

Abstract
Versatile photoresponsive gels based on tripodal low molecular weight gelators (LMWGs) are reported. A cyclohexane‐1,3,5‐tricarboxamide (CTA) core provides face‐to‐face hydrogen bonding and a planar conformation, inducing the self‐assembly of supramolecular polymers. The CTA core was substituted with three arylazopyrazole (AAP) arms. AAP is a molecular photoswitch that isomerizes reversibly under alternating UV and green light irradiation. The E isomer of AAP is planar, favoring the self‐assembly, whereas the Z isomer has a twisted structure, leading to a disassembly of the supramolecular polymers. By using tailor‐made molecular design of the tripodal gelator, light‐responsive organogels and hydrogels were obtained. Additionally, in the case of the hydrogels, AAP was coupled to the core through hydrazones, so that the hydrogelator and, hence, the photoresponsive hydrogel could also be assembled and disassembled by using dynamic covalent chemistry.
Keywords: arylazopyrazole, dynamic covalent chemistry, gels, light-responsive materials, supramolecular polymers
Introduction
Low molecular weight gelators (LMWGs) represent a viable alternative for the development of soft materials.1, 2, 3, 4 They can self‐assemble through noncovalent interactions, such as hydrogen bonding,5, 6 host–guest (e.g. crown ether–cation),7 metal–ligand,8, 9, 10, 11 and π–π interactions,12, 13 forming extended and entangled supramolecular polymers.14, 15, 16 Depending on the solvents used in the gels, they can be classified as organogels (organic solvent) or hydrogels (aqueous media). Gelation of LMWGs typically occurs from the highest soluble state, for example, high temperature and ionized form. Owing to the dynamic properties of LMWGs, their formation can be triggered by pH,17, 18 solvent,19 temperature,20 enzyme,21, 22 light,23, 24 or addition of salt.25, 26 As a consequence, the solubility of the gelator is gradually reduced. Nucleation occurs and the growth of nanofibrils is initiated. Thus, self‐assembly of LMWGs is most often a kinetic trapping process.27, 28 Recent applications of LMWGs include tissue engineering,29, 30 optoelectronic devices,31, 32, 33 sensors,34, 35, 36 and actuators.37, 38
Materials stimulated by light are of particular interest, because light is a non‐invasive stimulus. Also, light irradiation can spatially and temporally target a specific area, or spot, on a surface or inside a material.39, 40 Photoresponsive gelators have gained increasing attention in recent years.41 LMWGs comprising coumarins,42, 43 spiropyrans,44 stilbenes,45, 46 azobenzenes,47, 48 and dithienylethenes49, 50 were reported. Among them, azobenzenes are without doubt the most investigated class of photoswitches. However, it is known that the photo‐isomerization of azobenzenes results in an equilibrium, the photostationary state (PSS), which is typically around 80 % in both directions E to Z and Z to E.51 This phenomenon of incomplete photoisomerization can be even worse in solid‐phase materials due to the reduced dynamics required for isomerization of the azobenzene.52, 53 To address this limitation, arylazopyrazoles (AAPs) have been recently introduced as improved light‐responsive molecular switches.54, 55, 56 In further studies, we have investigated, with others, the properties of AAPs57 as molecular switches and developed several supramolecular systems ranging from colloidal host–guest complexes,58, 59 foams,60 adhesives,61 and DNA complexes.62, 63 Recently, also a light‐responsive peptide hydrogel featuring host–guest interactions between AAP peptides and cyclodextrin vesicles was introduced by our group.64
A particularly versatile class of supramolecular polymers assembles from tripodal “core–arm” small molecules. In most cases, these supramolecular polymers are based on cyclohexane,65, 66 or benzene67, 68 cores. Benzene‐1,3,5‐tricarboxamide (BTA) derivatives established by Meijer and co‐workers are distinguished examples of tripodal monomers with benzene as the core and variable functionalities connected to the core by amide bonds.68 The intermolecular hydrogen bonding provided by the amides initiates the self‐assembly. By appending different arms to the BTA core, diverse soft materials and applications, including hydrogels,69, 70 protein recognition,71, 72 imaging,67 and catalysis73 were developed. Similar to BTA, cyclohexane‐1,3,5‐tricarboxamide (CTA) also affords intermolecular face‐to‐face hydrogen bonding and in addition, displays conformational flexibility. The flexible cyclohexane core permits the optimal stacking of the LMWGs and forms an extended supramolecular polymer. More interestingly, if hydrazones are applied to connect the arms and the core, dynamic and catalytic controlled in situ formation of hydrogels can be performed.74, 75, 76 In this regard, a hydrazide core and aldehyde arms have been combined.77 A detailed study of acid‐catalyzed dynamic covalent gels has been investigated by van Esch, Eelkema, and co‐workers with negatively charged liposomes78 and patterned sulfonic acid surfaces.79 The introduction of dynamic covalent bonds, such as hydrazone, can not only facilitate the synthesis, but also broaden the possible application of dynamic molecular systems and materials, by simply mixing hydrazide core and aldehyde arms with diverse functions.80, 81, 82, 83
Zhou et al. have reported photoresponsive organogels based on a CTA core and arms comprising azobenzene units.65 In their study, the light‐induced sol–gel transition and thermal stability of the organogelator were explored. However, a rheological study of the organogel was not reported. Also, the overlapping UV/Vis absorption of E and Z azobenzene potentially limits the reversibility of the sol–gel transition. Very recently, Venkataramani and co‐workers have reported tripodal AAP derivatives comprising a BTA core and demonstrated light‐controlled rewritable imaging in the solid state.67 From these preceding studies, we conclude that the combination of a CTA core and AAP arms is highly promising to develop versatile light‐responsive LMWGs for both organic solvents and aqueous solutions.
Here, we report a family of light‐responsive tripodal LMWGs, in which the molecular design was pursued by the core–arm approach. The general design and the functions of each molecular component are described in Scheme 1. Firstly, the CTA core allows for optimal stacking by intermolecular hydrogen bonding. Secondly, the AAP moieties in the arms stabilize the self‐assembly through π–π interactions and provide photoresponse. Thirdly, the functionalities at the end of the arms increase solubility in the desired solvent and potentially give rise to higher‐ordered self‐assembly, such as bundles, enhancing gelation. Finally, the connections between the core and the arms are designed to be either permanent (amide) or dynamic covalent (hydrazone). The dynamic covalent approach can not only facilitate the in situ synthesis of hydrogelator, but also be used as a chemical stimulus for gel formation and disassembly. Light‐response, rheology, and dynamic properties of tripodal AAP‐based LMWGs are explored in this article.
Scheme 1.
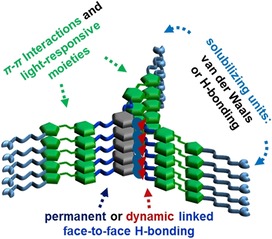
Molecular design of tripodal cyclohexane‐1,3,5‐tricarboxamide (CTA)‐based gelators and the function of each molecular component to form supramolecular polymers and gels.
Results and Discussion
Molecular design and synthesis
A light‐responsive organogelator (G1, Figure 1 A) was synthesized by using cis,cis‐cyclohexane‐1,3,5‐tricarboxylic acid as the core allowing for optimal stacking. An amine‐functionalized AAP was selected as the arm. In addition, the AAP was equipped with a long alkyl chain at the end of each arm. This design offers excellent gelation properties through three kinds of interactions. 1) The amide link between core and arms forms hydrogen bonds and allows for the planar orientation of the AAP arms. 2) The AAP unit contributes π–π interactions of the aromatic rings and, more interestingly, provides light‐responsive control of the stacking properties of the LMWG. 3) The aliphatic chain at the end of the arms creates a nonpolar exterior of the LMWG, yielding good solubility in organic solvents and additional dense packing by van der Waals interactions.
Figure 1.
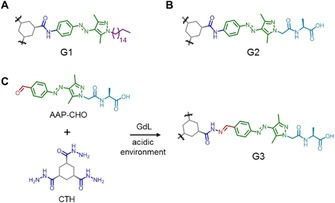
Molecular structure of amide‐linked A) organogelator (G1) and B) hydrogelator (G2) as well as the in situ formation of C) hydrazone‐linked hydrogelator G3) from CTH and AAP‐CHO triggered by the hydrolysis of GdL.
To enable biomimetic applications, hydrogels rather than organogels are desirable and, hence, the enhancement of water solubility is required. One straightforward approach is to maintain the molecular design of the organogelator G1 and attach the amino acid alanine at the end of the arms (G2, Figure 1 B). The terminal amino acid not only improves water solubility, but also presents additional hydrogen bonding between the supramolecular polymers. Furthermore, by using a closely similar molecular design, it was possible to introduce the dynamic covalent properties of hydrazones in the hydrogelator. For this purpose, a cyclohexanetrishydrazide (CTH) core and an aldehyde terminated AAP peptide (AAP‐CHO) were prepared (Figure 1 C). The hydrazone can be assembled by hydrazide and aldehyde under acidic (pH 4–6) or catalytic (e.g., aniline) conditions.74 By exploiting the hydrolysis of glucono‐δ‐lactone (GdL) to generate acidic conditions, the hydrazone‐linked hydrogelator (G3, Figure 1 C) should potentially form in situ. Experimental details of the preparation of the precursors and gelators can be found in the Supporting Information.
Photoisomerization of gelators
The photoisomerization of gelators G1–G3 was investigated by UV/Vis absorption spectroscopy. It should be noted that these measurements were carried out far below the critical gelation concentrations (see below). Thus, the spectrum of a solution of G1 (6.7 μm, as prepared) in toluene was measured (Figure 2 A). Subsequently, the solution was irradiated with UV light (λ=365 nm). Upon irradiation, the typical changes in the absorbance spectra for the E–Z isomerization of AAPs could be observed.54 The strong absorbance at 340 nm corresponding to the π–π* transition is reduced and blueshifted, whereas the absorbance around 440 nm (n–π* transition) has increased in intensity and shows a characteristic redshift (Figure 2 A). Upon irradiation with green light (λ=520 nm), the original spectrum was observed with slightly higher intensity at 340 nm, indicating that in gelator G1 as prepared by chemical synthesis a small percentage of Z isomer is present (Figure 2 A). The photoisomerization of G1 is reversible over at least four cycles without any sign of loss of efficiency (Figure 2 B).
Figure 2.
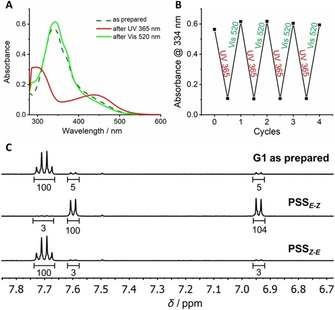
Photoisomerization of G1 confirmed by A) UV/Vis absorption spectroscopy (6.7 μm in toluene). B) Reversible E–Z‐photoisomerization for four cycles. C) Quantification of the E–Z‐photoisomerization under UV and visible light irradiation. PSS measurements were carried out using G1‐ref in [D8]‐THF (500 μm).
Quantification of the photoisomerization was carried out determining the PSS by NMR spectroscopy (Figure 2 C). Due to the intermolecular stacking, G1 showed rather broad signals in the proton NMR spectrum. Therefore, a one‐armed reference molecule G1‐ref was used for NMR experiments (structure and analysis of G1‐ref are provided in the Supporting Information). By integration of the proton signals in the aromatic region, the PSS for both the photoisomerization from E to Z and from Z to E could be determined. It was found that in the solution of G1‐ref as prepared the isomer ratio was 91:9 (E/Z), which changed drastically to 2:98 (E/Z) after irradiation with UV light (λ=365 nm). After irradiation with green light (λ=520 nm), the E isomer was reobtained with 94:6 (E/Z). Again, this value is higher compared to the percentage of E isomer in the initial spectrum, which is in a good agreement to the results obtained by UV/Vis spectroscopy. In summary, G1 showed an excellent light response, identical to the AAP derivatives reported previously by our group.54
Similar characteristics were found for the hydrogelators through the same approach by using a combination of UV/Vis absorption and NMR measurements. Both G2 and G3 exhibited typical AAP UV/Vis absorption spectra and showed fully reversible photoswitchability for at least five cycles (Figures S1 and S2, Supporting Information). In addition, PSS measurements confirmed the excellent photoisomerization efficiency of both hydrogelators. For G2, it showed PSS of 93 % for E to Z and 94 % for Z to E (Figure S3, Supporting Information). In the case of hydrazone based gelator G3, cyclohexane monohydrazide (CMH) was applied instead of CTH to give G3‐ref to avoid broad signals due to stacking in D2O. After mixing CMH and AAP‐CHO in the presence of GdL overnight, PSS of 90 % for E to Z and 89 % for Z to E were observed (Figure S4, Supporting Information). The spectroscopic data and PSS of gelators G1–G3 are summarized in Table 1.
Table 1.
Absorbance maxima (λ max) and photostationary state PSS of gelators G1–G3.
| π–π* λ max [nm] | n–π* λ max [nm] | PSSE–Z [%] | PSSZ–E [%] | |||
|---|---|---|---|---|---|---|
| E | Z | E | Z | |||
| G1 | 336 | 303 | 427 | 437 | 98[a] | 94[a] |
| G2 | 347 | 314 | 419 | 437 | 93[b] | 94[b] |
| G3 | 346 | 321 | 421 | 434 | 90[c] | 89[c] |
[a] NMR using G1‐ref in [D8]‐THF. [b] NMR in [D6]‐DMSO. [c] NMR using G3‐ref in D2O.
