

Abstract
Biocatalysis is attracting interest in the chemical industry as a sustainable alternative in large‐scale chemical transformations. However, low operational stability of naturally evolved enzymes is a challenge and major efforts are required to engineer protein stability, usually by directed evolution. The development of methods for protein stabilization based on rational design is of great interest, as it would minimize the efforts needed to generate stable enzymes. Here we present a rational design strategy based on proline substitutions in flexible areas of the protein identified by analyzing B‐factors. Several proline substitutions in the amine transaminase from Chromobacterium violaceum were shown to have a positive impact on stability with increased half‐life at 60 °C by a factor of 2.7 (variant K69P/D218P/K304P/R432P) as well as increased melting temperature by 8.3 °C (variant K167P). Finally, the presented method utilizing B‐factor analysis in combination with the proline rule was deemed successful at increasing the stability of this enzyme.
Keywords: biocatalysis, enzyme stabilization, enzymes, molecular modeling, protein engineering
Introduction
The amount of biocatalytic applications in the chemical industry is constantly increasing as the many benefits of enzymes compared to other catalysts are recognized.1 Enzymes have been optimized by evolution to perform very specific tasks in nature and therefore often catalyze reactions with high activity and selectivity (chemo‐, regio‐ and stereoselectivity). These are important characteristics of an industrial catalyst and modern techniques have now made it straightforward to engineer these naturally evolved catalysts into tailor‐made enzymes for any unnatural reaction of choice.2
However, since enzymes are evolved by nature in natural conditions, which often consist of neutral pH and moderate temperatures, their stability in industrial settings can be limited.3 Methods to improve enzyme stability include protein engineering (directed evolution, rational design or combinations thereof), immobilization and medium engineering.4 Directed evolution is a Nobel prize‐awarded commonly applied strategy to create new robust biocatalysts that can withstand the harsh conditions of an industrial process.5 It is an attractive option as it does not require any knowledge about the enzyme, but even though the efficiency of the method is constantly increasing, it can still be a time‐ and resource‐consuming process due to extensive library screening and data analysis. Methods for stability improvement based on rational design are of great interest, as it would require less work and resources as only small amounts of variants are created. Protein engineering methods based on rational design require structural information of the enzyme to be available. The need for structural information is however becoming less of an issue, since the number of published crystal structures is constantly increasing in parallel with advances in homology and molecular modeling.
Rational design approaches for improvement of enzyme stability are often focused on introducing amino acid interactions that promote enzyme rigidity, such as covalent disulfide bonds between cysteine residues, ionic salt‐bridges or hydrophobic interactions.6 These mutations are hard to predict and require trial and error, which leads to difficulties in developing a general strategy. An alternative approach is the previously formulated “Proline rule”, which states that introduction of proline residues in certain positions in a protein leads to general stabilization.7 The unique structure of proline, where the side‐chain is covalently bound to the backbone amide nitrogen makes the role of proline in protein folding especially interesting. The structure of proline gives rise to two specific features in the protein. Firstly, the covalent bond to the amide nitrogen means that this nitrogen cannot take part in hydrogen bonding and secondly, the five‐membered ring of proline makes the bond between the α‐carbon (Cα) and the backbone nitrogen more rigid and less prone to rotation.8 Early work on oligo‐1,6‐glucosidases showed that proline content in enzymes from thermophilic organisms is generally higher than in mesophilic organisms.9 This observation then led to an early version of the Proline rule stating that mutagenesis of the second position (i+1) in β‐turns increases protein stability.7a The theory was later experimentally confirmed and it was shown that these mutations were indeed effective and cumulative.10 It was also shown that proline substitutions in the N‐terminal caps of α‐helices had a similar effect. This finally led to what is known as the Proline rule. It states that proline substitutions in: 1) the i+1 position of β‐turns, and 2) the N1 position of α‐helices generally increases thermostability.7b Proline, which is generally defined as a “helix‐breaker”, has actually been shown to have a rather high occurrence in the N1 position of α‐helices.11 The Proline rule has been applied with varying success in several cases with the goal of increasing protein stability.12
Amine transaminases13 (ATAs) have gained increased industrial attention due to their ability to catalyze asymmetric transamination of ketones with excellent enantioselectivity and yield.14 Wild‐type ATAs are however rarely stable enough for industrial applications and improvement of stability is therefore required.15 Here, the bacterial amine transaminase from Chromobacterium violaceum (Cv‐ATA) was applied as a model enzyme to evaluate the Proline rule as a strategy for enzyme stabilization. The stability of Cv‐ATA has previously been improved through immobilization16 and medium engineering.15, 17 Here we present a method for improvement of the stability of Cv‐ATA using B‐factor analysis in combination with the Proline rule. B‐factors describe atom fluctuation caused by thermal motion.18 B‐factor data are generally available in X‐ray crystal structures of proteins and can be used to localize flexible regions that can be stabilized. The Proline rule was applied to stabilize some flexible regions in Cv‐ATA and all mutations were analyzed with regards to melting point (T m), inactivation temperature (T 50) and half‐life values (t 1/2). Molecular dynamics simulations have also been performed in order to understand the individual mutation's impact on stability. The combined results of this study were used to evaluate the potential of B‐factor guided proline substitution as a strategy for protein stabilization.
Results and Discussion
Flexible regions in Cv‐ATA were identified by accessing B‐factors of all α‐carbons (Cα) from a previously published crystal structure of holo Cv‐ATA (PDB ID: 4A6T).19 This crystal structure contains four subunits. Therefore, a mean value of the four available Cα B‐factors for each residue was calculated and plotted against residue number (Figure 1). Peaks in the B‐factor plot were deemed as flexible regions and the relative degree of flexibility was assessed by difference in peak height. This approach has previously been successfully applied to an R‐selective amine transaminase from Aspergillus terreus in order to identify positions important for improving stability.20
Figure 1.
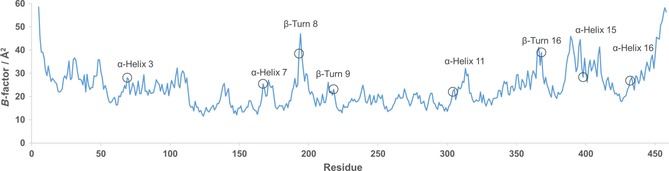
Cv‐ATA Cα B‐factor/residue plotted against amino acid residue number. Each secondary structure element chosen for proline substitution is denoted with the label centered above it. Residues chosen for mutation are marked with a circle.
The protein structure was divided into secondary structure elements of interest (α‐helices and β‐turns; Table S1 in the Supporting Information). In accordance with the Proline rule, most of the peaks in the B‐factor plot belonged to α‐helices and β‐turns. Seven surface‐exposed secondary‐structure elements (Figure S1) were selected for proline substitution based on their elevated B‐factors in relation to nearby residues (Table 1, Figure 1). In addition, the C‐terminal α‐helix 16 was also included to see if it could be stabilized. It is a long α‐helix in which the residue chosen for mutation has a low B‐factor, but the end of the α‐helix is very flexible. Peaks that were discarded either belonged to loops or already contained prolines in the appropriate (or adjacent to the appropriate) positions. Some peaks were also discarded in order to keep down sample size.
Table 1.
Amino acid residues chosen for mutation based on a combination of elevated B‐factors and the proline rule.
| Mutation | Secondary structure element | B‐factor [Å2] |
|---|---|---|
| K69P | α‐helix 3 | 28.1 |
| K167P | α‐helix 7 | 25.5 |
| K193P | β‐turn 8 | 38.5 |
| D218P | β‐turn 9 | 23.1 |
| K304P | α‐helix 11 | 22.1 |
| E368P | β‐turn 16 | 39.0 |
| G398P | α‐helix 15 | 28.3 |
| R432P | α‐helix 16 | 26.9 |
Initially, the eight selected proline mutations (Table 1) were combined into one eight‐point mutant designated as Pro8. However, this variant generated an inactive (Table 2, based on spectrophotometric analysis using the acetophenone assay21) and mostly insoluble (Figure S2) protein product. The eight substitutions of Pro8 were therefore divided into four double mutations (K69P/K304P, K167P/E368P, K193P/G398P and D218P/R432P) to find out which proline substitutions were responsible for the inactivation of the protein. Three of these variants were produced as soluble and active proteins (Table 2, Figure S3) with specific activities in the same range as wild type (WT) that has a specific activity of 4.05 U mg−1 (1 unit is hereby defined as 1 μmol of formed product per min), while K193P/G398P remained insoluble (Figure S3) and inactive (Table 2).
Table 2.
Kinetic parameters of Cv‐ATA WT and all variants at 25 °C.
| Specific activity |
|
|
/ [b] | |||
|---|---|---|---|---|---|---|
| [U mg−1][a] | [mm][b] | [U mg−1][b] | ||||
| WT | 4.05 | 7.60 | 8.18 | 1.08 | ||
| Pro8 | n.d.[c] | – | – | – | ||
| K69P/K304P | 6.67 | 11.0 | 19.7 | 1.79 | ||
| K167P/E368P | 3.04 | 10.6 | 8.42 | 0.794 | ||
| K193P/G398P | n.d.[c] | – | – | – | ||
| D218P/R432P | 5.07 | 6.65 | 9.35 | 1.41 | ||
| K193P | 3.99 | 8.80 | 8.78 | 1.00 | ||
| G398P | n.d.[c] | – | – | – | ||
| Pro7 | 4.28 | 6.48 | 7.84 | 1.21 | ||
| K69P | 3.82 | 8.18 | 8.25 | 1.01 | ||
| K167P | 2.59 | 6.39 | 4.80 | 0.751 | ||
| D218P | 2.33 | 9.48 | 5.93 | 0.626 | ||
| K304P | 3.99 | 9.33 | 9.43 | 1.01 | ||
| E368P | 4.97 | 8.14 | 10.5 | 1.29 | ||
| R432P | 3.65 | 5.50 | 6.00 | 1.09 | ||
| Pro4 | 5.73 | 7.26 | 11.5 | 1.58 | ||
| Pro5 | 5.10 | 7.89 | 11.9 | 1.51 |
[a] Specific activity based on a model reaction with (S)‐1‐phenylethylamine and pyruvate where substrate concentrations are fixed at 2.5 mm. [b] Apparent kinetic constants were determined under pseudo‐one‐substrate conditions. [c] Not detected.
Two single‐point variants, K193P and G398P, were then generated. Variant G398P was shown to be inactive (Table 2) and it was therefore concluded that G398P was the mutation causing the insolubility. To explore the amino acid position 398 further, the amino acid sequence of Cv‐ATA was aligned with the 500 most similar sequences in the IMG database.22 The negative effect of the G398P substitution can most likely be explained by the high conservation of glycine (97 %) in that position (Table S2). In addition, when looking back at the residue G398, it was realized that it might actually not be in the N1 position of the α‐helix. It is more likely that the N1 position is occupied by E399.
With the detrimental mutation G398P removed, a seven‐point variant, denoted Pro7, was created. The Pro7 variant was soluble and active (Table 2, Figure S4) with a similar specific activity as WT. However, it showed no significant improvement regarding stability compared to the WT enzyme (data not shown). This prompted us to generate all seven single‐point mutants in order to investigate each substitution′s effect on stability. Initially, stability of the single‐point mutants was investigated by measuring residual activity over time at two different temperatures (37 and 60 °C; Figure 2). Significant stability improvement of several variants was shown at 37 °C. After approximately seven days, K69P, K304P, E368P and R432P retained 11–31 % residual activity, while the WT enzyme retained only 7 % activity (Figure 2 A). At 60 °C (Figure 2 B), three variants (D218P, K304P and R432P) performed better than WT while two variants (K69P and K167P) showed a decrease in stability. However, variant K69P showed an interesting pattern at 60 °C where it initially was more stable than WT. However, after a while the rate of inactivation increased and at the end‐point, residual activity of K69P was lower than the WT.
Figure 2.
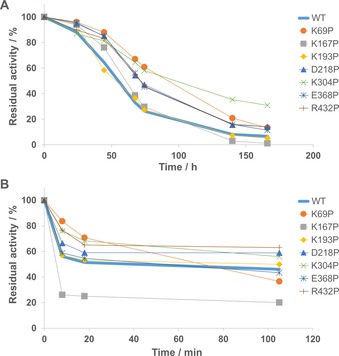
Residual activity of Cv‐ATA WT and all active single‐point mutants plotted against time at: A) 37 °C, and B) 60 °C.
To further investigate stability of the variants, T 50 and t 1/2 values were determined for WT and all variants (Table 3, Figure S5). The values show consistency with the previous experiment as variants D218P, K304P, E368P and R432P all show increased T 50 and t 1/2 while K167P has a five times lower t 1/2 at 60 °C compared to the WT.
Table 3.
Stability values determined for Cv‐ATA WT and all active variants.
| ΔT 50 [a] | t 1/2 (60 °C) | ΔT m [a] | |
|---|---|---|---|
| ratio to WT[b] | |||
| WT | 0.0 | 1.0 | 0.0 |
| K69P | 0.8 | 0.8 | −2.4 |
| K167P | −4.7 | 0.2 | 8.3 |
| K193P | 0.5 | 0.8 | 7.0 |
| D218P | 1.1 | 1.6 | 1.3 |
| K304P | 1.8 | 1.3 | −0.9 |
| E368P | 2.4 | 1.6 | −0.7 |
| R432P | 2.1 | 1.6 | 5.5 |
| K69P/K304P | 1.9 | 2.0 | −1.3 |
| K167P/E368P | −1.7 | 0.6 | 8.9 |
| D218P/R432P | 3.4 | 2.2 | 1.7 |
| Pro4 | 2.6 | 2.7 | −0.4 |
| Pro5 | 3.5 | 2.2 | 0.3 |
| Pro7 | 1.3 | 1.0 | −1.8 |
[a] Presented as the difference between the variant and WT. The degree of inactivation for calculating T 50 was measured after 15 min. [b] Presented as the ratio between the variant and WT: t 1/2(variant)/t 1/2(WT).
The Michaelis–Menten kinetics was also investigated for WT and all variants at 25 °C. The WT enzyme has a of 7.60 mm for (S)‐1‐phenylethylamine and a of 8.18 U mg−1 (Table 2). The kinetic constants and specific activity for all variants (except for the inactive ones) display no significant changes and it can therefore be concluded that the mutations do not have an impact on catalysis.
Since it was previously shown that proline substitutions following the Proline rule are cumulative,10 the previously created double mutants were also investigated in order to see any possible cumulative effects of these mutations. It is however not straightforward identifying a cumulative pattern within the double‐mutants. The T 50 values show a cumulative pattern while the t 1/2 values do not (Table 3). It is therefore not possible to draw any conclusions about any cumulative effects based on these double mutants. In order to further investigate this, all the best single‐point mutants from the stability over time experiments at different temperatures (Figure 2) were incorporated into one five‐point mutant designated as Pro5 (K69P/D218P/K304P/E368P/R432P). The four‐point mutant Pro4 (K69P/D218P/K304P/R432P) was generated in the process of making Pro5 and was therefore included in the subsequent experiments. As shown in Table 3, Pro4 and Pro5 are among the best performing variants with regards to T 50 and t 1/2. The effects of the mutations are however not cumulative since T 50 and t 1/2 of Pro5 are almost identical to the double mutant D218P/R432P. These two mutations are both in Pro5 and if the effects of the mutations were cumulative, a positive contribution from the other mutations would also be seen.
Thermal unfolding of the variants was investigated by differential scanning fluorimetry (DSF)23 where protein unfolding can be monitored over time as temperature is increasing. The T m was determined and large improvements could be seen for K167P, K193P and R432P. The highest ΔT m at 8.3 °C was shown for K167P (Table 3). The other single‐point mutations showed no significant change in T m value, except for K69P for which the T m value dropped by 2.4 °C. The double mutant K167P/E368P has the highest increase in T m (8.9 °C) out of all variants while the other combined mutations show no significant change except for Pro7 that, interestingly, has a ΔT m of −1.8 °C (Table 3). The T m values do not display a cumulative pattern and while some mutations seem to be deleterious, they are not so in every case.
It can be seen that many variants showing an increased T m do not display an increased T 50 or t 1/2 (Table 3). This can be explained by the fact that T m is related to thermodynamic (conformational) stability while T 50 and t 1/2 are related to kinetic (long‐term) stability.3, 24 These two are related to different processes of inactivation and a mutation showing an effect on one cannot be expected to show an effect on the other.
An unfolding profile was obtained by measuring increase in fluorescence over time at 70 °C (Figure 3). These results correlate well to the T m results, as the variants with improved T m unfolded slower than the WT.
Figure 3.
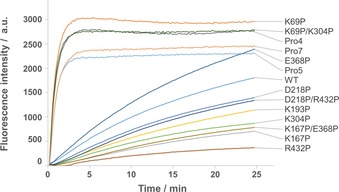
Temperature‐induced enzyme unfolding of Cv‐ATA WT and all active variants.
To investigate and visualize the individual effects of the mutations on the stability of the protein, molecular dynamics (MD) simulations were performed on single‐point mutants with improved T 50 and t 1/2. The double mutant D218P/R432P and Pro5 was also included in the MD simulation. By monitoring the mobility of each atom during the course of a MD simulation, protein flexibility over time can be analyzed. This is important since highly flexible residues can trigger protein unfolding due to their large fluctuations.25 The MD simulations were carried out at 95 °C over the course of 10 ns. The elevated temperature was chosen in order to be able to observe an effect on protein motion within a shorter time. Snapshots were saved every 0.25 ns and were subsequently aligned against the starting structure for analysis of RMSD values. The results of the MD simulations are presented as total RMSD of the protein over the duration of simulation (Figure 4) as well as mean RMSD/residue (Figure 5). As can be seen in Figure S6, it takes approximately 2 ns for the structure to equilibrate during the MD simulation and the RMSD/residue is therefore calculated in the period of 2–10 ns. The total RMSD over time is shown both for Cα (Figure 4 A) and for all atoms (Figure S6). For all atoms, a difference between the variants cannot be seen. This is not surprising since this includes the RMSD for all sidechains, including mobile surface exposed residues, and we assume that a clear difference in this graph would indicate a drastic improvement in protein stability. However, when RMSD for only the Cα atoms is calculated, a clear difference between the variants can be seen. By excluding the first time point, a linear regression could be performed (Figure 4 B) and here it can be seen that the WT reaches a higher RMSD in the beginning of the MD simulation compared to the variants. Interestingly, two variants (E368P and Pro5) initially have a slower increase in RMSD but after 10 ns they are at the same level as the WT. Variants K304P, R432P and D218P/R432P all have a slower initial increase of RMSD but the increase during the linear phase is lower as well, indicating a decreased backbone flexibility in these variants. The only variant showing higher RMSD than the WT is D218P that has a similar initial increase as WT but the linear phase has a higher increase. When looking at mean RMSD/residue (Figure 5), stability effects on single amino acids or separate secondary structure elements can be visualized. The results show that large parts of the protein retain a similar flexibility before and after mutation. Obvious deviations from this observation are for example, D218P where a large unsuspected peak arises in the area of residues 24–30. This area corresponds to a short surface exposed α‐helix that is in no way close to residue 218. The same variant shows a slightly increased flexibility in the area of residues 90–110 which is again an area that is not near the mutation. However, it is next to the previously mentioned area and the increased flexibility can possibly be explained by this. Another outlier is the peak around residue 173 in Pro5. This peak is relatively close to the α‐helix that should be affected by the K304P mutation and a small increase in flexibility is observed in the same area when looking at the single‐point mutant K304P. All variants, including the WT, has a peak of similar height at position 316 except for the double mutant D218P/R432P that seems to display a drastic decrease at this position. Also in this case, the peak is not near the mutations. One area of interest is the C‐terminal α‐helix that in theory should be stabilized by the mutation R432P in the beginning of the helix. The WT shows the highest flexibility in this area indicating that all mutations partly stabilize this α‐helix and interestingly, the two most rigid variants in this area both include the mutation R432P (R432P and D218P/R432P). Concluding this experiment, one can see that most mutations do not show an immediate effect on flexibility at the point of mutation (except for R432P). There are however distant effects of many mutations that are difficult to explain and this further highlights the challenge of improving stability in proteins by rational design.
Figure 4.
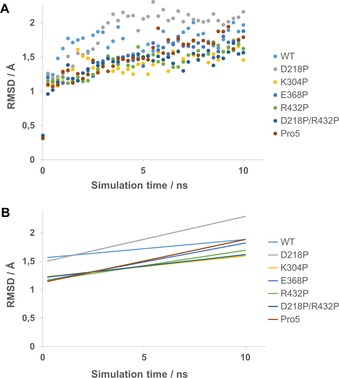
A) Total Cα‐RMSD over time for Cv‐ATA WT and variants taken from MD‐simulations. B) Linear regressions of results from Figure 4 A.
Figure 5.
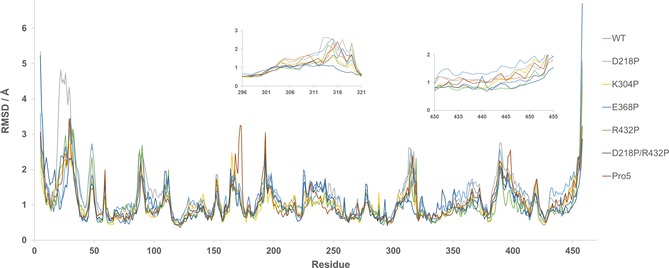
Mean RMSD/residue during the period of 2–10 ns of a MD simulation.
Conclusions
The stability of Cv‐ATA has been successfully improved by employing a B‐factor guided proline substitution strategy. Flexible regions were identified by analyzing B‐factors from the Cv‐ATA crystal structure and the Proline rule was thereafter used to predict proline substitutions that in theory would stabilize said regions. Successful variants include a four‐point mutant (Pro4) and a double mutant (D218P/R432P) that increased the half‐life at 60 °C by a factor of 2.7 and 2.2, respectively. Some proline substitutions were also shown to drastically increase the melting point (T m) with the most successful single‐point variant increasing T m with 8.3 °C (K167P). Molecular dynamics simulations were also performed in order to understand the effects of the mutations on protein flexibility. Clear differences could be seen when comparing variants, but mostly in regions that were not close to the mutated residues.
Finally, the presented B‐factor guided proline substitution strategy was shown to be an effective method for improving stability of Cv‐ATA in a rational fashion. Even though the Proline rule did not exert the same predictability and additivity as has been previously shown, it is still a useful method in the toolbox for rational design strategies aiming at increasing enzyme stability.
Experimental Section
Materials: All chemicals were purchased from Sigma–Aldrich and used without further purification. The Ni‐NTA Sepharose resin used for His‐trap purification was purchased from IBA (Sweden). PD10 desalting columns were purchased from GE Healthcare Life Science (Sweden).
Enzyme cloning and generation of enzyme variants: The C. violaceum amine transaminase gene (GenBank accession WP_011135573) was previously inserted into pET28a(+) with a N‐terminal His6 tag.26 The plasmid sequence was verified (Eurofins MWG Operon, Ebersberg, Germany) before transformation into Escherichia coli BL21(DE3). The enzyme variant Pro8 was synthesized, codon optimized and cloned into pET28a(+) using restriction sites NcoI and BamHI by GenScript. The rest of the enzyme variants were generated by site‐directed mutagenesis. Site‐directed mutagenesis was performed by the Quick Change method with modifications according to Zheng et al.,27 by using Phusion High‐Fidelity DNA Polymerase from Thermo Fisher. Alternatively, site‐directed mutagenesis using the MEGAWHOP28 strategy was performed to obtain the double mutants. The primers used in this work are listed below, with the mutations indicated in lower case:
CvATA_Pro8 P398G_fw: 5′‐GTTCCCGGACTTTggtGAAATCGGCAC‐3′
CvATA_Pro8 P398G_rv: 5′‐GATTTCaccAAAGTCCGGGAACAGCTCAC‐3′
CvATA_K69P_fw: 5′‐GTTATGGTCGCccgGATTTTGCAGAAG‐3′
CvATA_K69P_ rv: 5′‐CAAAATCcggGCGACCATAACCAACATTAAC‐3′
CvATA_K167P_fw: 5′‐GGTGGTATGccgTATATGCATGAACAGGG‐3′
CvATA_K167P_rv: 5′‐GCATATAcggCATACCACCCAGGCTGG‐3′
CvATA_K193P_fw: 5′‐GTATAAACATGGCccgGATATGACACCG‐3′
CvATA_K193P_rv: 5′‐CATATCcggGCCATGTTTATACCACCACC‐3′
CvATA_D218P_fw: 5′‐GGTGCCccgAAAGTTGCAGCATTTG‐3′
CvATA_D218P_rv: ′‐GCAACTTTcggGGCACCAATTTCCAG‐3′
CvATA_K304P_fw: 5′‐GTTGGTccgCGTGTTGCAGAAGGTCTG‐3′
CvATA_K304P_rv: 5′‐GCAACACGcggACCAACAAAAACTGC‐3′
CvATA_E368P_fw: 5′‐GTTTTccgCATGTTGATGATGTTCGTGG‐3′
CvATA_E368P_rv: 5′‐CATCAACATGcggAAAACGGCTAAAGG‐3′
CvATA_G398P_fw: 5′‐GATTTTccTGAAATTGGCACCCTGTG‐3′
CvATA_G398P_rv: 5′‐GCCAATTTCAggAAAATCCGGAAAC‐3′
CvATA_R432P_fw: 5′‐GACCCcgGCCGAAGTTGATGAAATG‐3′
CvATA_R432P_rv: 5′‐CAACTTCGGCcgGGGTCATCAC‐3′
Enzyme expression and purification: One BL21(DE3) E. coli colony, containing WT Cv‐ATA or variants in pET28a(+) was incubated in lysogeny broth (5 mL) at 37 °C and 200 rpm, overnight. The overnight culture was used to inoculate terrific broth (TB 400 mL) medium. The culture was further incubated at 200 rpm and 37 °C. At OD600 0.7–0.9, isopropyl β‐d‐1‐thiogalactopyranoside (IPTG, 1 mm) was added and the temperature was decreased to 20 °C. Both media were supplemented with 50 mg L−1 kanamycin. After 16 h of induction, cells were harvested by centrifugation (Beckman coulter Avanti J‐26XP) at 11 500 g for 30 min at 4 °C. The collected cell pellet was resuspended in binding buffer for disruption by sonication (6 min; Branson Sonifier 250) at 30 duty cycles % and with a 5–6 Microtip limit. After 30 min of centrifugation at 3500 g and 4 °C, the supernatant was collected and filtered (0.45 μm). Purification was performed on a Ni‐NTA His‐trap column, according to the IBA manufacturer′s manual with binding buffer (50 mm NaH2PO4, 300 mm NaCl, 20 mm imidazole, pH 8.2) or elution buffer (50 mm NaH2PO4, 300 mm NaCl, 500 mm imidazole, pH 8.2).
Samples were buffer exchanged to HEPES buffer (50 mm, pH 8.2) with 5 mm pyridoxal‐5′‐phosphate (PLP) in the supernatant by using a PD10 desalting column. The samples were incubated at 4 °C, overnight, followed by 1 h at 37 °C the next day to obtain re‐binding PLP to the enzyme. Additional desalting with a PD10 column was performed to remove the unbound PLP.
Enzyme activity assay and determination of kinetic parameters: The acetophenone assay21 was performed at 25 °C to assay enzyme activity. The reaction mixture (1 mL) generally composed of 10 μg of enzyme, (S)‐1‐phenylethylamine (2.5 mm) and pyruvate (2.5 mm) in 50 mm HEPES pH 8.2. The formation of acetophenone was monitored at 245 nm on a Cary50 UV/Vis spectrophotometer. In order to obtain the kinetic parameters, the same model reaction was performed in microplates with a final volume of 100 μL and 1 μg of enzyme. In this case, acetophenone production was monitored on a FLUOstar Omega plate reader (BMG Labtech). To obtain the Michaelis–Menten constant ( ), six different concentrations of (S)‐1‐phenylethylamine, ranging from 0.1 to 15 mm, were used. Each test was carried out with at least three replicates. The and values were calculated by nonlinear regression using GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, CA, USA).
Enzyme stability measurement: temperature of half inactivation: Residual activity of Cv‐ATA WT and variants incubated at different temperatures was evaluated in order to obtain the temperature of half inactivation (T 50). For this, 0.5 mg mL−1 of enzyme solution in HEPES (50 mm, pH 8.2) was incubated from 50 to 80 °C in 5 °C intervals, and after 15 min, the activity assay was performed. All measurements were performed in duplicate. To calculate T 50, the obtained data was fitted to the Boltzmann equation in GraphPad Prism 6.0.
Enzyme stability measurement: half‐life of thermal inactivation: To obtain the half‐life of thermal inactivation (t 1/2), 0.5 mg mL−1 of Cv‐ATA WT and variants in HEPES (50 mm, pH 8.2) were incubated at 60 °C for different time intervals, and the activity assay was applied to obtain the residual activity values. All measurements were performed in duplicates. To calculate t 1/2, the obtained data were fitted to the one‐phase decay equation in GraphPad Prism 6.0.
Enzyme stability measurement: Melting temperature measurements: Melting temperature (T m) values were measured using differential scanning fluorimetry (DSF).23 The measurement was performed on a CFX96 real‐time PCR detection system and C1000 thermal cycler at 569 nm. The reaction sample (20 μL) consisted of 0.5 mg mL−1 enzyme and 3.75×SYPRO Orange protein gel stain (Sigma Aldrich, S5692) in HEPES (50 mm, pH 8.2). The temperature range was set to 25–95 °C, with an increase of 1 °C min−1. Every measurement was performed in duplicate. The T m value was determined by fitting the original data to the Boltzmann equation in GraphPad Prism 6.0.
Enzyme stability measurement: temperature‐induced enzyme unfolding: SYPRO Orange dye was used to monitor enzyme unfolding at 70 °C. A CFX96 real‐time PCR detection system and C1000 thermal cycler were utilized to record fluorescence emission at 569 nm every 5 s at 70 °C. The reaction mixture (20 μL), consisted of enzyme (0.5 mg mL−1) and SYPRO Orange dye (3.75×) in HEPES buffer (50 mm, pH 8.2). Every measurement was performed in duplicate.
Bioinformatics: In order to identify similar proteins to Cv‐ATA (WP_011135573), a BLASTp search against the Integrated Microbial Genomes (IMG) database22 of genome sequences was performed. A total number of 500 proteins were identified with an E‐value lower than 1e−10, and those were selected for sequence alignment using Clustal Omega to determine residue conservation.
Molecular dynamics simulation: MD simulations were performed using YASARA29 structure version 18.3.23. The 3D structure of Cv‐ATA WT (PDB ID: 4A6T) was obtained from the PDB database30 and prepared for modeling by deleting one of the dimers and adding all missing hydrogens. The simulations were performed inside a water‐filled simulation cell (cuboid shape, 20 Å around all atoms) with periodic boundaries at pH 8.2. An energy minimization was performed using the AMBER14 force field.31 The energy‐minimized structure was subjected to a MD simulation at 338 K using a water density of 0.981 g mL−1. The simulation duration was 10 ns and snapshots were saved every 0.25 ns.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Maria Svedendahl Humble studied chemistry and chemical engineering at KTH Royal Institute of Technology in Stockholm. In 2010, she obtained her PhD in biotechnology, focusing on biocatalysis, from KTH. She did her postdoctoral work, at both KTH and Stockholm University, on the protein engineering of amine transaminases and lipases. Thereafter, she worked for three years at KTH Industrial Biotechnology as an Assistant Professor in Biochemistry with a focus on biocatalysis. Currently, Maria is working at Pharem Biotech AB, developing enzyme‐based products for waste water treatment.

Biographical Information
Per Berglund is Professor of Biochemistry at Department of Industrial Biotechnology at KTH Royal Institute of Technology, Stockholm. He studied chemistry and obtained his PhD in organic chemistry from Mid Sweden University. He was then a postdoctoral fellow at University of Toronto, with Prof. J. Bryan Jones and returned to Sweden in 1997 to start his career at KTH where he was made Full Professor in Biochemistry 2009. His research interests include stereoselective biocatalysis and enzyme engineering and his group now focuses on transaminase biocatalysis and engineering for cascade synthesis. In 2010, he was elected Vice Dean of KTH.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was funded by Carl Tryggers Foundation for Scientific Research (CTS 16:456 and CTS 18:803) and KTH Royal Institute of Technology.
H. Land, J. C. Campillo-Brocal, M. Svedendahl Humble, P. Berglund, ChemBioChem 2019, 20, 1297.
Contributor Information
Dr. Maria Svedendahl Humble, Email: maria@pharem.se.
Prof. Per Berglund, Email: per.berglund@biotech.kth.se.
References
- 1.
- 1a. Choi J.-M., Han S.-S., Kim H.-S., Biotechnol. Adv. 2015, 33, 1443–1454; [DOI] [PubMed] [Google Scholar]
- 1b. Hughes G., Lewis J. C., Chem. Rev. 2018, 118, 1–3; [DOI] [PubMed] [Google Scholar]
- 1c. Chapman J., Ismail A., Dinu C., Catalysts 2018, 8, 238. [Google Scholar]
- 2.
- 2a. Bornscheuer U. T., Huisman G. W., Kazlauskas R. J., Lutz S., Moore J. C., Robins K., Nature 2012, 485, 185–194; [DOI] [PubMed] [Google Scholar]
- 2b. Bornscheuer U. T., Philos. Trans. R. Soc. London Ser. A 2018, 376, 20170063. [Google Scholar]
- 3. Polizzi K. M., Bommarius A. S., Broering J. M., Chaparro-Riggers J. F., Curr. Opin. Chem. Biol. 2007, 11, 220–225. [DOI] [PubMed] [Google Scholar]
- 4. Ó′Fágáin C., in Protein Chromatography: Methods and Protocols (Eds.: D. Walls, T. S. Loughran), Humana, Totowa, 2011, pp. 103–136. [Google Scholar]
- 5. Bornscheuer U. T., Hauer B., Jaeger K. E., Schwaneberg U., Angew. Chem. Int. Ed. 2019, 58, 36–40; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 36–41. [Google Scholar]
- 6.
- 6a. Yang H., Liu L., Li J., Chen J., Du G., ChemBioEng Rev. 2015, 2, 87–94; [Google Scholar]
- 6b. Yu H., Huang H., Biotechnol. Adv. 2014, 32, 308–315. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Suzuki Y., Proc. Jpn. Acad. Ser. B 1989, 65, 146–148; [Google Scholar]
- 7b. Watanabe K., Suzuki Y., J. Mol. Catal. B 1998, 4, 167–180. [Google Scholar]
- 8. MacArthur M. W., Thornton J. M., J. Mol. Biol. 1991, 218, 397–412. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Suzuki Y., Aoki R., Hayashi H., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1982, 704, 476–483; [Google Scholar]
- 9b. Suzuki Y., Tomura Y., Eur. J. Biochem. 1986, 158, 77–83; [DOI] [PubMed] [Google Scholar]
- 9c. Suzuki Y., Fujii H., Uemura H., Suzuki M., Starch/Staerke 1987, 39, 17–23; [Google Scholar]
- 9d. Suzuki Y., Oishi K., Nakano H., Nagayama T., Appl. Microbiol. Biotechnol. 1987, 26, 546–551. [Google Scholar]
- 10. Watanabe K., Masuda T., Ohashi H., Mihara H., Suzuki Y., Eur. J. Biochem. 1994, 226, 277–283. [DOI] [PubMed] [Google Scholar]
- 11. Richardson J. S., Richardson D. C., Science 1988, 240, 1648–1652. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Tian J., Wang P., Gao S., Chu X., Wu N., Fan Y., FEBS J. 2010, 277, 4901–4908; [DOI] [PubMed] [Google Scholar]
- 12b. Yu H., Zhao Y., Guo C., Gan Y., Huang H., Biochim. Biophys. Acta Proteins Proteomics 2015, 1854, 65–72; [DOI] [PubMed] [Google Scholar]
- 12c. Trevino S. R., Schaefer S., Scholtz J. M., Pace C. N., J. Mol. Biol. 2007, 373, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Steffen-Munsberg F., Vickers C., Kohls H., Land H., Mallin H., Nobili A., Skalden L., van den Bergh T., Joosten H. J., Berglund P., Höhne M., Bornscheuer U. T., Biotechnol. Adv. 2015, 33, 566–604. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Guo F., Berglund P., Green Chem. 2017, 19, 333–360; [Google Scholar]
- 14b. Gomm A., O'Reilly E., Curr. Opin. Chem. Biol. 2018, 43, 106–112; [DOI] [PubMed] [Google Scholar]
- 14c. Kelly S. A., Pohle S., Wharry S., Mix S., Allen C. C. R., Moody T. S., Gilmore B. F., Chem. Rev. 2018, 118, 349–367. [DOI] [PubMed] [Google Scholar]
- 15. Chen S., Land H., Berglund P., Humble M. S., J. Mol. Catal. B 2016, 124, 20–28. [Google Scholar]
- 16.
- 16a. Engelmark Cassimjee K., Kadow M., Wikmark Y., Humble M. S., Rothstein M. L., Rothstein D. M., Bäckvall J. E., Chem. Commun. 2014, 50, 9134–9137; [DOI] [PubMed] [Google Scholar]
- 16b. Abaházi E., Sátorhelyi P., Erdélyi B., Vértessy B. G., Land H., Paizs C., Berglund P., Poppe L., Biochem. Eng. J. 2018, 132, 270–278. [Google Scholar]
- 17. Chen S., Berglund P., Humble M. S., Mol. Catal. 2018, 446, 115–123. [Google Scholar]
- 18. Yuan Z., Bailey T. L., Teasdale R. D., Proteins Struct. Funct. Bioinf. 2005, 58, 905–912. [DOI] [PubMed] [Google Scholar]
- 19. Humble M. S., Cassimjee K. E., Håkansson M., Kimbung Y. R., Walse B., Abedi V., Federsel H.-J., Berglund P., Logan D. T., FEBS J. 2012, 279, 779–792. [DOI] [PubMed] [Google Scholar]
- 20. Huang J., Xie D. F., Feng Y., Biochem. Biophys. Res. Commun. 2017, 483, 397–402. [DOI] [PubMed] [Google Scholar]
- 21. Schätzle S., Höhne M., Redestad E., Robins K., Bornscheuer U. T., Anal. Chem. 2009, 81, 8244–8248. [DOI] [PubMed] [Google Scholar]
- 22. Chen I. M. A., Markowitz V. M., Chu K., Palaniappan K., Szeto E., Pillay M., Ratner A., Huang J., Andersen E., Huntemann M., Varghese N., Hadjithomas M., Tennessen K., Nielsen T., Ivanova N. N., Kyrpides N. C., Nucleic Acids Res. 2017, 45, D507–D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Niesen F. H., Berglund H., Vedadi M., Nat. Protoc. 2007, 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- 24. Iyer P. V., Ananthanarayan L., Process Biochem. 2008, 43, 1019–1032. [Google Scholar]
- 25. Ruvinsky A. M., Kirys T., Tuzikov A. V., Vakser I. A., J. Bioinf. Comput. Biol. 2012, 10, 1241002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cassimjee K. E., Humble M. S., Miceli V., Colomina C. G., Berglund P., ACS Catal. 2011, 1, 1051–1055. [Google Scholar]
- 27. Zheng L., Baumann U., Reymond J.-L., Nucleic Acids Res. 2004, 32, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miyazaki K., Takenouchi M., Biotechniques 2002, 33, 1033–1034, 1036–1038. [DOI] [PubMed] [Google Scholar]
- 29. Krieger E., Vriend G., Bioinformatics 2014, 30, 2981–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berman H., Henrick K., Nakamura H., Nat. Struct. Biol. 2003, 10, 980. [DOI] [PubMed] [Google Scholar]
- 31.D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham, T. A. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, S. LeGrand, T. Luchko, R. Luo, B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, P. A. Kollman, Amber 14 2014.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary