

Abstract
Objective
Solriamfetol (JZP‐110) is a selective dopamine and norepinephrine reuptake inhibitor with wake‐promoting effects. This phase 3 study (NCT02348593) evaluated the safety and efficacy of solriamfetol in narcolepsy.
Methods
Patients with narcolepsy with mean sleep latency <25 minutes on the Maintenance of Wakefulness Test (MWT), Epworth Sleepiness Scale (ESS) score ≥10, and usual nightly sleep ≥6 hours were randomized to solriamfetol 75, 150, or 300 mg, or placebo for 12 weeks. Coprimary endpoints were change from baseline to week 12 in MWT and ESS. Improvement on the Patient Global Impression of Change (PGI‐C) was the key secondary endpoint.
Results
Safety and modified intention‐to‐treat populations included 236 and 231 patients, respectively. Solriamfetol 300 and 150 mg were positive on both coprimary endpoints. Least squares mean (standard error [SE]) changes from baseline were 12.3 (SE = 1.4) and 9.8 (SE = 1.3) minutes for solriamfetol 300 and 150 mg on the MWT, respectively, versus 2.1 (SE = 1.3) minutes for placebo, and −6.4 (SE = 0.7) for 300 mg and −5.4 (SE = 0.7) for 150 mg on the ESS versus −1.6 (SE = 0.7) for placebo (all p < 0.0001). At week 12, higher percentages of patients treated with solriamfetol 150 mg (78.2%) and 300 mg (84.7%) reported PGI‐C improvement relative to placebo (39.7%; both p < 0.0001). Adverse events ≥5% across all solriamfetol doses included headache (21.5%), nausea (10.7%), decreased appetite (10.7%), nasopharyngitis (9.0%), dry mouth (7.3%), and anxiety (5.1%).
Interpretation
Solriamfetol has the potential to be an important therapeutic option for the treatment of impaired wakefulness and excessive sleepiness in patients with narcolepsy. ANN NEUROL 2019;85:359–370.
Narcolepsy is a chronic neurological disorder resulting from the dysregulation of neurophysiologic pathways that control the stability of sleep and wake states.1 Excessive sleepiness (ES) and impaired daytime wakefulness are essential and somewhat independent symptoms of narcolepsy.2, 3 These symptoms contribute to reductions in function, productivity, and quality of life, and an increased risk for motor vehicle accidents.4, 5, 6, 7, 8, 9 Although ES and impaired wakefulness are present in all individuals with narcolepsy,10 2 types of narcolepsy are recognized: type 1 and type 2, formerly known as narcolepsy with and without cataplexy, respectively. Narcolepsy type 1 is characterized by the presence of cataplexy and/or hypocretin deficiency as measured by reduced hypocretin levels in the cerebrospinal fluid, and type 2 is characterized by the absence of cataplexy but the presence of relevant findings on the Multiple Sleep Latency Test (ie, ≥2 sleep onset rapid eye movement periods and sleep latency ≤8 minutes) that are indicative of narcolepsy.10
Treatment of narcolepsy is symptomatically driven, and management of ES generally relies on pharmacologic intervention from several medication classes including the central nervous system depressant sodium oxybate, stimulants such as methylphenidate and amphetamines, and the wake‐promoting agents modafinil and armodafinil.2, 11 Additionally, the wake‐promoting agent pitolisant has been recently approved for the treatment of narcolepsy in Europe.12 However, current pharmacologic options may be associated with factors such as poor tolerability, tolerance over time, abuse potential, and suboptimal response that may preclude or limit their use in some patients.2, 11
Solriamfetol (formerly known as JZP‐110 and ADX‐N05) is a selective dopamine and norepinephrine reuptake inhibitor that binds to dopamine and norepinephrine transporters in vitro at concentrations in the micromolar range, inhibiting reuptake without promoting the release of monoamines.13 Solriamfetol is distinguished from other wake‐promoting agents by its dual reuptake inhibition at dopamine and norepinephrine transporters, and is distinguished from the amphetamine stimulants by its lack of release of monoamines.14, 15 Together, these differences may account for the robust wake‐promoting effects and the lack of rebound hypersomnia that have been observed in rodent models of narcolepsy.13, 16 In 2 phase 2 controlled clinical trials in adult patients with narcolepsy, solriamfetol reduced patient‐reported ES as measured on the Epworth Sleepiness Scale (ESS) and improved wakefulness as measured by the objective Maintenance of Wakefulness Test (MWT).17, 18
This phase 3 trial was initiated to demonstrate the efficacy and safety of solriamfetol for the treatment of ES and impaired wakefulness in patients with narcolepsy type 1 or type 2.
Patients and Methods
Study Design and Study Participants
This was a clinical trial from the Treatment of Obstructive Sleep Apnea and Narcolepsy Excessive Sleepiness (TONES) phase 3 program, the TONES 2 study. This phase 3, double‐blind, randomized, placebo‐controlled, parallel‐group study (ClinicalTrials.gov identifier NCT02348593, EudraCT number 2014‐005487‐15) was performed at 50 study centers in the United States and Canada, and 9 centers in Finland, France, Germany, and Italy. The study was approved by the institutional review board or independent ethics committee for each study center, and was performed in accordance with the Declaration of Helsinki; all patients provided written informed consent.
Eligible patients were 18 to 75 years old, with a diagnosis of narcolepsy type 1 or type 2 according to criteria in either the International Classification of Sleep Disorders, 3rd edition (ICSD‐3)10 or Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM‐5).19 The DSM‐5 criteria include patients who have been diagnosed with narcolepsy based on the presence of cataplexy and were applied in this study to include such patients who had been diagnosed with narcolepsy on the basis of cataplexy under ICSD‐2 but who no longer meet diagnostic criteria based on a history of cataplexy under ICSD‐3. Additional inclusion criteria were baseline mean sleep latency <25 minutes on the first 4 trials of a 5‐trial, 40‐minute MWT,20 baseline ESS21 score ≥10, usual nightly total sleep time ≥6 hours (by self‐report), and a body mass index between 18 and 45 kg/m2. The presence of any clinically relevant untreated medical, psychiatric, or behavioral disorder or medical condition other than narcolepsy that is associated with ES (ie, night‐time or variable shift work) was reason for exclusion, as was a history or presence of any acutely unstable medical or psychiatric disorder, or surgical history that could affect the safety of the patient; female patients who were pregnant or lactating were also excluded. Although use of medications that could affect the evaluation of ES or cataplexy was also reason for exclusion, patients could be enrolled if prior use had stopped for >5 half‐lives of the drug and the patient had returned to baseline level of daytime sleepiness ≥7 days prior to the baseline visit.
Randomization and Masking
Stratified randomization was performed based on the presence or absence of cataplexy, with patients assigned in a 1:1:1:1 ratio to treatment with solriamfetol 75, 150, or 300 mg, or placebo. The investigator accessed an Interactive Voice Response System or an Interactive Web Response System to randomize eligible patients.
All study personnel were blinded to the study treatments, and a double‐blind approach was used whereby all study drugs were prepared in identical opaque gelatin capsules to ensure adequate blinding.
Interventions and Outcomes
Patients who met all inclusion and no exclusion criteria and were randomized to the treatment groups received oral placebo or solriamfetol 75, 150, or 300 mg once daily for 12 weeks. Patients who were randomized to the 150 and 300 mg doses received 75 and 150 mg, respectively, on days 1 through 3 of the first week, with the full dose commencing on day 4.
The coprimary endpoints were change from baseline to week 12 on (1) MWT mean sleep latency on the first 4 trials of the MWT and (2) ESS score. The MWT was performed after in‐clinic overnight polysomnography at baseline and weeks 1, 4, and 12 or early termination, and the ESS was completed by the patients at baseline and weeks 1, 4, 8, and 12 or early termination. If data from ≥2 of the first 4 individual MWT trials were missing at a given time point, the mean sleep latency on the MWT was set to missing at that specific time point. If data from only 1 of the first 4 individual MWT trials were missing, the mean of the remaining 3 of the first 4 MWT trials was used for calculating the mean sleep latency on the MWT for that individual at that specific time point. The ESS total score is the sum of 8 item scores. If ≥3 item scores on the ESS were missing at a specific time point, the ESS total score was set to missing. If only 1 or 2 ESS items were missing at a specific time point, the mean of the remaining 7 or 6 nonmissing ESS items for that individual at that time point was used to impute the missing ESS items. In these cases, the ESS total score was calculated as the sum of the observed and the imputed item scores.
The percentage of patients who reported improvement on the Patient Global Impression of Change (PGI‐C)22 at week 12 was the key secondary endpoint. Other secondary endpoints were time course on the MWT as measured by the change in sleep latency on each of the 5 MWT trials; change in the mean sleep latency from baseline to week 4; change in ESS from baseline to weeks 1, 4, and 8; percentage of patients who reported improvement on the PGI‐C at weeks 1, 4, and 8; and percentage of patients reported improved at weeks 1, 4, 8, and 12 on the Clinical Global Impression of Change (CGI‐C).22 Improvements on the global scales were defined by responses reflecting “minimal,” “much,” or “very much” improved. Change in the mean and median weekly number of cataplexy attacks was an exploratory endpoint among the subgroup of patients who reported the presence of cataplexy. These patients completed a cataplexy frequency diary to record the number of cataplexy attacks that they had each day, beginning after discontinuation of narcolepsy medication and through week 12.
Safety and tolerability evaluation included the incidence of all adverse events reported by the patients, who were queried about adverse events at each clinic visit and during telephone contact calls for the weeks where no clinic visits were scheduled, as well as those adverse events noted by the investigator. Weight, vital signs, electrocardiograms, and the Columbia‐Suicide Severity Rating Scale23 were assessed at each clinic visit. On the days when MWT was performed, blood pressure and heart rate measurements were collected at 7 time points, with the average of these time points from predose to 9 hours postdose used for evaluating change from baseline. Additionally, blood pressure and heart rate were assessed at 30‐minute intervals using 24‐hour ambulatory blood pressure monitoring at baseline and at week 8. Mean values for blood pressure and heart rate were calculated over the 24‐hour period for each individual, and mean changes from screening to week 8 were summarized.
Statistical Analyses
Accounting for withdrawals, approximately 240 patients were planned for enrollment, approximately 60 for each treatment group. This sample size was based on an estimate of 54 patients per group to provide at least 80% power to detect a difference of 6 minutes in the mean sleep latency time as determined from the MWT (mean of the first 4 trials) and a difference of 4 points on the ESS changes from baseline to week 12 between each solriamfetol treatment dose group and placebo. These estimates were based on the effects observed at the 150 and 300 mg doses in 2 phase 2 studies.17, 18 This calculation also assumed standard deviations (SDs) in the changes from baseline of 10 minutes for the MWT and 6 points for the ESS, and a 2‐sided significance level of 0.05 using a t test.
Efficacy analyses were based on the modified intention‐to‐treat (mITT) population, defined as all randomized patients who received at least 1 dose of study drug, and had baseline and at least 1 postbaseline evaluation by MWT or ESS. Both MWT and ESS endpoints were analyzed using a mixed‐effect repeated measures model, including fixed effects for treatment (ie, dose group), visit (as a discrete and repeated factor), treatment‐by‐visit interaction, baseline value of the corresponding endpoint (as a continuous covariate), and randomization stratification factor (ie, presence or absence of cataplexy). The response variable was the change from baseline to each postbaseline visit, and the coprimary endpoints were the changes from baseline to week 12. The least squares (LS) mean and the standard error (SE) of treatment difference versus placebo and their 95% confidence intervals (CIs) were presented. The PGI‐C and CGI‐C were analyzed using chi‐squared tests. A fixed hierarchical testing procedure was used to correct for multiplicity, starting with the highest solriamfetol dose for the coprimary endpoints and followed by the key secondary endpoint. Both coprimary endpoints (MWT and ESS) had to be significant at the 0.05 level in the primary analysis for testing to proceed to the key secondary endpoint (PGI‐C); testing proceeded in the order specified above for each subsequent lower dose with statistical significance claimed only for those outcomes above the break in the hierarchy. Nominal p values are presented for differences below the hierarchical break.
In addition to the primary analyses described above, 4 sensitivity analyses were performed to assess the impact of missing data on the coprimary endpoints. For the MWT and ESS, 2 single imputation approaches (last observation carried forward and mean imputation) and 2 multiple imputation approaches (Markov chain Monte Carlo with regression method24 and Pattern Mixture model using dropout pattern imputation method25) were conducted. In addition, for the key secondary endpoint of PGI‐C, 2 single imputation approaches (worst case and varies by early termination reason) were conducted to assess the impact of missing data. All analyses were performed using SAS v9.3 or later (SAS Institute, Cary, NC).
Results
Patient Population
Of the 239 patients who were randomized, 236 received at least 1 dose of study drug and were included in the safety population (Fig 1). The mITT population consisted of 231 patients; 1 patient randomized to placebo and 4 patients randomized to solriamfetol 150 mg did not have baseline or at least 1 postbaseline efficacy assessment of MWT and ESS. The discontinuation rate was highest in the solriamfetol 300 mg group (27.1%, with lack of efficacy [10.2%, n = 6] and adverse events [8.5%, n = 5] as the most common reasons for discontinuation), followed by the solriamfetol 75 mg (16.9%), placebo (10.3%), and solriamfetol 150 mg (7.3%) groups (see Fig 1). Discontinuation due to lack of efficacy did not appear to be dose‐related. Three of the 6 patients who discontinued due to lack of efficacy in the 300 mg group and 3 of the 4 patients who discontinued due to lack of efficacy in the 75 mg group had cataplexy at screening and had discontinued their anticataplectic medication(s) prior to starting study drug on day 1 of the current study.
Figure 1.
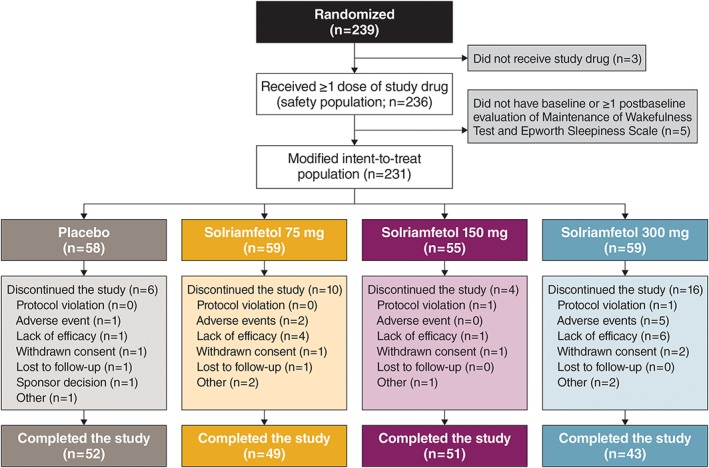
Patient disposition. [Color figure can be viewed at www.annalsofneurology.org]
Demographic and clinical characteristics were similar across treatment groups (Table 1). Overall, the majority of patients (64.4%) were rated by clinicians as moderately or markedly ill and were characterized by impaired wakefulness and ES, as indicated by baseline MWT mean sleep latency of 7.5 (SD = 5.7) minutes and ESS scores of 17.2 (SD = 3.2), respectively. Most patients (90.7%) had prior use of psychostimulants; prior use of sodium oxybate and antidepressants was reported for 25.8% and 34.7% of patients, respectively. Cataplexy was present in 50.8% of patients overall, with similar percentages of patients with cataplexy in each of the treatment groups (see Table 1); demographic and baseline characteristics were similar between patients with cataplexy and those without cataplexy (data not shown).
Table 1.
Baseline Demographic and Clinical Characteristics of the Safety Population
| Characteristic | Placebo, n = 59 | Solriamfetol | Total, N = 236 | ||
|---|---|---|---|---|---|
| 75 mg, n = 59 | 150 mg, n = 59 | 300 mg, n = 59 | |||
| Age, yr | 36.0 ± 15.2 | 36.5 ± 12.8 | 38.1 ± 13.0 | 34.3 ± 11.5 | 36.2 ± 13.2 |
| Sex, n | |||||
| M | 24 (40.7%) | 22 (37.3%) | 17 (28.8%) | 19 (32.2%) | 82 (34.7%) |
| F | 35 (59.3%) | 37 (62.7%) | 42 (71.2%) | 40 (67.8%) | 154 (65.3%) |
| Race, n | |||||
| Asian | 0 | 0 | 3 (5.1%) | 3 (5.1%) | 6 (2.5%) |
| Black or African American | 10 (16.9%) | 12 (20.3%) | 6 (10.2%) | 5 (8.5%) | 33 (14.0%) |
| White | 47 (79.7%) | 46 (78.0%) | 48 (81.4%) | 48 (81.4%) | 189 (80.1%) |
| Other | 2 (3.4%) | 1 (1.7%) | 2 (3.4%) | 3 (5.1%) | 8 (3.4%) |
| BMI, kg/m2 | 29.1 ± 6.0 | 27.9 ± 5.4 | 27.9 ± 5.8 | 28.1 ± 6.3 | 28.3 ± 5.9 |
| Presence of cataplexy, n | 29 (49.2%) | 31 (52.5%) | 30 (50.8%) | 30 (50.8%) | 120 (50.8%) |
| MWT sleep latency, minutesa | 6.1 ± 5.6 | 7.5 ± 5.4 | 7.7 ± 5.6 | 8.7 ± 6.2 | 7.5 ± 5.7 |
| ESS scoreb | 17.3 ± 2.8 | 17.3 ± 3.5 | 16.9 ± 3.7 | 17.2 ± 2.8 | 17.2 ± 3.2 |
| Baseline CGI‐S score, n | |||||
| 1 = Normal, not at all ill | 0 | 0 | 0 | 0 | 0 |
| 2 = Borderline ill | 0 | 0 | 0 | 0 | 0 |
| 3 = Mildly ill | 1 (1.7%) | 3 (5.1%) | 3 (5.1%) | 1 (1.7%) | 8 (3.4%) |
| 4 = Moderately ill | 14 (23.7%) | 14 (23.7%) | 16 (27.1%) | 17 (28.8%) | 61 (25.8%) |
| 5 = Markedly ill | 26 (44.1%) | 20 (33.9%) | 24 (40.7%) | 21 (35.6%) | 91 (38.6%) |
| 6 = Severely ill | 13 (22.0%) | 17 (28.8%) | 13 (22.0%) | 12 (20.3%) | 55 (23.3%) |
| 7 = Among the most extremely ill | 4 (6.8%) | 5 (8.5%) | 3 (5.1%) | 8 (13.6%) | 20 (8.5%) |
| Missing | 1 (1.7%) | 0 | 0 | 0 | 1 (0.4%) |
Values are mean ± standard deviation or n (%).
MWT measures a patient's ability to stay awake for a given period of time. Patients were included if their baseline mean sleep latency was <25 minutes on the first 4 trials of a 5‐trial, 40‐minute MWT.
ESS scores range from 0 to 24, with scores of 16–24 indicating severe excessive sleepiness.
BMI = body mass index; CGI‐S = Clinical Global Impression of Severity; ESS = Epworth Sleepiness Scale; F = female; M = male; MWT = Maintenance of Wakefulness Test.
Efficacy
Solriamfetol 300 mg and 150 mg doses met the coprimary endpoints of MWT and ESS as well as the percentage of patients who reported improvement on the PGI‐C (all p < 0.0001; Table 2). Significance was not achieved for solriamfetol 75 mg on the MWT.
Table 2.
Hierarchical Testing of Coprimary and Key Secondary Efficacy Endpoints in the Modified Intention‐to‐Treat Population
| Endpoint | Solriamfetol, Treatment Difference from Placebo, Least Squares Mean (95% CI) | ||
|---|---|---|---|
| 300 mg | 150 mg | 75 mg | |
| Maintenance of Wakefulness Test, min | 10.14 (6.39, 13.90), p < 0.0001 | 7.65 (3.99, 11.31), p < 0.0001 | 2.62 (−1.04, 6.28), p = 0.1595 |
| Epworth Sleepiness Scale | −4.7 (−6.6, −2.9), p < 0.0001 | −3.8 (−5.6, −2.0), p < 0.0001 | −2.2 (−4.0, −0.3), p = 0.0211 |
| Patient Global Impression of Change (%)a | 45.1 (29.51, 60.67), p < 0.0001 | 38.5 (21.86, 55.19), p < 0.0001 | 28.1 (10.80, 45.48), p = 0.0023b |
A fixed hierarchical testing procedure was used to correct for multiplicity, starting with the highest solriamfetol dose for the coprimary endpoints and followed by the key secondary endpoint; testing proceeded in that order for each subsequent lower dose, with statistical significance claimed only for those outcomes above the break in the hierarchy.
Patient Global Impression of Change is a patient self‐reported, 7‐point assessment, ranging from “very much worse” to “very much improved.”
Nominal p value, because it is below the hierarchical break.
The LS mean change from baseline at week 12 on the MWT showed an increase in mean sleep latency of 12.3 (SE = 1.4) and 9.8 (SE = 1.3) minutes with solriamfetol 300 mg and 150 mg, respectively, which was significant compared with 2.1 (SE = 1.3) minutes for placebo (both p < 0.0001; Fig 2A). These changes resulted in LS mean differences versus placebo of 10.1 (95% CI = 6.4–13.9) minutes for solriamfetol 300 mg and 7.7 (95% CI = 4.0–11.3) minutes for solriamfetol 150 mg. Although the LS mean increase in sleep latency of 4.7 (SE = 1.3) minutes at the 75 mg dose was approximately 2‐fold higher than that of placebo, significance was not met (p = 0.1595).
Figure 2.
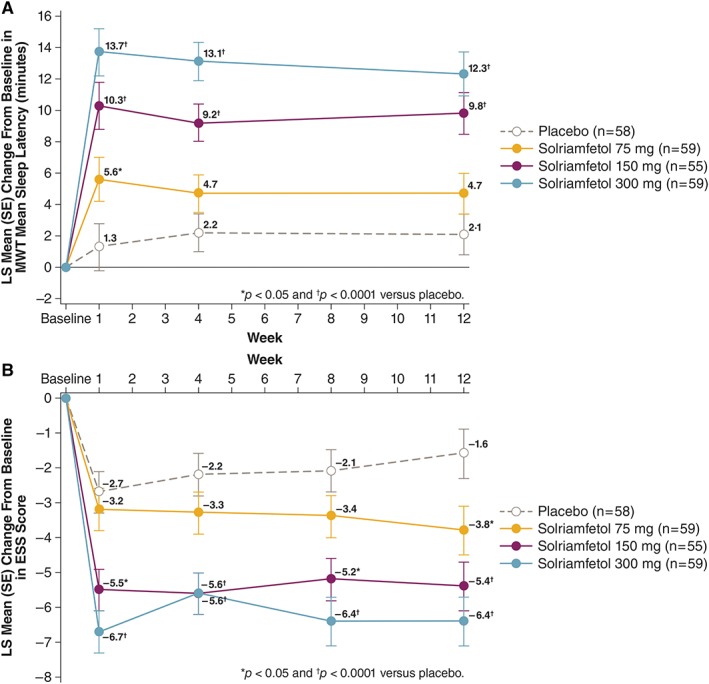
Change from baseline in coprimary endpoints. (A) Least squares (LS) mean change in Maintenance of Wakefulness Test (MWT) sleep latency from baseline in minutes for all treatment groups. (B) LS mean change in Epworth Sleepiness Scale (ESS) score for all treatment groups. All p values are nominal at weeks 1, 4, and 8. SE = standard error. [Color figure can be viewed at www.annalsofneurology.org]
For the ESS score (see Fig 2B), the LS mean change from baseline at week 12 was −6.4 (SE = 0.7), −5.4 (SE = 0.7), and −3.8 (SE = 0.7) for the 300 mg, 150 mg, and 75 mg doses of solriamfetol, respectively, and − 1.6 (SE = 0.7) with placebo. These reductions resulted in significant LS mean differences versus placebo of −4.7 (95% CI = −6.6 to −2.9, p < 0.0001) for solriamfetol 300 mg, −3.8 (95% CI = −5.6 to −2.0, p < 0.0001) for solriamfetol 150 mg, and −2.2 (95% CI = −4.0 to −0.3, p = 0.0211) for solriamfetol 75 mg.
Improvements were observed at all solriamfetol doses at week 1 on the MWT (see Fig 2A). The magnitude of effect remained stable over the 12 weeks of the study, and the 300 and 150 mg doses differed from placebo at weeks 1 and 4. Similar patterns were observed on the ESS (see Fig 2B), with reductions in ESS score relative to placebo observed as early as week 1 with the 300 and 150 mg doses, and effects remained stable over the study duration.
Evaluation of mean sleep latency on each of the 5 individual MWT trials at week 12 showed efficacy beginning at 1 hour after dosing through 9 hours after dosing for solriamfetol 150 and 300 mg (Fig 3).
Figure 3.
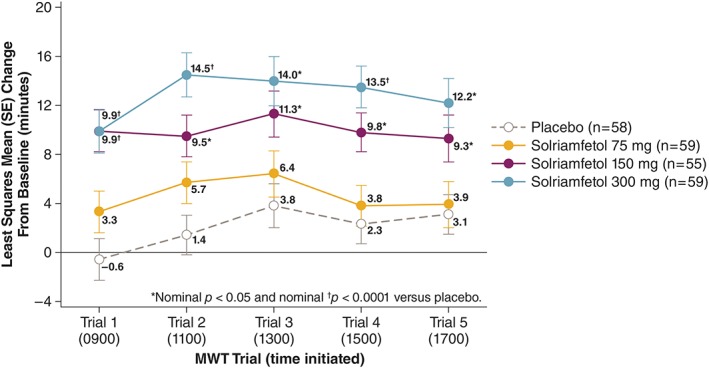
Change from baseline in sleep latency for each of the 5 individual Maintenance of Wakefulness Test (MWT) trials at week 12 (modified intent‐to‐treat population). Individual MWT trials, each of 40‐minute duration, were performed at 2‐hour intervals at the times shown in parentheses, starting 1 hour after dosing. SE = standard error. [Color figure can be viewed at www.annalsofneurology.org]
Solriamfetol increased the percentage of patients who reported improvement in their overall condition on the PGI‐C (Fig 4A). At week 12, these increases were dose‐dependent and were significant for solriamfetol 300 mg (84.7%) and 150 mg (78.2%) doses versus placebo (39.7%; both p < 0.0001); the 75 mg dose was nominally significant (67.8%) compared with placebo (p = 0.0023, but the comparison was below the hierarchical break). Effects were observed at all doses by week 1 and remained stable over the course of the study.
Figure 4.
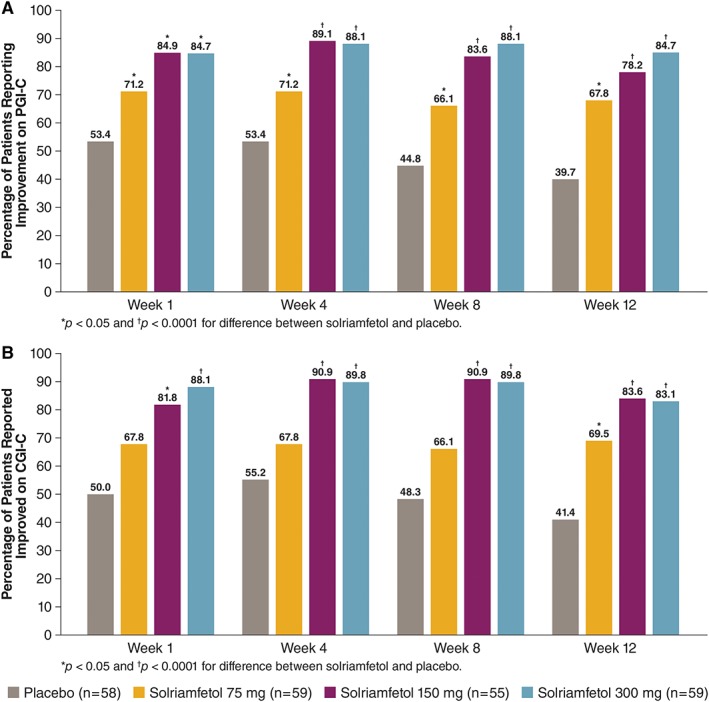
Percentage of patients with global improvements. (A) Percentage of patients who reported improvement on the Patient Global Impression of Change (PGI‐C). (B) Percentage of patients reported to be improved on the Clinical Global Impression of Change (CGI‐C). Improvements on the global scales were defined by responses reflecting “minimal,” “much,” or “very much” improved. [Color figure can be viewed at www.annalsofneurology.org]
Similar patterns and percentages were observed on the CGI‐C (see Fig 4B). All doses resulted in higher percentages of patients who improved as early as week 1, with effects at 300 mg and 150 mg maintained over the study. The results of each of the sensitivity analyses across each of the endpoints (MWT, ESS, and PGI‐C) yielded similar results and conclusions as the primary analyses of those endpoints (Supplementary Table).
There was no clear effect of solriamfetol on the number of cataplexy attacks per week among patients with cataplexy, although this study was not powered or designed to rigorously evaluate the effects of solriamfetol on cataplexy (data not shown). Preliminary analyses26 did not suggest any clinically meaningful differences in efficacy or safety between narcolepsy patients with and without cataplexy (manuscript in preparation).
Safety
The incidence of any adverse event was higher with solriamfetol (68.4%) than with placebo (45.8%; Table 3). Discontinuations due to adverse events were greater in the solriamfetol 150 (5.1%) and 300 mg (8.5%) groups than with placebo (1.7%); other than cataplexy, which resulted in discontinuation in 2 patients, none of the adverse events leading to study discontinuation occurred in >1 patient. One patient in the solriamfetol 150 mg group had 2 serious adverse events of noncardiac chest pain and anxiety that were deemed by the investigator not to be related to study medication; this patient continued the study without recurrence of the events.
Table 3.
TEAEs in the Safety Population
| TEAE | Placebo, n = 59 | Solriamfetol | |||
|---|---|---|---|---|---|
| All Doses, n = 177 | 75 mg, n = 59 | 150 mg, n = 59 | 300 mg, n = 59 | ||
| Any TEAE | 27 (45.8%) | 121 (68.4%) | 34 (57.6%) | 47 (79.7%) | 40 (67.8%) |
| Serious TEAE | 0 | 1 (0.6%) | 0 | 1 (1.7%) | 0 |
| TEAE leading to study drug discontinuation | 1 (1.7%) | 9 (5.1%) | 1 (1.7%) | 3 (5.1%) | 5 (8.5%) |
| Most common TEAEsa | |||||
| Headache | 3 (5.1%) | 38 (21.5%) | 6 (10.2%) | 14 (23.7%) | 18 (30.5%) |
| Nausea | 1 (1.7%) | 19 (10.7%) | 3 (5.1%) | 6 (10.2%) | 10 (16.9%) |
| Decreased appetite | 1 (1.7%) | 19 (10.7%) | 5 (8.5%) | 5 (8.5%) | 9 (15.3%) |
| Nasopharyngitis | 3 (5.1%) | 16 (9.0%) | 5 (8.5%) | 8 (13.6%) | 3 (5.1%) |
| Dry mouth | 2 (3.4%) | 13 (7.3%) | 3 (5.1%) | 4 (6.8%) | 6 (10.2%) |
| Anxiety | 1 (1.7%) | 9 (5.1%) | 1 (1.7%) | 3 (5.1%) | 5 (8.5%) |
| Diarrhea | 1 (1.7%) | 8 (4.5%) | 2 (3.4%) | 3 (5.1%) | 3 (5.1%) |
| Dyspepsia | 0 | 6 (3.4%) | 1 (1.7%) | 2 (3.4%) | 3 (5.1%) |
| Dizziness | 2 (3.4%) | 6 (3.4%) | 2 (3.4%) | 1 (1.7%) | 3 (5.1%) |
| Fatigue | 0 | 5 (2.8%) | 0 | 2 (3.4%) | 3 (5.1%) |
| Weight decreased | 0 | 5 (2.8%) | 1 (1.7%) | 1 (1.7%) | 3 (5.1%) |
| Upper respiratory tract infection | 1 (1.7%) | 5 (2.8%) | 1 (1.7%) | 4 (6.8%) | 0 |
| Insomnia | 0 | 5 (2.8%) | 2 (3.4%) | 0 | 3 (5.1%) |
| Constipation | 1 (1.7%) | 4 (2.3%) | 3 (5.1%) | 1 (1.7%) | 0 |
| Influenza | 3 (5.1%) | 4 (2.3%) | 2 (3.4%) | 1 (1.7%) | 1 (1.7%) |
| Heart rate increased | 0 | 4 (2.3%) | 0 | 0 | 4 (6.8%) |
| Weight increased | 3 (5.1%) | 3 (1.7%) | 2 (3.4%) | 0 | 1 (1.7%) |
Most common TEAEs in this table were those reported by ≥5% in any treatment group.
TEAEs = treatment‐emergent adverse events.
Adverse events with an incidence ≥5% in combined solriamfetol dose groups included headache (21.5%), nausea (10.7%), decreased appetite (10.7%), nasopharyngitis (9.0%), dry mouth (7.3%), and anxiety (5.1%; see Table 3). A post hoc analysis was conducted of the incidence of headache‐related events, including headache and migraine, in patients who either had or who did not have headache, such as headache, migraine, tension headache, or cluster headache, in their medical history. This analysis showed that patients who had a medical history of headache had a higher rate of headache during the study relative to those without a medical history of headache (14.3% vs 4.4% in the placebo group, and 31% vs 19.3% in the combined solriamfetol group). However, there did not appear to be a differential increase in the incidence of headache or migraine in the solriamfetol group compared with the placebo group between the subgroups of patients with and without a medical history of headache. Insomnia was reported in 2.8% of patients with solriamfetol and none with placebo; no patient discontinued the study due to insomnia.
No drug‐related effects were found on clinical laboratory assessments. The average of vital signs taken from predose to 9 hours postdose showed an increase from baseline in systolic and diastolic blood pressure (1–2 mmHg) and heart rate (2–4 beats per minute) for solriamfetol 150 and 300 mg dose relative to minimal changes (<1 mmHg or beats per minute) for placebo (Table 4). These effects on blood pressure and heart rate from baseline to week 8 using 24‐hour ambulatory blood pressure monitoring were similar to what was observed during the days on which MWT was performed (see Table 4). No patient had a treatment‐emergent adverse event of hypertension, and 2 patients had a treatment‐emergent adverse event of blood pressure increase (1 in the 150 mg group and 1 in the 300 mg group). Study drug was not interrupted in either case, and both patients completed the study.
Table 4.
Change from Baseline to Last Assessment in Vital Signs (Patients with Nonmissing Values)
| Vital Sign | Placebo, n = 59 | Solriamfetol | |||
|---|---|---|---|---|---|
| 75 mg, n = 59 | 150 mg, n = 59 | 300 mg, n = 59 | All Doses, n = 177 | ||
| Patients with clinical laboratory assessment data at week 12, na | 50 | 48 | 49 | 43 | 140 |
| Heart rate, beats/min | 0.5 ± 6.7 | 0.6 ± 6.6 | 2.5 ± 4.7 | 4.3 ± 7.6 | 2.4 ± 6.5 |
| Blood pressure, mmHg | |||||
| Systolic | 0.6 ± 8.1 | 0.3 ± 6.8 | 1.2 ± 7.4 | 2.0 ± 7.4 | 1.2 ± 7.2 |
| Diastolic | −0.6 ± 5.2 | 1.0 ± 4.4 | 1.4 ± 4.9 | 2.1 ± 5.0 | 1.5 ± 4.8 |
| Ambulatory blood pressure monitoring in safety population at week 8, nb | 50 | 46 | 46 | 41 | 133 |
| Heart rate, beats/min | −0.6 ± 7.0 | 1.0 ± 8.0 | 0.7 ± 7.1 | 5.3 ± 7.6 | 2.2 ± 7.8 |
| Blood pressure, mmHg | |||||
| Systolic | −0.3 ± 9.3 | 1.8 ± 6.5 | −0.5 ± 5.5 | 2.4 ± 6.0 | 1.2 ± 6.1 |
| Diastolic | −0.1 ± 7.2 | 1.4 ± 5.1 | 0.4 ± 4.5 | 3.0 ± 5.0 | 1.6 ± 4.9 |
Values are mean ± standard deviation.
Vital signs averaged across predose to 9 hours postdose.
Vital signs matched by time point at baseline and week 8.
Discussion
This phase 3 study demonstrated the robust effects of solriamfetol for improving wakefulness on the MWT and reducing ES on the ESS in a large population of patients with narcolepsy. This study also supports the safety and tolerability profile of solriamfetol. Solriamfetol demonstrated dose‐dependent efficacy that was significantly superior to placebo on both coprimary endpoints at the 150 and 300 mg doses. The 75 mg dose resulted in significantly greater improvement than placebo on the ESS but not on the MWT.
Importantly, improvements on both coprimary endpoints were observed at week 1, and these effects were maintained at the 150 and 300 mg doses over the study duration, indicating that there was no apparent tolerance over the 12 weeks of the study. Onset of efficacy at week 1 is consistent with what was observed previously in individuals with narcolepsy.18 Furthermore, on the MWT, significant effects were observed across all 5 of the individual MWT trials at the 150 and 300 mg doses, effects that were maintained over a 9‐hour duration following dosing at the 12‐week time point. This effect across the day may be longer in duration than what was observed in prior studies of the wake‐promoting agents armodafinil and modafinil in patients with narcolepsy, where the effect appeared to diminish during the 4th and 5th time trials later in the day.27, 28 However, definitive conclusions cannot be drawn in the absence of comparative trials.
Differences from placebo for changes on the MWT in the current study were 7.7 and 10.1 minutes for solriamfetol 150 and 300 mg, respectively. These values are substantially larger than what has been reported for modafinil, a dopamine reuptake inhibitor that is currently one of the most widely prescribed drugs for the treatment of ES in narcolepsy. Differences from placebo of 2.81 minutes (95% CI = 2.10–3.53) and 2.82 minutes (95% CI = 2.40–3.24) were reported in a meta‐analysis of modafinil parallel‐group studies in narcolepsy and all modafinil narcolepsy studies (ie, parallel‐group and crossover combined), respectively,29 and the effects of modafinil were shown to be similar (ie, noninferior) to pitolisant.30 Although the current clinical trial utilized a 40‐minute MWT, whereas other reports used 20‐ and 40‐minute measurements,31, 32, 33, 34, 35, 36, 37 the apparently larger effects of solriamfetol in this study may be related to its activity as a dual dopamine/norepinephrine reuptake inhibitor that lacks the serotonergic effects or norepinephrine‐releasing effects of other drugs.38 The differences in ESS between solriamfetol and placebo were −4.7 for 300 mg and −3.8 for 150 mg, whereas a difference of −2.73 (95% CI = −3.39 to −2.00) was reported for modafinil in the meta‐analysis.29 However, in the absence of head‐to‐head studies of solriamfetol and modafinil/armodafinil, outcomes from this study cannot be directly compared with results obtained with other agents.
The increased wakefulness and reduced ES observed with solriamfetol 150 and 300 mg paralleled global improvements assessed from both the patients’ and clinicians’ perspectives.
The overall safety and tolerability in this study were consistent with other studies of solriamfetol,17, 18 with the most common adverse events being headache, nausea, decreased appetite, nasopharyngitis, dry mouth, and anxiety; no serious adverse events were reported. Additionally, small increases in mean blood pressure and heart rate were observed, as previously reported with other wake‐promoting agents and psychostimulants.39
Conclusions with regard to the effect of solriamfetol on cataplexy are limited by this study not being designed to rigorously evaluate effects on cataplexy. The frequency of type 2 narcolepsy (ie, without cataplexy) in approximately 50% of the study population was also somewhat higher than reported in the narcolepsy literature.10 Whether the observed effects on efficacy in this study can be maintained without safety and tolerability concerns during long‐term treatment has been conducted, and results are currently under review.
In conclusion, once‐daily oral dosing of solriamfetol 150 and 300 mg resulted in major improvements in wakefulness and reductions in ES associated with narcolepsy together with patient‐ and clinician‐reported global improvements. The safety profile was consistent with other clinical studies of solriamfetol in narcolepsy. These results demonstrate that solriamfetol represents an important potential future therapeutic option for the treatment of impaired wakefulness and ES in individuals with narcolepsy.
Author Contributions
Study concept and design: all authors (with guidance from the authors, the study protocol was developed by the sponsor, Jazz Pharmaceuticals). Data acquisition and analysis: all authors. Drafting the manuscript: M.J.T. and Y.D with further input from all authors.
Potential Conflicts of Interest
In 2014, Jazz Pharmaceuticals acquired a license to develop and commercialize solriamfetol from Aerial Biopharma. Jazz Pharmaceuticals has worldwide development, manufacturing, and commercialization rights to solriamfetol, excluding certain jurisdictions in Asia. SK Biopharmaceuticals, the discoverer of the compound (also known as SKL‐N05), maintains rights in 12 Asian markets, including Korea, China, and Japan. Under the authors’ specific direction on content, organization, and interpretation of the data, E. Jay Bienen, PhD, of the Curry Rockefeller Group (CRG) provided medical writing assistance to the authors, including formatting, proofreading, copyediting, and fact checking, and is hereby acknowledged consistent with International Committee of Medical Journal Editors guidelines. Additional editorial assistance in formatting, proofreading, copyediting, and fact checking was also provided by CRG. The authors had full editorial control of the manuscript and provided their final approval of all content. Jazz Pharmaceuticals provided funding to CRG for editorial support. The following authors disclosed financial relationships with Jazz Pharmaceuticals. M.J.T. has received research/grant support and consultancy and speakers’ bureau fees from Jazz Pharmaceuticals. C.S. has served on the speakers’ bureau for Jazz Pharmaceuticals. H.E. has received research funding and consultancy, honoraria, and/or personal fees from Jazz Pharmaceuticals. G.P. has participated on an advisory board for Jazz Pharmaceuticals. Y.D. has received consultancy fees and/or honoraria and has been a speakers’ bureau member and/or an advisory board participant for Jazz Pharmaceuticals. D.C., L.P.C., and Y.L. are employees of Jazz Pharmaceuticals who, in the course of this employment, have received stock options exercisable for, and other stock awards of, ordinary shares of Jazz Pharmaceuticals. L.P.C. and Y.L. also have a patent pending. J.B. is a part‐time employee of Jazz Pharmaceuticals and shareholder of Jazz Pharmaceuticals. H.W. is a former employee of Jazz Pharmaceuticals. This clinical research was funded by Jazz Pharmaceuticals (the sponsor), which also took a leadership role in designing the study. All of the authors, including authors from Jazz Pharmaceuticals, assisted in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication. G.M. and B.C.C. report no conflicts of interest for the present study.
Supporting information
SUPPLEMENTARY TABLE Sensitivity Analyses of Change from Baseline to Week 12 in the Modified Intention‐to‐Treat Population
Appendix S1 Supporting Information.
Acknowledgment
This study was supported by Jazz Pharmaceuticals.
The authors would like to thank the patients, all the study investigators (listed in an online supplementary file), the study staff, and the nursing team for their participation in this research.
Contributor Information
Michael J. Thorpy, Email: michael.thorpy@einstein.yu.edu.
Yves Dauvilliers, Email: y-dauvilliers@chu-montpellier.fr.
References
- 1. Dauvilliers Y, Arnulf I, Mignot E. Narcolepsy with cataplexy. Lancet 2007;369:499–511. [DOI] [PubMed] [Google Scholar]
- 2. Thorpy MJ, Dauvilliers Y. Clinical and practical considerations in the pharmacologic management of narcolepsy. Sleep Med 2015;16:9–18. [DOI] [PubMed] [Google Scholar]
- 3. Erman M, Emsellem H, Black J, et al. Correlation between the Epworth Sleepiness Scale and the Maintenance of Wakefulness Test in patients with narcolepsy participating in two clinical trials of sodium oxybate. Sleep Med 2017;38:92–95. [DOI] [PubMed] [Google Scholar]
- 4. Dodel R, Peter H, Spottke A, et al. Health‐related quality of life in patients with narcolepsy. Sleep Med 2007;8:733–741. [DOI] [PubMed] [Google Scholar]
- 5. Ingravallo F, Gnucci V, Pizza F, et al. The burden of narcolepsy with cataplexy: how disease history and clinical features influence socio‐economic outcomes. Sleep Med 2012;13:1293–1300. [DOI] [PubMed] [Google Scholar]
- 6. Black J, Reaven NL, Funk S, et al. The Burden of Narcolepsy Disease (BOND) study: healthcare utilization and cost findings. Sleep Med 2014;15:522–529. [DOI] [PubMed] [Google Scholar]
- 7. Maski K, Steinhart E, Williams D, et al. Listening to the patient voice in narcolepsy: diagnostic delay, disease burden, and treatment efficacy. J Clin Sleep Med 2017;13:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smolensky MH, Di Milia L, Ohayon MM, Philip P. Sleep disorders, medical conditions, and road accident risk. Accid Anal Prev 2011;43:533–548. [DOI] [PubMed] [Google Scholar]
- 9. Pizza F, Jaussent I, Lopez R, et al. Car crashes and central disorders of hypersomnolence: a French study. PLoS One 2015;10:e0129386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. American Academy of Sleep Medicine . International classification of sleep disorders. 3rd ed. Darien, IL: American Academy of Sleep Medicine, 2014. [Google Scholar]
- 11. Barateau L, Lopez R, Dauvilliers Y. Treatment options for narcolepsy. CNS Drugs 2016;30:369–379. [DOI] [PubMed] [Google Scholar]
- 12. Kollb‐Sielecka M, Demolis P, Emmerich J, et al. The European Medicines Agency review of pitolisant for treatment of narcolepsy: summary of the scientific assessment by the Committee for Medicinal Products for Human Use. Sleep Med 2017;33:125–129. [DOI] [PubMed] [Google Scholar]
- 13. Baladi MG, Forster MJ, Gatch MB, et al. Characterization of the neurochemical and behavioral effects of solriamfetol (JZP‐110), a selective dopamine and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther 2018;366:367–376. [DOI] [PubMed] [Google Scholar]
- 14. Wisor J. Modafinil as a catecholaminergic agent: empirical evidence and unanswered questions. Front Neurol 2013;4:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rothman RB, Baumann MH, Dersch CM, et al. Amphetamine‐type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 2001;39:32–41. [DOI] [PubMed] [Google Scholar]
- 16. Hasan S, Pradervand S, Ahnaou A, et al. How to keep the brain awake? The complex molecular pharmacogenetics of wake promotion. Neuropsychopharmacology 2009;34:1625–1640. [DOI] [PubMed] [Google Scholar]
- 17. Bogan R, Feldman N, Emsellem HA, et al. Effect of oral JZP‐110 (ADX‐N05) treatment on wakefulness and sleepiness in adults with narcolepsy. Sleep Med 2015;16:1102–1108. [DOI] [PubMed] [Google Scholar]
- 18. Ruoff C, Swick T, Doekel R, et al. Effect of oral JZP‐110 (ADX‐N05) on wakefulness and sleepiness in adults with narcolepsy: a phase 2b study. Sleep 2016;39:1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. American Psychiatric Association . Diagnostic and statistical manual of mental disorders. 5th ed. Arlington, VA: American Psychiatric Association, 2013. [Google Scholar]
- 20. Littner MR, Kushida C, Wise M, et al. Practice parameters for clinical use of the multiple sleep latency test and the maintenance of wakefulness test. Sleep 2005;28:113–121. [DOI] [PubMed] [Google Scholar]
- 21. Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep 1991;14:540–545. [DOI] [PubMed] [Google Scholar]
- 22. Guy W. ECDEU assessment manual for psychopharmacology, revised. US Department of Health, Education, and Welfare publication (ADM 76‐338). Rockville, MD: National Institute of Mental Health, 1976. [Google Scholar]
- 23. Posner K, Brown GK, Stanley B, et al. The Columbia‐Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 2011;168:1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schafer JL. Analysis of incomplete multivariate data. New York, NY: Chapman & Hall, 1997. [Google Scholar]
- 25. Little RJA. Pattern‐mixture models for multivariate incomplete data. J Am Stat Assoc 1993;88:125–134. [Google Scholar]
- 26. Dauvillers Y, Shapiro C, Mayer G, et al. Solriamfetol (JZP‐110) for treatment of excessive sleepiness in narcoleptic patients with and without cataplexy: results from a randomized, phase 3, clinical trial. Sleep 2018;41(suppl):A229–A230 (Abstract). [Google Scholar]
- 27. Schwartz JR, Feldman NT, Bogan RK, et al. Dosing regimen effects of modafinil for improving daytime wakefulness in patients with narcolepsy. Clin Neuropharmacol 2003;26:252–257. [DOI] [PubMed] [Google Scholar]
- 28. Harsh JR, Hayduk R, Rosenberg R, et al. The efficacy and safety of armodafinil as treatment for adults with excessive sleepiness associated with narcolepsy. Curr Med Res Opin 2006;22:761–774. [DOI] [PubMed] [Google Scholar]
- 29. Golicki D, Bala MM, Niewada M, Wierzbicka A. Modafinil for narcolepsy: systematic review and meta‐analysis. Med Sci Monit 2010;16:RA177–RA186. [PubMed] [Google Scholar]
- 30. Dauvilliers Y, Bassetti C, Lammers GJ, et al. Pitolisant versus placebo or modafinil in patients with narcolepsy: a double‐blind, randomised trial. Lancet Neurol 2013;12:1068–1075. [DOI] [PubMed] [Google Scholar]
- 31. Besset A, Chetrit M, Carlander B, Billiard M. Use of modafinil in the treatment of narcolepsy: a long term follow‐up study. Neurophysiol Clin 1996;26:60–66. [DOI] [PubMed] [Google Scholar]
- 32. Broughton RJ, Fleming JA, George CF, et al. Randomized, double‐blind, placebo‐controlled crossover trial of modafinil in the treatment of excessive daytime sleepiness in narcolepsy. Neurology 1997;49:444–451. [DOI] [PubMed] [Google Scholar]
- 33. Randomized trial of modafinil for the treatment of pathological somnolence in narcolepsy. US Modafinil in Narcolepsy Multicenter Study Group. Ann Neurol 1998;43:88–97. [DOI] [PubMed] [Google Scholar]
- 34. Randomized trial of modafinil as a treatment for the excessive daytime somnolence of narcolepsy: US Modafinil in Narcolepsy Multicenter Study Group. Neurology 2000;54:1166–1175. [DOI] [PubMed] [Google Scholar]
- 35. Moldofsky H, Broughton RJ, Hill JD. A randomized trial of the long‐term, continued efficacy and safety of modafinil in narcolepsy. Sleep Med 2000;1:109–116. [DOI] [PubMed] [Google Scholar]
- 36. Black J, Houghton WC. Sodium oxybate improves excessive daytime sleepiness in narcolepsy. Sleep 2006;29:939–946. [DOI] [PubMed] [Google Scholar]
- 37. The US Xyrem Multi‐Center Study Group . A randomized, double blind, placebo‐controlled multicenter trial comparing the effects of three doses of orally administered sodium oxybate with placebo for the treatment of narcolepsy. Sleep 2002;25:42–49. [PubMed] [Google Scholar]
- 38. Nishino S, Okura M, Mignot E. Narcolepsy: genetic predisposition and neuropharmacological mechanisms. Sleep Med Rev 2000;4:57–99. [DOI] [PubMed] [Google Scholar]
- 39. Bosco A, Lopez R, Barateau L, et al. Effect of psychostimulants on blood pressure profile and endothelial function in narcolepsy. Neurology 2018;90:e479–e491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY TABLE Sensitivity Analyses of Change from Baseline to Week 12 in the Modified Intention‐to‐Treat Population
Appendix S1 Supporting Information.