Photoresponsive organogels
Gelation tests for organogelator G1 were performed by using a range of organic solvents and increasing concentrations of G1 to determine the critical gelation concentration (CGC, Table 2). A heating–cooling sequence was applied to obtain homogenous organogels. In each case, a known amount of G1 was added to 1 mL of solvent and heated until it was fully dissolved. Afterwards, the solution was allowed to cool down to room temperature and the gel formation was checked by an inverted vial test. If a solution was obtained, the amount of G1 was increased stepwise until a gel was achieved. The results displayed in Table 2 document the formation of a transparent orange organogel (G) in 9 out of 17 tested solvents with rather low CGC (<1 wt %). In most other solvents, the gelator did not show any significant solubility at room temperature and precipitated (P). An exception to this is chloroform, in which even at rather high amount of G1 (20 mg mL−1) a clear solution was obtained (S). The formation of a chloroform gel might be possible at higher concentrations of G1 but was not further investigated in this study. Table 2 shows that G1 gelates rather non‐polar and aprotic solvents with the exception of n‐alkanes. No gelation was observed in polar solvents and solvents that can function as hydrogen bond donors.
Table 2.
Solvent tests for gelation of G1 and CGC in each solvent (G: gel, P: precipitate, S: solution).
| Solvent | State | CGC [mg mL−1] |
Solvent | State | CGC [mg mL−1] |
|---|---|---|---|---|---|
| n‐decane | P | – | THF | G | 14 |
| n‐hexane | P | – | CHCl3 | S | – |
| 1‐bromohexane | G | 7 | CH2Cl2 | G | 4 |
| cyclohexane | G | 6 | EtOAc | P | – |
| methylcyclohexane | G | 14 | acetone | P | – |
| xylene | G | 8 | DMF | P | – |
| toluene | G | 8 | DMSO | P | – |
| bromobenzene | G | 6 | CH3CN | P | – |
| 1,4‐dioxane | G | 7 |
Next, the light response of the organogels was investigated in more detail. The organogels showed a distinct light‐induced sol–gel transition (Figure 3 A). For example, a heated solution of the planar E isomer of G1 in toluene (10 mg mL−1) formed an organogel upon heating then cooling due to intermolecular stacking of G1 as a result of hydrogen bonding, π–π, and van der Waals interactions. This gel fully liquefies upon irradiation with UV light due to the photoisomerization of AAP from the planar E isomer to the twisted Z isomer.53, 54, 55 The resulting twisted configuration of Z‐G1 prevents intermolecular stacking, resulting in a clear solution. Upon green light irradiation, the planar E isomer is re‐obtained, the intermolecular interactions are restored, and an organogel is reformed that passes the inverted vial test. As a control experiment a sample of the organogel was irradiated for 2.5 h with infrared light (980 nm), which does not induce isomerization of the AAP unit. As expected, in this experiment no gel‐to‐sol transition is observed. Thus, it can be concluded that the observed macroscopic changes are caused by the photoisomerization of the AAP units and cannot be attributed to for example, heating upon irradiation (Figure S10, Supporting Information). The microstructures of the obtained gels were visualized by scanning electron microscopy (SEM) of a cast‐dried organogel sample. Figure 3 B,C shows the formation of fibers in the scale of micrometers that entangle to a three‐dimensional network typical for LMWG.
Figure 3.
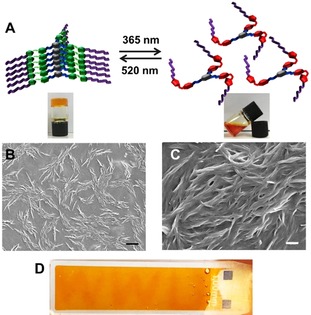
A) Schematic representation of the sol–gel transition of G1 under UV and green light irradiation in toluene. SEM images of 10 mg mL−1 G1 in toluene at B) 5k and C) 25k magnification [scale bars: B) 2 μm and C) 400 nm]. D) “WWU” was written in an organogel of 10 mg mL−1 G1 in toluene by UV light using a positive photomask.
Upon close inspection of Figure 3 A, it can be observed that in comparison to the organogel of E‐G1, the liquid state of Z‐G1 has a more reddish‐orange color, which is due to the increased and redshifted absorbance of the n–π* transition. Thus, the initials “WWU” of our university were written on the surface of the gel by UV light irradiation through a positive photomask (Figure 3 D). This observation indicates that these organogels can be applied for light‐responsive patterning by using a photomask, similar to what has been recently described by Venkataramani and co‐workers for tripodal AAP derivatives in the solid state.64
Rheological measurements confirmed the viscoelasticity and the photoresponse of the organogel prepared in toluene. Two different concentrations of the organogel, 10 and 15 mg mL−1, were tested by frequency sweep measurements (Figure 4 A). Both organogels displayed one order of magnitude higher storage modulus (G′) than loss modulus (G“), indicating the formation of viscoelastic organogels that are fully stable for the entire frequency sweep. Also, a concentration dependency to the stiffness was observed, which a 15 mg mL−1 sample exhibited higher moduli, both G′ and G′′, to the 10 mg mL−1 one. Then, the photo response of the organogel was further investigated (Figure 4 B). In the oscillatory rheological measurements, the original gel was first recorded. After irradiation with UV light for 5 min, another measurement was carried out. It showed a drastically reduced stiffness with almost three orders of magnitude. Moreover, randomly fluctuating points were observed for both moduli and even sometimes G′′ was over G′, suggesting that a liquid‐like material was obtained. As described above, we attribute this observation to disruption of intermolecular stacking by the twisted configuration of the Z isomer of G1. After irradiation with green light for 5 min, over two orders of magnitude of G′ was retrieved and again, stable moduli with G′ over G′′ were observed. This observation is in accordance with the restoration of intermolecular stacking of the planar E isomer of G1. Considering that the initial G′ is retrieved, it can be excluded that the initial drop of G′ can be attributed to mechanical disruption or heating of the gel during the rheological measurements. Two more irradiation cycles were carried out and a similar trend was observed, with high and stable moduli for the E isomer, and low and unstable moduli for the Z isomer (Figure 4 C).
Figure 4.
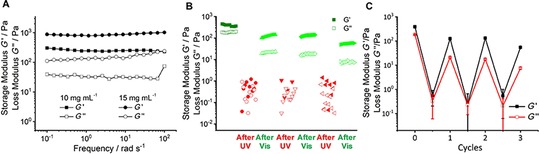
Rheological studies of G1 in toluene. A) Frequency sweep for increasing gelator concentrations (10 and 15 mg mL−1). B) Oscillatory rheological measurements for light‐responsive sol–gel transition of 15 mg mL−1 G1. C) Reversible sol–gel transition for three cycles.
Photoresponsive hydrogels
Considering that the formation of hydrogels from LMWGs is a kinetically controlled process, a smooth and homogeneous transition from the soluble and insoluble state of the gelator is critical. For peptide‐based hydrogelators, tuning the pH is a popular and useful trigger to make hydrogels. In particular, making use of the slow hydrolysis of GdL to generate an acidic environment, a more homogenous hydrogel can be obtained.84 Thus, G2 (2 or 5 mg mL−1) was first dissolved in a “highly soluble state” (fully deprotonated at pH≈8). Upon addition of GdL (5 or 10 mg mL−1), the hydrogelator was gradually transferred into a “poorly soluble state” (partially protonated at pH≈5), because the apparent pK a of alanine is around 5. Indeed, after standing in ambient conditions overnight, hydrogels were obtained that pass the inverted vial test. Again, irradiation of this sample with infrared light (980 nm) does not lead to any softening of the hydrogel (Figure S10, Supporting Information) The microstructure of the self‐supporting hydrogels was confirmed by SEM (Figure S5, Supporting Information).
The viscoelastic properties of the hydrogels were investigated by rheological measurements (Figure 5). Firstly, frequency‐sweep oscillation experiments were performed. Similar to organogels of G1, G′ values almost one order of magnitude higher than G′′ were found for hydrogels of G2, indicating the success of hydrogel preparation. In general, the stiffness of the gel is proportional to both the gelator concentration and the amount of GdL. The higher the concentration of hydrogelator, the more and longer the nanofibers obtained. This leads to an increasingly cross‐linked network and results in a stiffer gel. The amount of GdL determines the acidity of the gel solution. This is important for C‐terminal peptide amphiphiles, because they show higher stiffness under more acidic conditions.85 As shown in Figure 5 A, the highest G′ was found in 5 mg mL−1 G2 and 10 mg mL−1 GdL (solid triangles), whereas the lowest storage modulus was measured in the case of 2 mg mL−1 G2 and 5 mg mL−1 GdL (solid squares). By comparing the same hydrogelator concentration (2 mg mL−1) with different amount of GdL, the hydrogel had almost six‐fold higher stiffness by increasing the GdL concentration from 5 to 10 mg mL−1. This observation is due to the higher degree of protonation of G2 obtained in a more concentrated GdL solution. Thus, more and longer nanofibers form, which makes a stiffer hydrogel.
Figure 5.
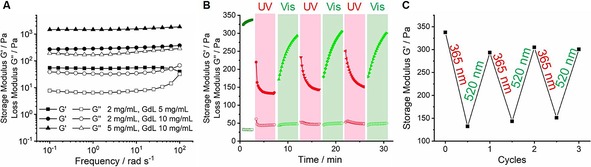
Rheological studies of G2. A) Frequency sweep for increasing G2 and GdL concentrations. B) Light‐induced stiffness modulation under UV and visible light irradiation (2 mg mL−1 G2 and 10 mg mL−1 GdL). C) Reversible storage modulus response to alternating UV and visible light irradiation for three cycles.
Next, the light‐responsive modulation of stiffness for hydrogels prepared from G2 was examined (Figure 5 B,C). The hydrogel formed with 2 mg mL−1 G2 and 10 mg mL−1 GdL was prepared one day before. The measurement started without any irradiation and the storage modulus showed a plateau value. Upon UV light irradiation (λ=365 nm), G′ dropped by over 50 % (Figure 5 B). We attribute this to the photoisomerization of the hydrogelator. Similar to G1, E‐G2 is expected to provide a stiffer material due to the face‐to‐face hydrogen bonding and the π–π interactions between the gelators. On the contrary, the Z‐G2 loses the additional π–π interactions between the AAPs due to their twisted structure.53, 54, 55 Thus, a diminished elastic modulus (G′) was observed. During the visible light irradiation (λ=520 nm), the storage modulus was restored. This can be explained by the recovery of the planar structure of AAP in E‐G2, which allows for the restoration of π–π interactions (Figure 5 B). Interestingly, during UV irradiation, the G′ reached a plateau faster, whereas upon visible light irradiation, a plateau was reached rather slowly. A reasonable explanation for this observation is that upon UV irradiation, the hydrogel softens, and the oscillation enhances this process. In contrast, when the material stiffens during the green light irradiation, the oscillation disturbs the recovery of the fibers and the network. Nevertheless, the photoisomerization of G2 induced a modulation of stiffness that can be repeated for at least three cycles (Figure 5 C). It is worth noting that during irradiation with either UV or green light the loss modulus remained almost constant. Additional photorheology experiments for different concentrations of samples (2 and 5 mg mL−1 G2 with 5 and 10 mg mL−1 GdL) can be found in the Supporting Information (Figure S6).
Photoresponsive and dynamic covalent hydrogels
Hydrogelator G3 containing dynamic covalent hydrazone bonds was synthesized in situ by employing GdL in the same fashion as describes above for G2, because the hydrazone formation is favored at pH 4–6, which is close to the apparent pK a of alanine. To ensure that a tripodal gelator will form, excess arms to core (6:1) were employed in this study.77 AAP‐CHO (15 mm) and CTH (2.5 mm) were dissolved at pH≈8 and subsequently, GdL was added. After gentle mixing, the solution was left overnight. A self‐supporting hydrogel was obtained and a schematic representation of the self‐assembly is depicted in Figure 6 A. Considering that the arms can be also considered as peptide amphiphiles (with AAP as the hydrophobic tail and alanine as the hydrophilic head group), a control experiment was carried out to ensure that the obtained hydrogels were achieved by the tripodal core–arm based hydrogelator G3. Indeed, when CTH was absent, a turbid solution with yellow precipitates of AAP‐CHO was observed (Figure 6 B). This finding supports our claim that the hydrogel is formed by the tripodal core–arm based hydrogelator G3. The self‐assembled structures of the hydrogels were examined by SEM. Again, typical fibrous and cross‐linked structures were found (Figure 6 C).
Figure 6.
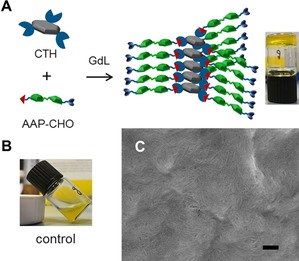
A) In situ formation of G3 from CTH (2.5 mm) and AAP‐CHO (15 mm) triggered by GdL (5 mg mL−1). B) Control experiment in the absence of CTH (AAP‐CHO: 15 mm and GdL: 5 mg mL−1). C) SEM image of 5 mm G3 (scale bar 200 nm).
To gain more information about the formation of G3, a kinetic study of the hydrazone formation was carried out. As a proof‐of‐principle, the formation of hydrazone between a hydrazide and an AAP‐CHO in presence of GdL was tracked by proton NMR (Figure 7 A). Similar to the determination of PSS for G1, in this study, a cyclohexane monohydrazide (CMH) was applied to avoid broadening proton signals due to intermolecular stacking of G3 in aqueous media. The decreasing signal of the aldehyde proton of AAP‐CHO (at ≈10 ppm) as well as the increasing signal of the hydrazone proton (red circle, 8.27 ppm) reveals the hydrazone formation. In addition, the large upfield shift of the ortho‐protons at the benzyl ring of AAP supports our claim for the formation of a hydrazone. The more downfield shifted proton (blue rhombus, 8.12 ppm) is attributed to the presence of a strong electron‐withdrawing formyl group at the ortho position, whereas, the more upfield shifted proton (purple pentagon, 7.86 ppm) is due to a more electron‐donating functional group, hydrazone. Integrating the hydrazone signal (red circle, 8.27 ppm) in 1 h intervals offers a quantitative measure of the hydrazone formation (Figure 7 and Figure S7 in the Supporting Information). Experimental details are provided in the Experimental Section.
Figure 7.
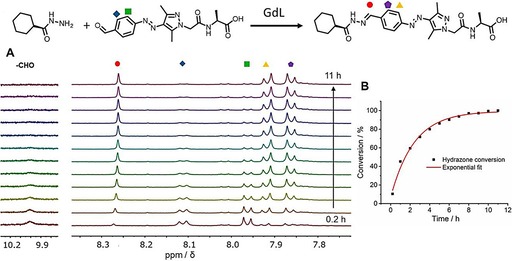
A) GdL triggered formation of hydrazane G3‐ref from CMH and AAP‐CHO was carried out using proton NMR in D2O (CMH: 500 μm, AAP‐CHO: 500 μm, and GdL: 1 mg mL−1). B) Formation of hydrazone in time based on the signal at 8.27 ppm.
The viscoelastic properties of the hydrogels of G3 were also investigated by rheology. Similar to G2, frequency‐sweep oscillation experiments were performed. Again, G′ values almost one order of magnitude higher then G′′ were found, indicating the success of hydrogel preparation. Again, the stiffness of the gel is proportional to both gelator concentration and the amount of GdL. The concentration of G3 refers to the concentration of CTH considering that an excess of AAP‐CHO was applied. The highest G′ was observed for 5 mm G3 with 10 mg mL−1 GdL and the lowest for 2.5 mm G3 with 5 mg mL−1 GdL (Figure S8, Supporting Information). Then, oscillatory rheological measurements were performed to examine the light response of the stiffness of G3. The hydrazone‐linked hydrogel formed with 5 mm CTH, 30 mm AAP‐CHO and 5 mg mL−1 GdL was prepared 24 h in advance. Similar to G1 and G2, E‐G3 is expected to give a stiffer gel, whereas Z‐G3 should give a softer gel. The measurements were carried out in the same fashion by recording firstly a non‐irradiated sample and subsequently irradiating with UV and visible light in an alternating fashion for three cycles. Indeed, a light‐responsive modulation of stiffness was observed similar to G2, that is, higher G′ for E‐G3 and lower G′ for Z‐G3. The same explanation can be applied to these findings: planar configuration and additional π–π interactions for the E isomer; twisted configuration and loss of π–π interactions for Z isomer. After the first cycle, around 30 % loss of storage modulus was found upon UV irradiation and in total three cycles of stiffness modulation were demonstrated (Figure 8 A,B). We conclude that the introduction of amides (G2) or hydrazones (G3) to conjugate the core to the AAP arms does not significantly affect the formation of hydrogels nor their photoresponse.
Figure 8.
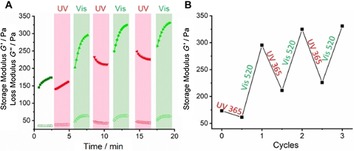
A) Light‐induced stiffness modulation of a hydrogel of 2.5 mm G3 triggered by 5 mg mL−1 GdL under UV and visible light. B) Storage modulus response to light.
The formation of G3 involves a dynamic reaction between hydrazide core and aldehyde arms. This means that when other hydrazides (or aldehydes) are added, they can be used to tune the self‐assembly of the gelator and the gel formation. Here, competitive experiments involving two different hydrazides were conducted. In the first assay, a neutral component, 4‐hydroxylbutyric acid hydrazide was applied (Figure 9 A). The addition of neutral hydrazide (NHy) suppresses the formation of G3 (prepared from 2.5 mm CTH, 15 mm AAP‐CHO and 5 mg mL−1 GdL), since the probability for AAP‐CHO to form a gelator (with CTH) or a non‐gelator (with NHy) are equal. Thus, after one day, a self‐supporting hydrogel was present in the absence of the competitor, whereas a liquid remained in the presence of NHy (5 or 25 mm). These findings are in agreement with our previous suggestions that the three‐armed structure is required for the hydrogel formation. Interestingly, if the concentration of NHy is rather low (5 mm), a hydrogel is obtained after two days (Figure 9 B). On the contrary, if the concentration of NHy is high enough (25 mm), the sample remains liquid‐like even after five days (Figure 9 C). This can be explained from the competing reactions of AAP‐CHO with NHy and CTH and the consideration that the self‐assembly of G3 is thermodynamically favoured. Only if sufficient three‐armed gelator G3 is formed, the self‐assembly will take place and as a result, a gel will form. When NHy is less concentrated, the formation of G3 is initially suppressed, but eventually wins out because the gel acts as a thermodynamic sink for G3. Also, a limited number of NHy arms will probably not disturb the self‐assembly process. Instead, if NHy is more concentrated, the formation of G3 is suppressed to such an extent that no nucleation points for the formation of self‐assembled nanofibrils are available, and no gelation occurs even after 5 days.
Figure 9.
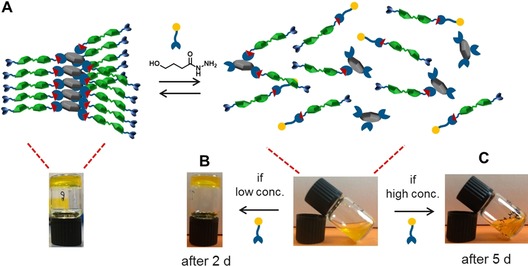
A) Schematic representation of the dynamic properties of G3 in the presence of a neutral hydrazide. Macroscopic pictures of G3 (2.5 mm CTH, 15 mm AAP‐CHO and 5 mg mL−1 GdL) in the presence of B) 5 mm and (C) 25 mm of NHy.
In a final experiment, if a cationic hydrazide (CatHy, 5 mm), betaine hydrazide, was applied, gel formation was completely suppressed even at low concentration (Figure S9, Supporting Information). This observation can be explained by considering the formation of cationic arms when CatHy reacts with CTH. The repulsion of the cationic arms disturbs the self‐assembly of G3 and, hence, it persists in a liquid state.
Conclusions
In this study, we demonstrated that a family of light‐responsive LMWGs can form gels in organic and aqueous media, and that the hydrogelator can have permanent (amide) or dynamic covalent (hydrazone) linkage. Tripodal LMWGs G1‐G3, comprising a CTA core and three AAP arms, were successfully synthesized and fully characterized. The excellent photoisomerization efficiency of all gelators were confirmed by UV/Vis absorption and NMR spectroscopy. Organogelator G1 forms organogels in nine organic solvents with rather low CGC (<1 wt %) and exhibits a reversible sol–gel transition under alternating UV and visible light irradiation. Homogeneous hydrogels prepared from G2 and G3 were obtained by tuning the pH through the hydrolysis of GdL. UV and visible light irradiation was utilized to modulate the stiffness of the hydrogels. In both cases, the E conformation of the hydrogelators represents the more stable and planar structure, which shows a higher storage modulus. On the contrary, the Z isomer of both G2 and G3 disrupt the planarity and the additional π–π interactions contributed by AAPs, resulting in a lower storage modulus and a softer gel. Finally, the dynamic formation of G3 and its influence on the hydrogel properties was explored. Dynamically linked G3 was obtained in situ from a trihydrazide core (CTH) and AAP aldehyde arms (AAP‐CHO). Neutral and cationic hydrazides were added to reverse the gel formation by hydrazone exchange. Our results demonstrate that molecular design of gelators with embedded photoswitches can lead to versatile light‐responsive and dynamic soft materials. The stiffness of these materials can be modulated by light, which is especially important when considering biomedical applications such as tissue engineering.86
Experimental Section
Organogels: A certain amount of G1 was added to 1 mL of organic solvent and heated until it was fully dissolved. Afterwards, the solution was allowed to cool down to RT and the gel formation was checked by an inverted vial test. If a solution was obtained, the amount of G1 was increased until a gel was achieved. The results displayed in Table 1 showed the formation of a transparent orange organogel (G) in 9 out of 17 tested organic solvents with different CGCs. Appearance of results was presented by gel (G), precipitate (P) and solution (S).
Hydrogels: Stock solutions of 10, 5 and 2 mg mL−1 of G2 were prepared in ddH2O and NaOH (1 m) was used to adjust the pH until the hydrogelators were fully dissolved (pH≈9). In a glass vial containing 2, 5, 10, 20 mg mL−1 of GdL, 200 μL of different concentrations of the stock solution was added. After no crystals were observed, the solution was left overnight for gelation. Samples were defined as gels if a reverse‐vial test was passed. Critical gelation concentration was found in 2 mg mL−1 and the minimum required GdL was 5 mg mL−1 in this concentration.
Dynamic covalent hydrogels: Stock solutions of 5 and 10 mm of CTH and the correspondingly 30 and 60 mm of AAP‐CHO were prepared. In a glass vial containing 2, 5, 10, 20 mg mL−1 of GdL, the core (CTH) and arm solutions (AAP‐CHO) were mixed with a 1:1 fashion. After a clear solution was obtained, it was then left untouched for gelation overnight. Critical gelation concentration was found in the presence of 2.5 mm of CTH and minimum required GdL was 5 mg mL−1 in this concentration.
Rheology and photoswitching experiments: Rheological measurements were performed by using a shear rheometer (MCR 102, Modular Compact Rheometer, Anton Paar) equipped with a glass plate (P‐PTD200/GL) and CP25‐2 cone plate (radius=12.5 mm, cone angle=2°, sample volume=0.16 mL). The gap between two plates were fixed at 0.106 mm during the measurements. The measurement temperature was maintained at 20 °C. Frequency sweep measurements were carried out with a 0.2 % strain amplitude. For irradiation experiments, the measurements were carried out with a 0.5 % strain amplitude and 10 Hz frequency. The photoisomerization was induced by in situ irradiation from beneath the glass plate by using LEDs as used in UV/Vis spectroscopy. Photoswitching experiments were performed for three cycles.
Hydrazone formation: NMR spectroscopy was used to track the hydrazone formation between cyclohexane monohydrazide (CMH) and AAP‐CHO triggered by GdL. Solutions of CMH and AAP‐CHO in D2O were prepared and mixed in a 1:1 fashion (final concentration: 500 μm). The above solution was then transferred to a vial containing 1 mg mL−1 GdL and well mixed before being transferred into an NMR tube. NMR spectra were recorded every hour for 12 spectra in total.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful for the financial support from EC H2020—Marie Skłodowska‐Curie Actions—Innovative Training Network, Multi‐App (project number: 642793) and the Deutsche Forschungsgemeinschaft (DFG EXC 1003). Lukas Ibing (MEET, WWU Münster) is acknowledged for SEM measurements.
C.-W. Chu, L. Stricker, T. M. Kirse, M. Hayduk, B. J. Ravoo, Chem. Eur. J. 2019, 25, 6131.
References
- 1. Terech P., Weiss R. G., Chem. Rev. 1997, 97, 3133–3160. [DOI] [PubMed] [Google Scholar]
- 2. Estroff L. A., Hamilton A. D., Chem. Rev. 2004, 104, 1201–1217. [DOI] [PubMed] [Google Scholar]
- 3. Tao K., Levin A., Adler-Abramovich L., Gazit E., Tao K., Chem. Soc. Rev. 2016, 45, 3935–3953. [DOI] [PubMed] [Google Scholar]
- 4. Fleming S., Ulijn R. V., Chem. Soc. Rev. 2014, 43, 8150–8177. [DOI] [PubMed] [Google Scholar]
- 5. Yagai S., Nakajima T., Kishikawa K., Kohmoto S., Karatsu T., Kitamura A., J. Am. Chem. Soc. 2005, 127, 11134–11139. [DOI] [PubMed] [Google Scholar]
- 6. Yagai S., Monma Y., Kawauchi N., Karatsu T., Kitamura A., Org. Lett. 2007, 9, 1137–1140. [DOI] [PubMed] [Google Scholar]
- 7. Yan X., Xu D., Chi X., Chen J., Dong S., Ding X., Yu Y., Huang F., Adv. Mater. 2012, 24, 362–369. [DOI] [PubMed] [Google Scholar]
- 8. Enomoto M., Kishimura A., Aida T., J. Am. Chem. Soc. 2001, 123, 5608–5609. [DOI] [PubMed] [Google Scholar]
- 9. Sreenivasachary N., Lehn J.-M., Proc. Natl. Acad. Sci. USA 2005, 102, 5938–5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weng W., Li Z., Jamieson A. M., Rowan S. J., Macromolecules 2009, 42, 236–246. [Google Scholar]
- 11. Piepenbrock M. M., Lloyd G. O., Clarke N., Steed J. W., Chem. Rev. 2010, 110, 1960–2004. [DOI] [PubMed] [Google Scholar]
- 12. Rest C., Mayoral M. J., Fucke K., Schellheimer J., Stepanenko V., Fernández G., Angew. Chem. Int. Ed. 2014, 53, 700–705; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 716–722. [Google Scholar]
- 13. Babu S. S., Praveen V. K., Ajayaghosh A., Chem. Rev. 2014, 114, 1973–2129. [DOI] [PubMed] [Google Scholar]
- 14. Brunsveld L., Folmer B. J. B., Meijer E. W., Sijbesma R. P., Chem. Rev. 2001, 101, 4071–4098. [DOI] [PubMed] [Google Scholar]
- 15. Aida T., Meijer E. W., Stupp S. I., Science 2012, 335, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu Z., Tantakitti F., Yu T., Palmer L. C., Schatz G. C., Stupp S. I., Science 2016, 351, 497–502. [DOI] [PubMed] [Google Scholar]
- 17. Grigoriou S., Johnson E. K., Chen L., Adams D. J., James T. D., Cameron P. J., Soft Matter 2012, 8, 6788–6791. [Google Scholar]
- 18. Frisch H., Besenius P., Macromol. Rapid Commun. 2015, 36, 346–363. [DOI] [PubMed] [Google Scholar]
- 19. Orbach R., Adler-Abramovich L., Zigerson S., Mironi-Harpaz I., Seliktar D., Gazit E., Biomacromolecules 2009, 10, 2646–2651. [DOI] [PubMed] [Google Scholar]
- 20. Kiyonaka S., Sugiyasu K., Shinkai S., Hamachi I., J. Am. Chem. Soc. 2002, 124, 10954–10955. [DOI] [PubMed] [Google Scholar]
- 21. Ulijn R. V., J. Mater. Chem. 2006, 16, 2217–2225. [Google Scholar]
- 22. Yang Z., Liang G., Xu B., Acc. Chem. Res. 2008, 41, 315–326. [DOI] [PubMed] [Google Scholar]
- 23. Haines L. A., Rajagopal K., Ozbas B., Salick D. A., Pochan D. J., Schneider J. P., J. Am. Chem. Soc. 2005, 127, 17025–17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muraoka T., Koh C. Y., Cui H., Stupp S. I., Angew. Chem. Int. Ed. 2009, 48, 5946–5949; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6060–6063. [Google Scholar]
- 25. Chen L., Pont G., Morris K., Lotze G., Squires A., Serpell L. C., Adams D. J., Chem. Commun. 2011, 47, 12071–12073. [DOI] [PubMed] [Google Scholar]
- 26. Cardoso A. Z., Mears L. L. E., Cattoz B. N., Griffiths P. C., Schweins R., Adams D. J., Soft Matter 2016, 12, 3612–3621. [DOI] [PubMed] [Google Scholar]
- 27. Raeburn J., Cardoso A. Z., Adams D. J., Chem. Soc. Rev. 2013, 42, 5143–5156. [DOI] [PubMed] [Google Scholar]
- 28. Mattia E., Otto S., Nat. Nanotechnol. 2015, 10, 111–119. [DOI] [PubMed] [Google Scholar]
- 29. Ryan D. M., Nilsson B. L., Polym. Chem. 2012, 3, 18–33. [Google Scholar]
- 30. Hartgerink J. D., Beniash E., Stupp S. I., Science 2001, 294, 1684–1688. [DOI] [PubMed] [Google Scholar]
- 31. Externbrink M., Riebe S., Schmuck C., Voskuhl J., Soft Matter 2018, 14, 6166–6170. [DOI] [PubMed] [Google Scholar]
- 32. Stone D. A., Tayi A. S., Goldberger J. E., Palmer L. C., Stupp S. I., Chem. Commun. 2011, 47, 5702–5704. [DOI] [PubMed] [Google Scholar]
- 33. Draper E. R., Walsh J. J., McDonald T. O., Zwijnenburg M. A., Cameron P. J., Cowan A. J., Adams D. J., J. Mater. Chem. C 2014, 2, 5570–5575. [Google Scholar]
- 34. Kiyonaka S., Sada K., Yoshimura I., Shinkai S., Kato N., Hamachi I., Nat. Mater. 2004, 3, 58–64. [DOI] [PubMed] [Google Scholar]
- 35. Qi Z., de Molina P. M., Jiang W., Wang Q., Nowosinski K., Schulz A., Gradzielski M., Schalley C. A., Chem. Sci. 2012, 3, 2073–2082. [Google Scholar]
- 36. Lin Q., Lu T.-T., Zhu X., Wei T.-B., Li H., Zhang Y.-M., Chem. Sci. 2016, 7, 5341–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goujon A., Mariani G., Lang T., Moulin E., Rawiso M., Buhler E., Giuseppone N., J. Am. Chem. Soc. 2017, 139, 4923–4928. [DOI] [PubMed] [Google Scholar]
- 38. Chen J., Leung F. K. C., Stuart M. C. A., Kajitani T., Fukushima T., van der Giessen E., Feringa B. L., Nat. Chem. 2018, 10, 132–138. [DOI] [PubMed] [Google Scholar]
- 39. Yagai S., Kitamura A., Chem. Soc. Rev. 2008, 37, 1520–1529. [DOI] [PubMed] [Google Scholar]
- 40. Khetan S., Burdick J. A., Soft Matter 2011, 7, 830–838. [Google Scholar]
- 41. Draper E. R., Adams D. J., Chem. Commun. 2016, 52, 8196–8206. [DOI] [PubMed] [Google Scholar]
- 42. Draper E. R., McDonald T. O., Adams D. J., Chem. Commun. 2015, 51, 12827–12830. [DOI] [PubMed] [Google Scholar]
- 43. Kim S. H., Sun Y., Kaplan J. A., Grinstaff M. W., Parquette J. R., New J. Chem. 2015, 39, 3225–3228. [Google Scholar]
- 44. Qiu Z., Yu H., Li J., Wang Y., Zhang Y., Chem. Commun. 2009, 3342–3344. [DOI] [PubMed] [Google Scholar]
- 45. Xu J. F., Chen Y. Z., Wu D., Wu L. Z., Tung C. H., Yang Q. Z., Angew. Chem. Int. Ed. 2013, 52, 9738–9742; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9920–9924. [Google Scholar]
- 46. Draper E. R., Eden E. G. B., McDonald T. O., Adams D. J., Nat. Chem. 2015, 7, 848–852. [DOI] [PubMed] [Google Scholar]
- 47. Sahoo J. K., Nalluri S. K. M., Javid N., Webb H., Ulijn R. V., Chem. Commun. 2014, 50, 5462–5464. [DOI] [PubMed] [Google Scholar]
- 48. Tiefenbacher K., Dube H., Ajami D., Rebek J., Chem. Commun. 2011, 47, 7341–7343. [DOI] [PubMed] [Google Scholar]
- 49. van Herpt J. T., Stuart M. C. A., Browne W. R., Feringa B. L., Chem. Eur. J. 2014, 20, 3077–3083. [DOI] [PubMed] [Google Scholar]
- 50. Wang S., Shen W., Feng Y., Tian H., Chem. Commun. 2006, 1497–1499. [DOI] [PubMed] [Google Scholar]
- 51. Nalluri S. K. M., Voskuhl J., Bultema J. B., Boekema E. J., Ravoo B. J., Angew. Chem. Int. Ed. 2011, 50, 9747–9751; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9921–9925. [Google Scholar]
- 52. Klajn R., Pure Appl. Chem. 2010, 82, 2247–2279. [Google Scholar]
- 53. Cheng S.-C., Chen K.-J., Suzaki Y., Tsuchido Y., Kuo T.-S., Osakada K., Horie M., J. Am. Chem. Soc. 2018, 140, 90–93. [DOI] [PubMed] [Google Scholar]
- 54. Stricker L., Fritz E.-C., Peterlechner M., Doltsinis N. L., Ravoo B. J., J. Am. Chem. Soc. 2016, 138, 4547–4554. [DOI] [PubMed] [Google Scholar]
- 55. Weston C. E., Richardson R. D., Haycock P. R., White A. J. P., Fuchter M. J., J. Am. Chem. Soc. 2014, 136, 11878–11881. [DOI] [PubMed] [Google Scholar]
- 56. Crespi S., Simeth N. A., König B., Nat. Rev. Chem. 2019, 3, 133–146. [Google Scholar]
- 57. Stricker L., Boeckmann M., Kirse T. M., Doltsinis N. L., Ravoo B. J., Chem. Eur. J. 2018, 24, 8639–8647. [DOI] [PubMed] [Google Scholar]
- 58. Engel S., Möller N., Stricker L., Peterlechner M., Ravoo B. J., Small 2018, 14, 1704287. [DOI] [PubMed] [Google Scholar]
- 59. Möller N., Hellwig T., Stricker L., Engel S., Fallnich C., Ravoo B. J., Chem. Commun. 2017, 53, 240–243. [DOI] [PubMed] [Google Scholar]
- 60. Schnurbus M., Stricker L., Ravoo B. J., Braunschweig B., Langmuir 2018, 34, 6028–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lamping S., Stricker L., Ravoo B. J., Polym. Chem. 2019, 10, 683–690. [Google Scholar]
- 62. Moratz J., Stricker L., Engel S., Ravoo B. J., Macromol. Rapid Commun. 2018, 39, 1700256. [DOI] [PubMed] [Google Scholar]
- 63. Adam V., Prusty D. K., Centola M., Skugor M., Hannam J. S., Valero J., Kloeckner B., Famulok M., Chem. Eur. J. 2018, 24, 1062–1066. [DOI] [PubMed] [Google Scholar]
- 64. Chu C.-W., Ravoo B. J., Chem. Commun. 2017, 53, 12450–12453. [DOI] [PubMed] [Google Scholar]
- 65. Zhou Y., Xu M., Wu J., Yi T., Han J., Xiao S., Li F., Huang C., J. Phys. Org. Chem. 2008, 21, 338–343. [Google Scholar]
- 66. Brizard A., Stuart M., van Bommel K., Friggeri A., de Jong M., van Esch J., Angew. Chem. Int. Ed. 2008, 47, 2063–2066; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2093–2096. [Google Scholar]
- 67. Devi S., Gaur A. K., Gupta D., Saraswat M., Venkataramani S., ChemPhotoChem 2018, 2, 806–810. [Google Scholar]
- 68. Kulkarni C., Meijer E. W., Palmans A. R. A., Acc. Chem. Res. 2017, 50, 1928–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Spitzer D., Marichez V., Formon G. J. M., Besenius P., Hermans T. M., Angew. Chem. Int. Ed. 2018, 57, 11349–11353. [DOI] [PubMed] [Google Scholar]
- 70. Seibt S., With S., Bernet A., Schmidt H., Langmuir 2018, 34, 5535–5544. [DOI] [PubMed] [Google Scholar]
- 71. Alemán García M. Á., Magdalena Estirado E., Milroy L.-G., Brunsveld L., Angew. Chem. Int. Ed. 2018, 57, 4976–4980; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5070–5074. [Google Scholar]
- 72. Müller M. K., Brunsveld L., Angew. Chem. Int. Ed. 2009, 48, 2921–2924; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2965–2968. [Google Scholar]
- 73. Neumann L. N., Baker M. B., Leenders C. M. A., Voets I. K., Lafleur R. P. M., Palmans A. R. A., Meijer E. W., Org. Biomol. Chem. 2015, 13, 7711–7719. [DOI] [PubMed] [Google Scholar]
- 74. Boekhoven J., Poolman J. M., Maity C., Li F., van der Mee L., Minkenberg C. B., Mendes E., van Esch J. H., Eelkema R., Nat. Chem. 2013, 5, 433–437. [DOI] [PubMed] [Google Scholar]
- 75. Zhou Y., Chen Y., Zhu P., Si W., Hou J., Liu Y., Chem. Commun. 2017, 53, 3681–3684. [DOI] [PubMed] [Google Scholar]
- 76. Trausel F., Versluis F., Maity C., Poolman J. M., Lovrak M., van Esch J. H., Eelkema R., Acc. Chem. Res. 2016, 49, 1440–1447. [DOI] [PubMed] [Google Scholar]
- 77. Poolman J. M., Boekhoven J., Besselink A., Olive A. G. L., van Esch J. H., Eelkema R., Nat. Protoc. 2014, 9, 977–988. [DOI] [PubMed] [Google Scholar]
- 78. Versluis F., van Elsland D. M., Mytnyk S., Perrier D. L., Trausel F., Poolman J. M., Maity C., Le Sage V. A. A., van Kasteren S. I., van Esch J. H., Eelkema R., J. Am. Chem. Soc. 2016, 138, 8670–8673. [DOI] [PubMed] [Google Scholar]
- 79. Olive A. G. L., Abdullah N. H., Ziemecka I., Mendes E., Eelkema R., van Esch J. H., Angew. Chem. Int. Ed. 2014, 53, 4132–4136; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4216–4220. [Google Scholar]
- 80. Poolman J. M., Maity C., Boekhoven J., van der Mee L., le Sage V. A. A., Groenewold G. J. M., van Kasteren S. I., Versluis F., van Esch J. H., Eelkema R., J. Mater. Chem. B. 2016, 4, 852–858. [DOI] [PubMed] [Google Scholar]
- 81. Rowan S. J., Cantrill S. J., Cousins G. R. L., Sanders J. K. M., Stoddart J. F., Angew. Chem. Int. Ed. 2002, 41, 898–952; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 938–993. [Google Scholar]
- 82. Wojtecki R. J., Meador M. A., Rowan S. J., Nat. Mater. 2011, 10, 14–27. [DOI] [PubMed] [Google Scholar]
- 83. Chakma P., Konkolewicz D., Angew. Chem. Int. Ed. 2019, DOI: 10.1002/anie.201813525. [DOI] [PubMed] [Google Scholar]
- 84. Adams D. J., Butler M. F., Frith W. J., Kirkland M., Mullen L., Sanderson P., Soft Matter 2009, 5, 1856–1862. [Google Scholar]
- 85. Tang C., Smith A. M., Collins R. F., Ulijn R. V., Saiani A., Langmuir 2009, 25, 9447–9453. [DOI] [PubMed] [Google Scholar]
- 86. Seliktar D., Science 2012, 336, 1124–1129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary