marked significant AI of 1,031 unique genes.
Subsequently, a meta‐analysis among heterozygous individuals of each SNP (null hypothesis: median φ = 0.49) and subsequent FDR correction for multiple testing revealed 2,070 SNPs that significantly marked AI among 1,031 genes (Figure 1B; also see Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract). To allow unambiguous annotation of the AI direction to a GWAS effector allele, for A>T, T>A, G>C, and C>G SNPs with MAFs surrounding 0.50 (n = 119), we supplied MAFs and SNPs in strong LD.
Intersection of genes subject to AI with those differentially expressed between preserved and paired OA‐lesioned cartilage
While articular cartilage genes subject to AI due to genetic variation could contribute to OA pathophysiology in various ways (e.g., in cartilage development or homeostasis), it can be expected that allelic imbalanced genes that additionally mark the articular cartilage’s disease state are more likely to contribute to or attenuate disease progression. Therefore, we went back to the original expression data and determined differential expression in patients for whom paired RNA sequencing data of both preserved and OA‐lesioned articular cartilage were generated (6 hip joints and 15 knee joints) (Table 1).
Of the 10,468 Ensembl gene identifiers with at least 5 counts per million, 137 and 86 were observed to be significantly (FDR <0.05) down‐regulated (fold change <0.5) and up‐regulated (fold change >2), respectively, in lesioned cartilage compared to preserved cartilage (see Supplementary Figure 1A and Supplementary Table 4, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract). As has been shown by microarray studies that have used a similar design, subsequent GO term enrichment analysis (see Supplementary Table 5, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract) revealed significant enrichment for inflammatory pathways (e.g., SCUBE1, CFH, and CXCL14) (see Supplementary Figures 1B–D, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract), pathways of response to wound healing (e.g., NOTCH3, BMP5, and SERPINE1) (see Supplementary Figures 1E–G), and joint development–associated pathways (e.g., SPP1, MMP3, and COL9A1) (see Supplementary Figures 1H–J).
Of the 223 differentially expressed genes, 49 were additionally subject to AI, marked by 128 SNPs (see Supplementary Table 6, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract). A notable example was the CRLF1 gene, which was subject to highly consistent AI, with the T allele of rs7256319 marking consistently lower expression of CRLF1 compared to the reference allele C (φ = 0.29, FDR = 4.02 × 10−21) (Figure 2A). Moreover, as shown in Figure 2B, the AI of CRLF1 was confirmed by custom TaqMan assay performed in 5 preserved and 5 lesioned articular cartilage samples, originating from 6 independent patients who underwent total knee replacement surgery. In parallel, expression of CRLF1 in the current data set differed significantly between preserved and OA‐lesioned cartilage, with significant up‐regulation in OA‐affected cartilage (fold change 4.6, FDR = 3.1 × 10−10) (Figure 2C).
Figure 2.
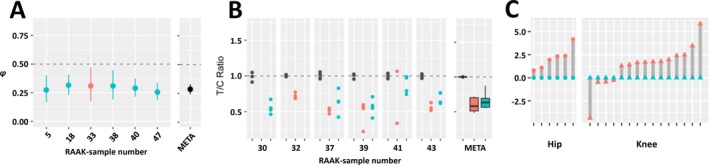
CRLF1 expression is subject to allelic imbalance (AI) and is modulated in articular cartilage with osteoarthritis (OA)–induced destruction. A, AI of rs7256319 in RNA sequencing data set, reported as the average fraction (φ) of the alternative allele reads among the total number of reads with 95% confidence interval, showing decreasing expression of CRLF1 transcript of the alternative T allele in preserved and lesioned OA cartilage relative to the reference C allele (φ = 0.29, false discovery rate [FDR] = 4.02 × 10−21). Preserved OA cartilage is depicted in blue, lesioned OA cartilage in red, and meta‐φ (META) in black. B, Replication of AI expression with rs7256319 by TaqMan genotyping in 6 additional knee samples, confirming the observed lower expression of the alternative allele T relative to the reference allele C of rs7256319 (T:C ratio = 0.63). Preserved OA cartilage is depicted in blue, lesioned OA cartilage in red, and genomic DNA (used as the reference ratio) in black. The meta‐φ of genomic DNA, lesioned cartilage complementary DNA (cDNA), and preserved cartilage cDNA is depicted in black, red, and blue, respectively. Data are shown as box plots. Each box represents the 25th to 75th percentiles. Lines inside the boxes represent the median. Lines outside the boxes represent the 10th and 90th percentiles. In A and B, horizontal dashed lines depict equal ratios of the CRLF1 rs7256319 alleles. C, Differential expression analyses of CRLF1 showing significantly up‐regulated expression in lesioned (red) compared to paired preserved (blue) OA articular cartilage (fold change 4.6, FDR‐corrected P = 3.1 × 10−10). RAAK = Research Arthritis and Articular Cartilage (study).
Cartilage AI SNPs that contribute to OA susceptibility
On the basis of the significant AI SNPs in articular cartilage (see Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract), it is to be expected that these SNPs are enriched for those conferring genetic susceptibility to OA. Hence, for 173 AI SNPs with FDR <5 × 10−8, not previously reported as OA risk genes, we went to the genome‐wide association catalogs of OA to look for their genetic association signal. We used the GWAS meta‐analysis for hip OA performed under the auspices of the Translational Research in Europe Applied Technologies in Osteoarthritis (TreatOA) consortium 32 and the GWAS meta‐analysis on cartilage thickness (measured by the OA endophenotype minimal joint space width [JSW]) 8. Given that the entire genome was not being assessed for genetic association, a nominal genetic association (P < 0.05) was considered. Moreover, we only checked for genetic association when the AI SNP could be used directly or when a clear proxy SNP was identified.
As shown in Table 2, we observed multiple AI SNPs conferring susceptibility to hip OA and/or minimal JSW. Notable examples in Table 2 are the AI SNP rs3133187 in the RPS3 gene conferring the most significant susceptibility to hip OA (Figure 3A) and the AI SNP rs1052429 in the WWP2 gene associated with minimal JSW (Figure 3B). Alternatively, in Supplementary Tables [Link], [Link] (http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract), we show the intersection of the 65 AI SNPs significantly associated with hip OA in the TreatOA GWAS meta‐analysis (FDR <0.05) and the 63 AI SNPs significantly associated with minimal JSW in the GWAS meta‐analysis on cartilage thickness (P < 0.05). Among these lists we find SNPs in compelling known (MGP, ALDH1A2, FRZB, COL11A1, PLEC) and potentially novel (ACAN, MATN, TNC, VEGFA, PLOD2) OA risk genes.
Table 2.
Intersection of highly significant allelic imbalance SNPs with published genome‐wide association data sets of minimal JSW and hip OAa
| Allelic imbalance | Genome‐wide association | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Positional gene | Meta‐φ | FDR | SNP | R2 | Minimal JSW | Hip OA | ||||||||||
| AA | AAF | EA | EAF | Beta | SEM | P | EA | EAF | Beta | SEM | P | ||||||
| rs4744 | PLA2G2A | A | 0.11 | 0.46 | 8.1 × 10−134 | rs11677 | 1.0 | A | 0.11 | −0.05 | 0.02 | 0.0091 | A | 0.12 | 0.00 | 0.04 | 0.9360 |
| rs4605 | FMOD | C | 0.70 | 0.49 | 0.000 | rs4605 | NA | C | 0.52 | −0.03 | 0.01 | 0.0679 | C | 0.46 | −0.09 | 0.03 | 0.0057 |
| rs3190 | PPP1CB | G | 0.56 | 0.59 | 6.3 × 10−10 | rs3190 | NA | G | 0.57 | 0.03 | 0.01 | 0.0076 | G | 0.57 | −0.00 | 0.02 | 0.8820 |
| rs1054629 | IBSP | T | 0.18 | 0.41 | 1.0 × 10−24 | rs13144371 | 1.0 | A | 0.24 | 0.03 | 0.01 | 0.0340 | A | 0.24 | −0.05 | 0.03 | 0.0801 |
| rs6546 | C1QTNF3 | G | 0.30 | 0.39 | 4.8 × 10−13 | rs840385 | 0.88 | C | 0.40 | −0.03 | 0.01 | 0.0168 | C | 0.39 | −0.03 | 0.03 | 0.2361 |
| rs3549 | SPARC | C | 0.39 | 0.45 | 1.1 × 10−58 | rs1059829 | 1.0 | A | 0.53 | 0.03 | 0.01 | 0.0284 | A | 0.53 | −0.02 | 0.02 | 0.3896 |
| rs3829078 | CA9 | G | 0.05 | 0.59 | 2.5 × 10−14 | rs3829078 | NA | G | 0.11 | 0.04 | 0.02 | 0.0495 | G | 0.11 | −0.04 | 0.04 | 0.2710 |
| rs13321 | TNC | G | 0.67 | 0.53 | 2.4 × 10−16 | rs12347433 | 0.1 | T | 0.72 | −0.02 | 0.01 | 0.1487 | T | 0.72 | 0.06 | 0.03 | 0.0256 |
| rs1871452 | CHST3 | A | 0.60 | 0.42 | 5.4 × 10−25 | rs731027 | 1.0 | T | 0.52 | 0.01 | 0.01 | 0.3073 | T | 0.50 | −0.06 | 0.02 | 0.0087 |
| rs3133187 | RPS3 | G | 0.12 | 0.42 | 5.0 × 10−9 | rs3133187 | NA | G | 0.06 | −0.04 | 0.03 | 0.1420 | G | 0.06 | 0.18 | 0.05 | 0.0011 |
| rs1800801 | MGP | T | 0.74 | 0.41 | 2.6 × 10−26 | rs4236 | 0.9 | T | 0.61 | −0.03 | 0.01 | 0.0433 | T | 0.60 | 0.03 | 0.03 | 0.2068 |
| rs3737548 | COL2A1 | T | 0.23 | 0.53 | 5.1 × 10−72 | rs1635553 | 0.4 | G | 0.46 | −0.02 | 0.01 | 0.2290 | G | 0.46 | −0.06 | 0.03 | 0.0161 |
| rs6647 | SERPINA1 | G | 0.75 | 0.41 | 7.2 × 10−283 | rs6647 | NA | G | 0.77 | −0.03 | 0.01 | 0.0848 | A | 0.76 | 0.06 | 0.03 | 0.0388 |
| rs1052429 | WWP2 | A | 0.86 | 0.56 | 3.4 × 10−55 | rs1566452 | 0.9 | A | 0.73 | −0.04 | 0.01 | 0.0028 | A | 0.71 | −0.05 | 0.03 | 0.0656 |
| rs2646108 | CRISPLD2 | A | 0.18 | 0.63 | 2.6 × 10−9 | rs2646108 | NA | A | 0.20 | −0.01 | 0.02 | 0.7410 | A | 0.19 | 0.09 | 0.03 | 0.0053 |
| rs6554 | UBA52 | T | 0.70 | 0.43 | 1.1 × 10−53 | rs6554 | NA | T | 0.60 | 0.00 | 0.01 | 0.9300 | T | 0.61 | 0.05 | 0.03 | 0.0416 |
| rs7499 | COL18A1 | A | 0.47 | 0.63 | 2.0 × 10−11 | rs7499 | NA | A | 0.40 | −0.01 | 0.01 | 0.3528 | A | 0.40 | −0.05 | 0.03 | 0.0332 |
| rs3088026 | COL6A2 | T | 0.07 | 0.53 | 5.5 × 10−3 | rs3088026 | NA | T | 0.09 | −0.02 | 0.02 | 0.4125 | T | 0.09 | −0.12 | 0.04 | 0.0055 |
SNPs = single‐nucleotide polymorphisms; JSW = joint space width; OA = osteoarthritis; AA = alternative allele; AAF = alternative allele frequency; φ = count fraction of the alternative allele reads among the total number of reads; FDR = false discovery rate relative to median φ = 0.49 as reference; R2 = measure of linkage disequilibrium of proxy SNP with allelic imbalance SNP; EA = effect allele; EAF = effect allele frequency; NA = not applicable.
Figure 3.
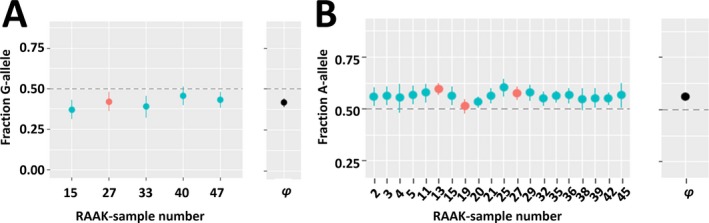
Compelling cartilage‐specific allelic imbalance (AI) genes. AI is reported as the average fraction (φ) of the alternative allele reads among the total number of reads with 95% confidence interval. A, Significant (P = 4.97 × 10−9) allelic imbalanced expression at rs3133187 with the G allele decreasing expression of RPS3 transcript in preserved and lesioned osteoarthritic (OA) cartilage relative to the A allele (meta‐φ = 0.42). The G allele of rs3133187 additionally confers significant association with hip OA (P = 1.1 × 10−3). B, Significant (P = 3.37 × 10−55) allelic imbalanced expression at rs1052429 with the A allele increasing expression of WWP2 transcript in preserved and lesioned OA cartilage relative to the G allele (meta‐φ = 0.56). The A allele of rs1052429, linked to the A allele of rs1566452 (r2 = 0.92, D′ = 1.0), additionally confers significant association with reduced minimal joint space width (P = 2.8 × 10−3), resulting in cartilage degeneration or OA. Horizontal dashed lines depict equal ratios of alleles. RAAK = Research Arthritis and Articular Cartilage (study).
Cartilage AI SNPs that contribute to expression quantitative trait loci (eQTLs)
To provide additional supporting data on the AI findings in the putative OA risk SNPs in Table 2, we combined the RNA sequencing data of the genes with genome‐wide SNP data (Illumina HumanOmniExpressExome) of RAAK study samples to extract gene‐targeted cartilage eQTLs on the basis of 50 samples that were overlapping. Additionally, we explored eQTL data of the Genotype‐Tissue Expression (GTEx) Project. The fact that GTEx data are merely from tissues other than those particularly relevant to OA indicates the generalizability of the identified AI to other tissues. As shown in Supplementary Table 9 (http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract), the GTEx eQTL data largely support (the direction of) the identified AI effects also in other tissues.
OA susceptibility SNPs that show AI in cartilage
Since identified OA risk SNPs have been demonstrated to frequently confer risk by modifying expression of positional genes in‐cis, the aforementioned genome‐wide AI data set of articular cartilage (see Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract) can function as a database to assess in silico the direction of effect of such identified susceptibility SNPs. Hence, we reviewed the literature to intersect the list of currently published robust genetic OA association signals with the identified AI SNPs. We included robustly identified susceptibility SNPs and their positional genes associated with knee, hand, and hip OA as well as OA‐associated phenotypes such as minimal JSW and markers of cartilage turnover (soluble cartilage oligomeric matrix protein). Of the 46 genes linked to the reported OA risk SNPs, 36 genes were detected in our data set (i.e., had detectable expression levels in cartilage) and were heterozygous carriers of a coding SNP. As shown in Supplementary Table 10 (http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract), we found 11 previously identified OA risk genes to contain SNPs that mark AI in articular cartilage. Of these, the respective risk alleles marked lower expression through AI in heterozygotes of FRZB (rs7775), COL11A1 (rs2615977 and rs1676486), IGFBP3 (rs788748 through double heterozygotes with rs6670), ALDH1A2 (rs3204689), and MGP (rs4764133 in LD with rs1800801). For SMAD3, CDCL5, COL12A1, BCAP29, PIK3R1, and COMP, AI was detected but not in relation to the reported risk alleles.
To provide additional functional data to these previously identified OA risk genes, level and differential expression data between preserved and lesioned OA articular cartilage are depicted in Supplementary Table 11 (http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract). For IGFBP3, PIK3R1, BCAP29, COL12A1, FRZB, ALDH1A2, and MGP, significant differential expression (FDR <0.05) was observed in addition to AI. Finally, to find supporting data on the AI findings in the OA risk SNPs, we combined the RNA sequencing data of the OA risk genes with genome‐wide association data (Illumina HumanOmniExpressExome) of RAAK study samples to extract gene‐targeted eQTL data on the basis of 50 cartilage samples that were overlapping. Additionally, to the eQTL data in Supplementary Table 11, we added data from the GTEx Project, although from tissues other than those particularly relevant to OA. For the OA genes GNL3, FTO, NCOA3, MICAL3, IFRD1, IGFBP3, TGFA, and MGP, we found significant eQTL effects (P < 0.05) of risk SNPs that substantiated the respective AI effects.
Discussion
Our approach in the current study comprises a concept framework for complex traits to identify disease‐relevant genetic variation, as reflected by allele‐associated transcription levels in cartilage, a pivotal tissue in the disease. We have aimed to present the reported observations as a legacy data set for researchers in the field to probe for their gene or SNP of interest. Herein, we highlight notable examples. Among our (highly) significant AI SNPs we confirmed well‐known genes that have previously been reported by others to confer robust risk of OA (e.g., MGP, ALDH1A2, IGFBP3, and FRZB) 17, 33, 34. We hypothesize that among our (highly) significant AI SNPs and particularly those that show additional, differential expression between preserved and lesioned OA cartilage (e.g., CRLF1 [Figure 2; also see Supplementary Table 6, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract]) or genetic association with OA phenotypes (e.g., WWP2 and RPS3 [Figure 3 and Table 2; also see Supplementary Tables [Link], [Link], http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract]) are putative novel compelling OA risk genes.
The CRLF1 gene, encoding for cytokine receptor–like factor 1 protein, harbors the rs7256319 C>T SNP that has marked imbalanced expression of its respective alleles in articular cartilage, reflected by consistent lower expression of the alternative allele T in comparison with the reference allele C among heterozygotes. As was also reported previously 25, 35, CRLF1 appeared to be significantly up‐regulated in lesioned compared to preserved OA articular cartilage (fold change 4.6, FDR = 3.1 × 10−10), as was its signaling partner CLCF1 (fold change 2.1, FDR = 1.0 × 10−6), while the protein complex signaling receptor gene CNTFR was significantly down‐regulated (fold change 0.3, FDR = 1.9 × 10−8) (see Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract). Additionally, it was shown by Tsuritani et al 35 that up‐regulation of the cytokine receptor–like factor 1/cardiotrophin‐like cytokine complex in ATDC5 cells disrupts cartilage homeostasis and promotes progression of OA by enhancing the proliferation of chondrocytes and suppressing the production of cartilage matrix. As such, we hypothesize that the alternative allele T of rs7256319 in heterozygote carriers may be able to mitigate CRLF1/CLCF1 signaling toward ongoing cartilage degradation due to primary OA.
Among the notable novel putative OA genes in Table 2 is WWP2, which showed (in addition to multiple coding SNPs marking consistent AI expression of WWP2 in cartilage) significant differential expression between preserved and lesioned OA cartilage (fold change 0.78, FDR = 0.0053; results not shown) and a signal of genetic association with minimal JSW (P = 0.0028) (Figure 3). Based on these data, we hypothesize that allele rs1052429 A located in WWP2 is an OA susceptibility allele that acts via higher expression of WWP2 in cartilage, which is associated with lower minimal JSW and thus with degeneration of cartilage. In addition, expression of WWP2 was previously shown to be consistently modified by methylation at CpG sites 36. The WWP2 protein is a member of the Nedd4 family of E3 ligases, which play an important role in protein ubiquitination 37. Moreover, the encoded protein was shown to physically interact with SOX9, thereby affecting the transcriptional activity of SOX9 via translocation to the nucleus 38.
Similarly, RPS3, encoding ribosomal protein S3, is subject to significant AI marked by rs3133187 with the G allele decreasing expression of RPS3 transcript in cartilage (FDR = 4.97 × 10−9) and significant association with hip OA (P = 1.1 × 10−3). Ribosomal protein S3 is a multifunctional DNA repair endonuclease and ribosomal protein, yet its role in inducing apoptosis through activation of CASP8/CASP3 may be relevant to mention with respect to OA 39. It was recently shown by Jeon et al 40 that selective removal of senescent cells may attenuate the development of OA. We hypothesize that decreasing RPS3 expression by the OA risk allele G of rs3133187 negatively affects removal of senescent chondrocytes by cytokine‐induced apoptosis. Nonetheless, functional studies are necessary to verify the exact mechanism by which the AI in CRLF1, WWP2, and RPS3 contributes to cartilage degeneration in humans. It would be preferable to investigate this using human in vitro micromass cultures in which the expression of the genes (e.g., by lentiviral induction) is modified in the direction of the risk alleles as previously found with, for example, GDF5 15 or DIO2 14.
Our RNA sequencing data set consisted of both preserved and lesioned OA samples, as well as knee and hip cartilage. Due to our focus on significant AI effects across our samples, we have ignored possible variation in the AI effects between preserved and lesioned OA cartilage or between knee and hip joints. Such variation could arise, e.g., due to local expression of transcription factors that exaggerate or attenuate the observed AI. By providing AI, defined by φ across all individuals heterozygous for any of the 13,435 SNPs (see Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract), possible differences in AI for preserved/OA or hip/knee cartilage may be explored. It should be mentioned, however, that the current sample size and the variable number of heterozygous individuals likely precludes a robust statement on either analyses and will require additional targeted AI measurements by, for example, TaqMan assay.
By combining reported genetic OA signals with AI SNPs that alter transcription of articular cartilage genes in ‐ cis, we also had the opportunity to address functionality of OA risk alleles. For example, the A allele of rs788748, located upstream of IGFBP3, is associated with lower odds of hip OA 41. Given that this SNP is not located in an exon, we assessed its potential revelance to AI through rs6670 double heterozygotes, which revealed that the protective rs788748 A allele marks lower expression of IGFBP3 compared to the G allele. In addition, significant up‐regulation of the gene was observed in OA‐affected cartilage compared to paired preserved cartilage, as well as a similar eQTL effect of the rs788748 SNP (see Supplementary Table 9, http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract), similar to what has been reported by others 8. These observations solidify the notion of IGFBP3's role in OA, and we propose that the reported protective effect of the rs788748 A allele is mediated through lower expression of the transcript.
For a selected number of comparable (although gene‐targeted) studies, we discuss the respective confirmations and/or discrepancies. A number of SNPs are known to mark AI in articular cartilage, as has been shown by gene‐targeted approaches. We were able to replicate the earlier observed AI of ALDH1A2 18 and MGP 20 and to a lesser extent that of DIO2 14 and GDF5 42. Furthermore, for rs11177 (GNL3) 16 and rs6617 (SPCS1) 16 in our data set, AI was not as obvious as previously reported, and we did not observe heterozygotes for rs143383 (GDF5) 23 or rs3815148 (HBP1) 24.
For COL11A1, AI was previously thoroughly investigated by Raine et al 17 in view of the OA risk SNP rs2615977 7 and the lumbar disc herniation SNP rs1676486 43. They showed that considerable AI of COL11A1 was correlated with the lumbar disc herniation risk allele of SNP rs1676486, while the observed AI of COL11A1 was not correlated with the OA risk allele of SNP rs2615977. The latter result was based on the AI effect observed in individuals double heterozygous for rs2615977 and rs9659030. As shown in Supplementary Figure 2 and Supplementary Table 8 (http://onlinelibrary.wiley.com/doi/10.1002/art.40748/abstract), we observed 2 independent COL11A1 SNPs (D′ = 0.2, r2 = 0.01) 44 with highly significant meta‐φ AI. The first was the abovementioned SNP rs9659030 with meta‐φ AI of 0.65 (P = 1.2 × 10−25) and the second was SNP rs2229783 with meta‐φ AI of 0.53 (P = 3.7 × 10−8). Notably, and in contrast to the results obtained by Raine et al 17, the extent and consistency of AI for rs9659030 were considerably higher than those for rs2229783 and rs1676486. The suggestive evidence of AI of the lumbar disc herniation risk allele of SNP rs1676486 as well as the AI of its proxy SNP rs2229783 (D′ = 1, r2 = 0.3) confirm that the lumbar disc herniation risk allele is likely associated with lower COL11A1 expression. Moreover, the AI of the noncoding OA risk SNP rs2615977, which is not in strong LD with a coding SNP, remains unclear. Yet, based on the proxy SNP rs1031820 (D′ = 0.77, r2 = 0.16), it may be that the OA risk allele of rs2615977 also acts via lower expression of COL11A1.
Taken together, these confirmations and/or discrepancies indicate first and foremost that additional replication to verify AI is required to increase confidence in the observed AI (e.g., by better preselection on heterozygous samples). Second, it stresses the fact that observed AI reflects regulatory properties of the respective LD block and does not per se identify genetic variation that affects respective gene expression levels mechanistically. Furthermore, despite the applied filtering steps and statistics, the list of significant AI SNPs potentially contains a number of false positives, some of which could have originated from alignment bias. While future novel alignment and other bioinformatic approaches 45 might address these issues from a more fundamental perspective, in the present study we have aimed to reduce false‐positive AI SNPs by including multiple filtering steps (0.1<φ<0.9, include SNPs with at least 2 heterozygotes, and null hypothesis adjustment).
While in canonical GWAS a strict genome‐wide significance level of 5 × 10−8 is imposed due to the vast amount of SNPs that are tested for, we postulate that providing SNPs that are more likely to affect expression of genes in‐cis in a disease‐relevant tissue could aid the search for the functional susceptibility SNPs and the putative OA risk gene. Nevertheless, among the significant AI SNPs, we did not necessarily obtain a clear enrichment of putative significant OA risk SNPs; among the SNPs for which we had both AI and GWAS data, 6.7% showed significant association with the OA phenotypes, while among the significant AI genes (FDR <0.05), 7.2% showed significant association with the OA phenotypes. Further downstream selection criteria, such as (but not limited to) significant differential expression between preserved and OA‐lesioned cartilage and/or trans‐eQTL analysis, will help tailor genetic association analyses even more and might attribute SNPs to specific disease facets, such as extent of cartilage degradation, as we have shown in the present study. Of note, inherent to our study design, we might have missed genes that affect joint morphology or cartilage integrity during development and/or that change expression in healthy cartilage or during early‐stage OA.
In summary, we present a framework and resulting data set for researchers in the OA research field to probe for disease‐relevant genetic variation that affects gene expression in pivotal disease‐affected tissue. This likely includes putative novel compelling OA risk genes such as CRLF1, WWP2, and RPS3.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Meulenbelt had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Den Hollander, Couthino de Almeida, Lakenberg, ‘t Hoen, Ramos, Meulenbelt.
Acquisition of data
Den Hollander, Bomer, Nelissen, Ramos, Meulenbelt.
Analysis and interpretation of data
Den Hollander, Pulyakhina, Boer, van der Breggen, Arindrarto, Couthino de Almeida, Sentner, Laros, ‘t Hoen, Slagboom, van Meurs, Ramos, Meulenbelt.
Supporting information
Supplementary Figures
Supplementary Table 1
Supplementary Table 2
Supplementary Table 3
Supplementary Table 4
Supplementary Table 5
Supplementary Table 6
Supplementary Table 7
Supplementary Table 8
Supplementary Table 9
Supplementary Table 10
Supplementary Table 11
Acknowledgments
We thank all RAAK study participants. We thank Eka Suchiman for her help in preparing DNA and RNA samples and collection of RAAK study specimens.
Supported in part by Treat OA (grant 200800 from the European Commission Seventh Framework Programme), the Dutch Arthritis Society (grant DAA 10‐1‐402), and the Dutch Scientific Research Council NWO ZonMW VICI (grant 91816631/528). The Research Arthritis and Articular Cartilage study is supported by Leiden University Medical Center.
Drs. van Meurs, Ramos, and Meulenbelt contributed equally to this work.
No potential conflicts of interest relevant to this article were reported.
References
- 1. Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 2012;64:1697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Man GS, Mologhianu G. Osteoarthritis pathogenesis—a complex process that involves the entire joint. J Med Life 2014;7:37–41. [PMC free article] [PubMed] [Google Scholar]
- 3. Saito T, Fukai AF, Mabuchi AF, Ikeda TF, Yano FF, Ohba S, et al. Transcriptional regulation of endochondral ossification by HIF‐2α during skeletal growth and osteoarthritis development. Nat Med 2010;16:678–86. [DOI] [PubMed] [Google Scholar]
- 4. Sherwood J, Bertrand J, Nalesso G, Poulet B, Pitsillides A, Brandolini L, et al. A homeostatic function of CXCR2 signalling in articular cartilage. Ann Rheum Dis 2015;74:2207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van der Kraan PM, Blaney Davidson EN, van den Berg WB. A role for age‐related changes in TGFβ signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res Ther 2010;12:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jeffries MA, Donica M, Baker LW, Stevenson ME, Annan AC, Humphrey MB, et al. Genome‐wide DNA methylation study identifies significant epigenomic changes in osteoarthritic cartilage. Arthritis Rheumatol 2014;66:2804–15. [DOI] [PubMed] [Google Scholar]
- 7. Zeggini E, Panoutsopoulou K, Southam L, Rayner NW, Day‐Williams AG, Lopes MC, et al. Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome‐wide association study. Lancet 2012;380:815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Castano‐Betancourt MC, Evans DS, Ramos YF, Boer CG, Metrustry S, Liu Y, et al. Novel genetic variants for cartilage thickness and hip osteoarthritis. PLoS Genet 2016;12:e1006260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meulenbelt I, Min JL, Bos S, Riyazi N, Houwing‐Duistermaat JJ, van der Wijk HJ, et al. Identification of DIO2 as a new susceptibility locus for symptomatic osteoarthritis. Hum Mol Genet 2008;17:1867–75. [DOI] [PubMed] [Google Scholar]
- 10. Xu Y, Barter MJ, Swan DC, Rankin KS, Rowan AD, Santibanez‐Koref M, et al. Identification of the pathogenic pathways in osteoarthritic hip cartilage: commonality and discord between hip and knee OA. Osteoarthritis Cartilage 2012;20:1029–38. [DOI] [PubMed] [Google Scholar]
- 11. Den Hollander W, Ramos YF, Bomer N, Elzinga S, van der Breggen R, Lakenberg N, et al. Transcriptional associations of osteoarthritis‐mediated loss of epigenetic control in articular cartilage. Arthritis Rheumatol 2015;67:2108–16. [DOI] [PubMed] [Google Scholar]
- 12. Ramos YF, Metrustry S, Arden N, Bay‐Jensen AC, Beekman M, de Craen AJ, et al. Meta‐analysis identifies loci affecting levels of the potential osteoarthritis biomarkers sCOMP and uCTX‐II with genome wide significance. J Med Genet 2014;51:596–604. [DOI] [PubMed] [Google Scholar]
- 13. Zhang L, Yang M, Marks P, White LM, Hurtig M, Mi QS, et al. Serum non‐coding RNAs as biomarkers for osteoarthritis progression after ACL injury. Osteoarthritis Cartilage 2012;20:1631–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bos SD, Bovee JV, Duijnisveld BJ, Raine EV, van Dalen WJ, Ramos YF, et al. Increased type II deiodinase protein in OA‐affected cartilage and allelic imbalance of OA risk polymorphism rs225014 at DIO2 in human OA joint tissues. Ann Rheum Dis 2012;71:1254–8. [DOI] [PubMed] [Google Scholar]
- 15. Reynard LN, Bui C, Canty‐Laird EG, Young DA, Loughlin J. Expression of the osteoarthritis‐associated gene GDF5 is modulated epigenetically by DNA methylation. Hum Mol Genet 2011;20:3450–60. [DOI] [PubMed] [Google Scholar]
- 16. Gee F, Clubbs CF, Raine EV, Reynard LN, Loughlin J. Allelic expression analysis of the osteoarthritis susceptibility locus that maps to chromosome 3p21 reveals cis‐acting eQTLs at GNL3 and SPCS1. BMC Med Genet 2014;15:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raine EV, Dodd AW, Reynard LN, Loughlin J. Allelic expression analysis of the osteoarthritis susceptibility gene COL11A1 in human joint tissues. BMC Musculoskelet Disord 2013;14:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Styrkarsdottir U, Thorleifsson G, Helgadottir HT, Bomer N, Metrustry S, Bierma‐Zeinstra S, et al. Severe osteoarthritis of the hand associates with common variants within the ALDH1A2 gene and with rare variants at 1p31. Nat Genet 2014;46:498–502. [DOI] [PubMed] [Google Scholar]
- 19. Bomer N, den Hollander W, Ramos YF, Bos SD, van der Breggen R, Lakenberg N, et al. Underlying molecular mechanisms of DIO2 susceptibility in symptomatic osteoarthritis. Ann Rheum Dis 2015;74:1571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Den Hollander W, Boer CG, Hart DJ, Yau MS, Ramos YF, Metrustry S, et al. Genome‐wide association and functional studies identify a role for matrix Gla protein in osteoarthritis of the hand. Ann Rheum Dis 2017;76:2046–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loughlin J. Osteoarthritis: all types of trouble–defining OA in the genomic era. Nat Rev Rheumatol 2011;7:200–1. [DOI] [PubMed] [Google Scholar]
- 22. Panoutsopoulou K, Thiagarajah S, Zengini E, Day‐Williams AG, Ramos YF, Meessen JM, et al. Radiographic endophenotyping in hip osteoarthritis improves the precision of genetic association analysis. Ann Rheum Dis 2017;76:1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Southam L, Rodriguez‐Lopez J, Wilkins JM, Pombo‐Suarez M, Snelling S, Gomez‐Reino JJ, et al. An SNP in the 5′‐UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum Mol Genet 2007;16:2226–32. [DOI] [PubMed] [Google Scholar]
- 24. Raine EV, Wreglesworth N, Dodd AW, Reynard LN, Loughlin J. Gene expression analysis reveals HBP1 as a key target for the osteoarthritis susceptibility locus that maps to chromosome 7q22. Ann Rheum Dis 2012;71:2020–7. [DOI] [PubMed] [Google Scholar]
- 25. Ramos YF, den HW , Bovee JV, Bomer N, van der Breggen R, Lakenberg N, et al. Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage: the RAAK study. PLoS One 2014;9:e103056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goya R, Sun MG, Morin RD, Leung G, Ha G, Wiegand KC, et al. SNVMix: predicting single nucleotide variants from next‐generation sequencing of tumors. Bioinformatics 2010;26:730–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robinson MD, McCarthy DJ, Smyth GK. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 29. Schoenmaker M, de Craen AJ, de Meijer PH, Beekman M, Blauw GJ, Slagboom PE, et al. Evidence of genetic enrichment for exceptional survival using a family approach: the Leiden Longevity Study. Eur J Hum Genet 2006;14:79–84. [DOI] [PubMed] [Google Scholar]
- 30. Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 2014;46:1173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wilkins JM, Southam L, Price AJ, Mustafa Z, Carr A, Loughlin J. Extreme context‐specificity in differential allelic expression. Hum Mol Genet 2007;16:537–46. [DOI] [PubMed] [Google Scholar]
- 32. Evangelou E, Kerkhof HJ, Styrkarsdottir U, Ntzani EE, Bos SD, Esko T, et al. A meta‐analysis of genome‐wide association studies identifies novel variants associated with osteoarthritis of the hip. Ann Rheum Dis 2014;73:2130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Raine EV, Reynard LN, van de Laar IM, Bertoli‐Avella AM, Loughlin J. Identification and analysis of a SMAD3 cis‐acting eQTL operating in primary osteoarthritis and in the aneurysms and osteoarthritis syndrome. Osteoarthritis Cartilage 2014;22:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gee F, Rushton MD, Loughlin J, Reynard LN. Correlation of the osteoarthritis susceptibility variants that map to chromosome 20q13 with an expression quantitative trait locus operating on NCOA3 and with functional variation at the polymorphism rs116855380. Arthritis Rheumatol 2015;67:2923–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsuritani K, Takeda J, Sakagami J, Ishii A, Eriksson T, Hara T, et al. Cytokine receptor‐like factor 1 is highly expressed in damaged human knee osteoarthritic cartilage and involved in osteoarthritis downstream of TGF‐β. Calcif Tissue Int 2010;86:47–57. [DOI] [PubMed] [Google Scholar]
- 36. Reynard LN. Analysis of genetics and DNA methylation in osteoarthritis: what have we learnt about the disease? Semin Cell Dev Biol 2017;62:57–66. [DOI] [PubMed] [Google Scholar]
- 37. Chen W, Jiang X, Luo Z. WWP2: a multifunctional ubiquitin ligase gene. Pathol Oncol Res 2014;20:799–803. [DOI] [PubMed] [Google Scholar]
- 38. Nakamura Y, Yamamoto K, He X, Otsuki B, Kim Y, Murao H, et al. WWP2 is essential for palatogenesis mediated by the interaction between Sox9 and mediator subunit 25. Nat Commun 2011;2:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jang CY, Lee JY, Kim J. RpS3, a DNA repair endonuclease and ribosomal protein, is involved in apoptosis. FEBS Lett 2004;560:81–5. [DOI] [PubMed] [Google Scholar]
- 40. Jeon OH, David N, Campisi J, Elisseeff JH. Senescent cells and osteoarthritis: a painful connection. J Clin Invest 2018;128:1229–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Evans DS, Cailotto F, Parimi N, Valdes AM, Castano‐Betancourt MC, Liu Y, et al. Genome‐wide association and functional studies identify a role for IGFBP3 in hip osteoarthritis. Ann Rheum Dis 2015;74:1861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Egli RJ, Southam L, Wilkins JM, Lorenzen I, Pombo‐Suarez M, Gonzalez A, et al. Functional analysis of the osteoarthritis susceptibility‐associated GDF5 regulatory polymorphism. Arthritis Rheum 2009;60:2055–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mio F, Chiba K, Hirose Y, Kawaguchi Y, Mikami Y, Oya T, et al. A functional polymorphism in COL11A1, which encodes the α1 chain of type XI collagen, is associated with susceptibility to lumbar disc herniation. Am J Hum Genet 2007;81:1271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Machiela MJ, Chanock SJ. LDassoc: an online tool for interactively exploring genome‐wide association study results and prioritizing variants for functional investigation. Bioinformatics 2018;34:887–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Castel SE, Levy‐Moonshine A, Mohammadi P, Banks E, Lappalainen T. Tools and best practices for data processing in allelic expression analysis. Genome Biol 2015;16:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Table 1
Supplementary Table 2
Supplementary Table 3
Supplementary Table 4
Supplementary Table 5
Supplementary Table 6
Supplementary Table 7
Supplementary Table 8
Supplementary Table 9
Supplementary Table 10
Supplementary Table 11