

Abstract
In the liver tissues of obese diabetic or nondiabetic patients, triggering receptor expressed on myeloid cells‐1 (TREM‐1) is usually found to be upregulated, thus leading to upregulation of various inflammatory cytokines and lipid accumulation. On the other hand, nonalcoholic fatty liver disease (NAFLD), characterized by excess lipid accumulation, and inflammatory injury in liver, is becoming an epidemic disease, globally. In the present study, we aimed to investigate the biological role and the underlying mechanisms of TREM‐1 in NAFLD. upregulation of TREM‐1 occurred in high‐fat diet (HFD)‐induced mice NAFLD model and oleic acid‐treated HepG2 and primary mouse hepatocytes cell model at messenger RNA and protein levels. Functional studies established that overexpression of TREM‐1 displayed hyperlipidemia, and increased in inflammatory indicators and lipid accumulation‐related genes, which was ameliorated by knockdown of TREM‐1. Our results also showed that obvious lipid accumulation and inflammatory injury occurred in the liver tissue of HFD‐fed mice, while treatment with lentiviral vector short hairpin TREM showed marked improvement in tissue morphology and architecture and less lipid accumulation, thus deciphering the mechanism through which knockdown of TREM‐1 ameliorated the inflammatory response and lipid accumulation of NAFLD mice through inactivation of the nuclear factor‐κB (NF‐κB) and PI3K/AKT signal pathways, respectively. In conclusion, TREM‐1/NF‐κB and TREM‐1/PI3K/AKT axis could be an important mechanism in ameliorating the inflammatory response and lipid accumulation, respectively, thus shedding light on the development of novel therapeutics to the treatment of NAFLD.
Keywords: inflammation, lipid accumulation, nonalcoholic fatty liver disease, TREM‐1
1. INTRODUCTION
Nonalcoholic fatty liver diseases (NAFLD) is a liver metabolic syndrome, affecting 24% of the worldwide population and making it the most common cause of chronic liver disease, both familiar in obese and normal‐weight people with markedly increasing incidence in recent years.1 Due to the fat deposited in the liver, NAFLD ranges from nonalcoholic fatty liver to nonalcoholic steatohepatitis (NASH), which typically occurs before liver fibrosis, cirrhosis, and hepatocellular carcinoma.2, 3, 4 Therefore, lipid accumulation is an important pathogenic factor for fatty liver diseases leading to hepatic steatosis,5, 6 and corn oil was successfully used to developed NAFLD models in rats.7, 8 In addition, steatosis combined with inflammation further develop the disease to NASH.9 Hence, antiglycemic drugs helping loss of liver fat10 and anti‐inflammation–ameliorating steatohepatitis11 may be the promising therapeutic approach for NAFLD. However, due to the unknown exact molecular pathogenesis of NAFLD, the primary task of improving NAFLD prognosis is to study its pathogenesis and effective therapeutic targets for treatment.
Triggering receptor expressed on myeloid cells‐1 (TREM‐1), member of TREM family, is a kind of immunoglobulin superfamily activation receptors,12 related to innate inflammatory response.13 The secretion of inflammatory factors triggered and amplified the inflammatory response in monocytes through TREM‐1.14 Activation of TREM‐1 induced phosphorylation of downstream target tyrosine‐protein kinase Lyn, nonreceptor tyrosine kinase Jak2, P13K/Akt, ERK1/2, phospholipase C (PLC), and the non‐T cell activation linker (NTAL).15, 16 Phosphorylation of Jak2 activated downstream of the signal transducer and activator of transcription 3/5 phosphorylation, which enhanced NF‐κB activity and an increase of NF‐κB subunits P50/P65, finally augmented expression of inflammatory factors.17, 18 Subramanian et al19 indicated that abnormal TREM‐1 expression was found in liver tissues of obese diabetic and nondiabetic patients. Zhou et al20 found that inhibition of TREM‐1 can inhibit inflammation and the occurrence of colon tumors. Tang et al21 found that knockdown of TREM‐1 can inhibit the activity of the nuclear factor‐κB (NF‐κB) pathway, thereby inhibiting interleukin‐1β (IL‐1β)‐induced cartilage cell damage. The expression of TREM‐1 in bone marrow cell membrane was induced by dyslipidemia in hyperlipidemic model mice, which resulted in increased monocytes, cytokines, and foam cells, and promoted lipid accumulation by regulating genes related to cholesterol metabolism.22 So far, the studies of TREM‐1 mainly focused on the direction of inflammation, while the mechanism of action in NAFLD is not clear.
This study aimed to study the effect of TREM‐1 on inflammatory response and lipid accumulation induced by NAFLD. In our present study, we first investigated the effect of TREM‐1 on NAFLD; second, found the downstream regulator NF‐κB/PI3K/AKT. Last, we investigated the functional roles of TREM‐1/NF‐κB/PI3K/AKT in the progression of NAFLD and further explored their potential mechanisms. This study provides a basis and new target for individualized treatment of NAFLD by exploring the regulatory mechanisms of TREM‐1/NF‐κB and TREM‐1/PI3K/AKT in the occurrence and development of NAFLD.
2. MATERIALS AND METHODS
2.1. Cell lines and transfection
Primary mouse hepatocytes (PMH) was isolated from the livers of male C57/BL6 mice as before.23 Cells were then seeded into six‐well plates precoated with 400 µL 1 mg/mL collagen solution with 6 × 105 cells/well. Hepatocytes were cultured in William's medium E (Pan Biotech GmbH, Aidenbach, Germany) containing 100 U/mL penicillin, 100 µg/mL streptomycin, 0.00001% insulin‐transferrin‐selenium, and 100 nM dexamethasone. Human hepatocellular carcinoma cell line HepG2 was maintained in cultured in a Dulbecco's modified Eagle's medium‐based medium (Gibco BRL, Grand Island) with 10% fetal bovine serum, 100 mg/mL streptomycin and 100 U/mL penicillin. The 37°C constant temperature incubator was used to incubating cells with 5% CO2.
The full‐length complementary DNA (cDNA) of human TREM‐1 was amplified by polymerase chain reaction (PCR) with specific primers: 5′‐CGAATGGTCAACCTTCAAG‐3′ (forward) and 5′‐CTGGTATAGAGTGGGCACAA‐3′ (reverse). Expression plasmids pcDNA3/V5‐TREM‐1 (Invitrogen, Carlsbad, CA) were then constructed and sequenced with V5 epitope tag was fused to the C‐terminus of TREM‐1. The vector was then transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
The small interfering RNAs (siRNAs) targeting TREM‐1 were designed and chemically synthesized as follows: 5′‐CCGGAAGTGTATGTGATCAGAGTAATCAAGAGATTACTCTGATCACATACACTTTTTTTG‐3′ (siTREM‐1 1#); 5′‐AATTCAAAAAAAGTGTATGTGATCAGAGTAATCTCTTGAATTACTCTGATCACATACACTT‐3′ (siTREM‐1 2#). The negative control was as follows: 5′‐GGUUUGGCUGGGGUGUUAUdTdT‐3′. Transfection was also performed using Lipofectamine 2000 according to the manufacturer's protocol.
2.2. Cell steatosis model
When PMH and HepG2 cells were grown to approximately 80% confluence, the cells were cultured in FBS‐free medium for 24 hours in 96‐well culture plate. The cells were then treated with 200 μL of 5 mM of oleic acid (OA) solution for 24 hours. After the medium was removed, 100 μL of fixative solution was added and incubated at room temperature for 10 minutes. Control cells were treated with OA‐free medium containing albumin.
2.3. Lentivirus infection
Three TREM‐1 short hairpin RNA (shRNA) containing lentiviral vectors (Lv‐shTREM‐1, Lv‐shTREM‐2, Lv‐shTREM‐3; National RNAi Core Facility, Academia Sinica, Taiwan) were obtained and target sequences 1: GTCAACCTTCAAGTGGAAGAT, 2: CCAGAAAGCTTGGCAGATAAT, and 3: CCTGACTCTGAAATCAACCTT. In addition, a scrambled sequence (5′‐CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG‐3′) was also used as the control for the knockdown study. Lentivirus was harvested 48 hours after cotransfection of pLL3.7‐shRNA with psPAX2 and pMD2.G into HEK‐293T cells using the Lipofectamine 2000 reagent.
2.4. Animal experimental model
Healthy male C57/BL6 mice, weighing between 18 and 22 g and 8 to 12‐week‐old, were used for experimentation, and each mouse was housed separately with standard pellet diet and water for acclimatization. After 1 week, mice were randomly divided into two individual groups, one group of mice were fed with normal control diet (NCD; n = 6; 10 kcal% fat, 20 kcal% protein, and 70 kcal% carbohydrate; Medicience Diets Co Ltd, Yangzhou, China) and the other with high‐fat diet (HFD; n = 36; 60 kcal% fat, 20 kcal% protein, and 20 kcal% carbohydrate) for 20 weeks. At 15th week of feeding, HFD‐fed mice were further divided into five groups: intraperitoneal injection with 50 mg/kg pyrrolidine dithiocarbamate (PDTC) HFD group (n = 6), intraperitoneal injection with 60 mg/kg MK‐2206‐2HCl HFD mice (MK‐2206; n = 6), HFD mice treated with lentiviral vector 2 for knockdown TREM‐1 (Lv‐shTREM‐2; n = 6), HFD mice treated with lentiviral vector 3 for knockdown TREM‐1 (Lv‐shTREM‐3; n = 6), and HFD mice treated with lentiviral vector control (Lv‐shRNA; n = 6). Treatments with different vectors were carried out by tail‐vein injection. At 20th week, the mice were killed by an overdose of sodium pentobarbital, and the liver tissues were collected. Livers were dissected and fixed in 4% paraformaldehyde solution, and then processed by paraffin tissue processing machine. After dehydration, 5‐μm sections were stained with hematoxylin and eosin (H&E) and following routine techniques. To visualize lipids, frozen sections were stained using Oil Red O. The stained sections were then viewed under a bright field microscope (Nikon, Tokyo, Japan).
2.5. RNA preparation and quantitative reverse‐transcription PCR
Sample RNAs were extracted by the means of TRIzol reagent (Invitrogen). The cDNAs were synthesized using the Reverse Transcription System Bestar qPCR RT Kit according to the manufacturer's instruction with ABI 7500 Real‐Time PCR System (Applied Biosystems, Foster City, CA). Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was used as the internal reference. The primer sequences are as shown: TREM‐1, 5′‐CGGAATTCGAGCTTGAAGGATGAGGAAGGC‐3′ (forward) and 5′‐AATCCAGAGTCTGTCACTTGAAGGTCAGTC‐3′ (reverse); IL‐6, 5′‐TCCAGTTGCCTTCTTGGGAC‐3′ (forward) and 5′‐GTACTCCAGAAGACCAGAGG‐3′ (reverse); IL‐1β, 5′‐CCAGCTTCAAATCTCACAGCAG ‐3′ (forward) and 5′‐CTTCTTTGGGTATTGCTTGGGATC‐3′ (reverse); tumor necrosis factor‐α (TNF‐α), 5′‐CACAGAAAGCATGATCCGCGA‐3′ (forward) and 5′‐CGGCAGAGAGGAGGTTGACTTTCT‐3′ (reverse); interferon‐γ (IFN‐γ), 5′‐GCGCAAAGCCATAAATGAAC‐3′ (forward) and 5′‐CTCAGAAAGCGGAAGAGAAG‐3′ (reverse); monocyte chemoattractant protein‐1 (MCP‐1), 5′‐ACTGAAGCTCGTACTCTC‐3′ (forward) and 5′‐CTTGGGTTGTGGAGTGAG‐3′ (reverse); macrophage inflammatory protein‐1α (MIP‐1α), 5′‐GCTGACTACTTTGAGACGAGC‐3′ (forward) and 5′‐CCAGTCCATAGAAGAGGTAGC‐3′ (reverse); macrophage scavenger receptor 1 (MSR1), 5′‐TGAACGAGAGGATGCTGACTG‐3′ (forward) and 5′‐TGTCATTGAACGTGCGTCAAA‐3′ (reverse); low‐density lipoprotein receptor (LDLR), 5′‐ACCCCAAGACGTGCTCCCAGGATG‐3′ (forward) and 5′‐CGCAGTGCTCCTCATCTGACTTGTC‐3′ (reverse); ATP‐binding cassette transporter‐1 (ABCA1), 5′‐GGACATGCACAAGGTCCTGA‐3′ (forward) and 5′‐CAGAAAATCCTGGAGCTTCAAA‐3′ (reverse); ATP‐binding cassette sub‐family G member 1 (ABCG1), 5′‐CCCTCAAAGCCGTATCTGAC‐3′ (forward) and 5′‐TTGACACCATCCCAGCCTAC‐3′ (reverse); Niemann‐Pick disease, type C1 (NPC1), 5′‐TCTGAATGCGGTCTCCTTG‐3′ (forward) and 5′‐TATGGCTGCAGAACTCCACA‐3′ (reverse); NPC intracellular cholesterol transporter 2 (NPC2), 5′‐TATCCACGATGCGTTTTCTG‐3′ (forward) and 5′‐TCAGGCT‐CAGGAATAGGGAA‐3′ (reverse); StAR‐related lipid transfer protein 4 (STARD4), 5′‐AGAAGTGTCGGGAAGGCAATG‐3′ (forward) and 5′‐AACTGGCTTTATGCAATCCCA‐3′ (reverse); GAPDH, 5′‐TGTTCGTCATGGGTGTGAAC‐3′ (forward) and 5′‐ATGGCATGGACTGTGGTCAT‐3′ (reverse).
2.6. Immunohistochemistry
Paraffin‐embedded 5‐µm‐thick sections of liver tissues were deparaffinized and heated in 0.01 mol/L citrate buffer. Sections were then blocked for endogenous peroxidase by incubating in 3% H2O2 and then washed in phosphate‐buffered saline (PBS) containing 0.05 M ethylenediaminetetraacetic acid (EDTA) followed by 4% paraformaldehyde. Tissues were incubated in 4% dry milk and 0.3% goat serum in PBS solution for 20 minutes to block nonspecific binding. Then, 4‐μm sections were incubated overnight at room temperature with the anti‐TREM‐1 antibody (AF1278; 1:80; R&D Systems, Minneapolis, MN). After washing, sections were then incubated for another 2 hours with horseradish peroxidase goat anti‐rabbit immunoglobulin G secondary antibody. Slides were counterstained with hematoxylin to stain cell nuclei to identify cells, be dehydrated and examined under a light microscope.
2.7. Western blot analysis
We collected the cells and flash‐froze them by liquid nitrogen; ultrasonic cell‐break method was adopted with twice for 5 seconds in 50 mM lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 10 mM β‐glycerophosphate, 1% Triton X‐100, 5 mM EDTA, 0.2 mM Na3VO4, 2 µg/mL leupeptin, 2 µg/mL pepstatin A) on ice. Homogenates were centrifuged at 12 000g at 4°C for 30 minutes, supernatants were collected. Protein lysates (30 μg) were loaded onto the sodium dodecyl sulfate‐polyacrylamide gels for electrophoresis and transferred to polyvinylidene difluoride (PVDF) membranes. PVDF membranes were incubated overnight with the primary antibody as follows: monoclonal antibodies (1:1000; Santa Cruz, CA) against TREM‐1, AKT, p‐AKT, p65, and p‐p65 solute in PBS‐Tween 20, followed by 5% bovine serum albumin blocking. Washed with Tris‐buffered saline with Tween 20 (10 minutes × 3 times), the membranes were then probed with the appropriate secondary antibody (1:5000; Abcam, Cambridge, English). Immunoreactivity was determined and observed using enhanced chemiluminescence (Millipore, Billerica, MA). Actin was used as a control.
2.8. Statistical analysis
All data are presented as mean ± SEM. All experiments were performed at least in three independent times. By the means of one‐way analysis of variance followed by Duncan's multiple‐comparison test using SPSS 19.0 (SPSS Inc, Chicago, IL) we calculated the statistical significance. P < 0.05, P < 0.01 or P < 0.001 were regarded as statistically significant.
2.9. Ethical Statement
All animal experiment were approved by the Institutional Research Ethics Committee of Union Hospital of HUST.
3. RESULTS
3.1. TREM‐1 was part of physiological response to lipotoxicity in NAFLD
To investigate the potential correlation between TREM‐1 expression and metabolic homeostasis in the fatty liver, we examined hepatic TREM‐1 expression in HFD‐fed mice by real‐time PCR and immunohistochemistry. As depicted in Figure 1A, the expression of TREM‐1 messenger RNA (mRNA) was significantly higher in steatotic livers from HFD‐fed mice than normal control diet (NCD)‐fed mice (P < 0.001). Consistent with our observation, analysis of immunohistochemistry showed increased protein expression of TREM‐1 in HFD compared with NCD (P < 0.001) (Figure 1B). We then incubated hepatocyte HepG2 and PMH cells to a pathophysiologically relevant concentration of free fatty acids (FFAs; 5 mmol/L OA) for 24 hours to simulate the excessive uptake of fatty acids (FAs). Consistent with the upregulation of TREM‐1 in steatotic livers in vivo, TREM‐1 was also rapidly increased after OA stimulation in both HepG2 and PMH cells by the means of Western blot analysis (P < 0.001) (Figure 1C). Western blot analysis of TREM‐1 expression in in vivo HFD‐fed mice and in vitro HepG2/PMH showed as a doublet as shown in Figure 1A and 1C, thus indicating that TREM‐1 was part of physiological response to lipotoxicity in NAFLD.
Figure 1.
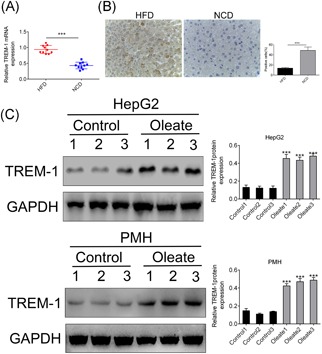
TREM‐1 was part of physiological response to lipotoxicity in NAFLD. A, qRT‐PCR analysis of TREM‐1 in the livers of HFD‐fed vs NCD‐fed mice. B, Immunohistochemistry analysis of TREM‐1 in the livers of HFD‐fed vs NCD‐fed mice. C, Western blot analysis of TREM‐1 in the OA‐induced HepG2/PMH vs control cells. ***P < 0.001. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HFD, high‐fat diet; NAFLD, nonalcoholic fatty liver disease; NCD, normal control diet; OA, oleic acid; PMH, primary mouse hepatocytes; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; TREM‐1, triggering receptor expressed on myeloid cells‐1
3.2. TREM‐1 regulated inflammatory cytokines and lipid accumulation
To detect whether TREM‐1 could affect the inflammatory response and lipid accumulation in NAFLD or not, we successfully generated four stable cell lines for overexpression with pcDNA‐TREM‐1 or knockdown TREM‐1 with siTREM‐1 1# and siTREM‐1 2# in both HepG2 and PMH cell lines confirmed by the means of qRT‐PCR (Figure 2A) and Western blot analysis (Figure 2B). We then measured the messenger RNA (mRNA) levels of inflammatory factors IL‐1β, IL‐6, TNF‐α, IFN‐γ, MCP‐1, and MIP‐1α in the four stable cell lines using qRT‐PCR. After treatment with 5 mmol/L OA for the indicated times, overexpression of TREM‐1 promoted proinflammatory cytokines IL‐1β, IL‐6, TNF‐α, IFN‐γ, MCP‐1, and MIP‐1α secretion in both HepG2 and PMH cells (P < 0.001) (Figure 2C), while knockdown of TREM‐1 decreased them (Figure 2C). These results were in line with the well‐established proinflammation function of TREM‐1, and knockdown TREM‐1 could also inhibit the inflammatory reaction in response to OA stimulation too.
Figure 2.
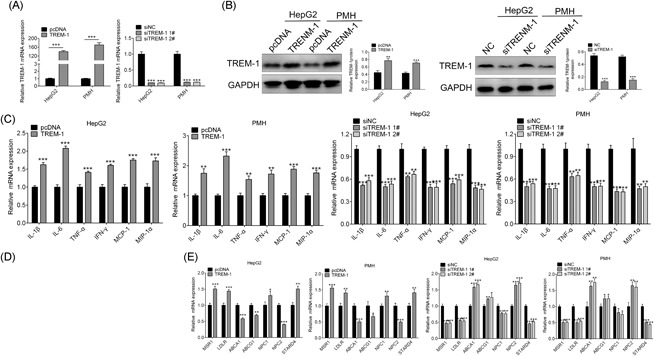
TREM‐1 regulated inflammatory cytokines and lipid accumulation. qRT‐PCR analysis of TREM‐1 mRNA in stable cell lines of HepG2 and PMH treated with OA that either overexpression or knockdown of TREM‐1. Western blot analysis of TREM‐1 protein in stable cell lines of HepG2 and PMH treated with OA that either overexpression or knockdown of TREM‐1. qRT‐PCR analysis of IL‐1β, IL‐6, TNF‐α, IFN‐γ, MCP‐1, and MIP‐1α mRNA in stable cell lines of HepG2 and PMH treated with OA that either overexpression or knockdown of TREM‐1. Oil Red O staining of HepG2 and PMH cells treated with OA that either overexpression or knockdown of TREM‐1. qRT‐PCR analysis of MSR1, LDLR, ABCA1, ABCG1, NPC1/2, and STARD4 mRNA in stable cell lines of HepG2 and PMH treated with OA that either overexpression or knockdown of TREM‐1. **P < 0.01 and ***P < 0.001. ABCA1, ATP‐binding cassette transporter‐1; ABCG1, ATP‐binding cassette sub‐family G member 1; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HFD, high‐fat diet; IFN, interferon; IL, interleukin; LDLR, low‐density lipoprotein receptor; MCP‐1, monocyte chemoattractant protein‐1; MIP‐1α, macrophage inflammatory protein‐1α; mRNA, messenger RNA; MSR1, macrophage scavenger receptor 1; NAFLD, nonalcoholic fatty liver disease; NC, negative control; NCD, normal control diet; NPC1, Niemann‐Pick disease, type C1; NPC2, NPC intracellular cholesterol transporter 2; OA, oleic acid; PMH, primary mouse hepatocytes; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; siNC, small interfering negative control; STARD4, StAR‐related lipid transfer protein 4; TNF‐α, tumor necrosis factor‐α; TREM‐1, triggering receptor expressed on myeloid cells‐1
We then studied whether TREM‐1 could increase lipids accumulation in HepG2 and PMH cells or not. Overexpression of TREM‐1 in HepG2 and PMH cells showed more lipid droplet accumulation compared with the control cells, while less lipid droplet in siTREM‐1 treated cells through Oil Red O staining (Figure 2D). Along with their morphological presentation, overexpression of TREM‐1 increased the expression of MSR1 and LDLR, which was involved in mediating the endocytosis of modified low‐density lipoproteins (LDL)24 or cholesterol‐rich LDL25 for the amplified uptake of fatty acid, as well as NPC1, which mediated intracellular cholesterol trafficking, STARD4, which linked to the movement of cholesterol to the endoplasmic reticulum (Figure 2E), while decreased in knockdown of TREM‐1 by siTREM‐1 (Figure 2E). ABCA1 and ABCG1, involved in cholesterol and phospholipids export and may regulate cellular lipid homeostasis,26 and NPC2 were decreased in overexpression of TREM‐1, and increased in siTREM‐1 (Figure 2E). These findings demonstrated that TREM‐1 could induce lipids deposition in hepatocytes and plays a role as positive regulator for lipogenesis and dysregulated cholesterol metabolism in hepatocytes.
3.3. TREM‐1 is a positive regulator of OA‐induced PI3K/AKT and NF‐κB activation
We tried to analyze PI3K/AKT and NF‐κB signal pathways involved in NAFLD in HepG2 and PMH cells where TREM‐1 was stably overexpressed. Overexpression of TREM‐1 showed activation of PI3K/AKT signal pathway in response to OA with the significant upregulation of p‐AKT compared with the control (P < 0.001) (Figure 3A). On the other hand, treatment with AKT inhibitor (1 μmol/L; MK‐2206) relieved the promotion of PI3K/AKT by overexpression of TREM‐1 (Figure 3A). In addition, activation of NF‐κB in response to overexpression of TREM‐1 was indicated by higher phospho‐p65 (p‐p65) levels (Figure 3B). Treatment with NF‐κB inhibitor (10 μmol/L PDTC) also relieved the promotion of NF‐κB by overexpression of TREM‐1 (Figure 3B). These findings support the canonical role played by TREM‐1 as an accelerator of PI3K/AKT and NF‐κB signal pathways, even in response to OA stimulation. Taken together, TREM‐1 induced OA‐induced inflammatory response and lipid accumulation via enhancing PI3K/AKT and NF‐κB activation.
Figure 3.
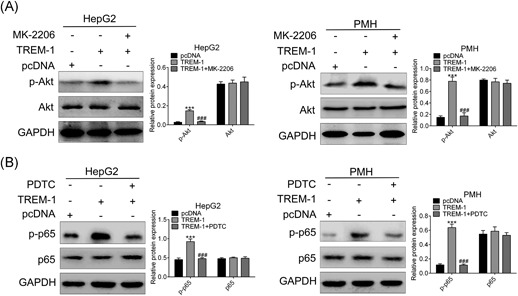
TREM‐1 is a positive regulator of OA‐induced PI3K/AKT and NF‐κB activation. The effect of AKT inhibitor MK‐2206 and TREM‐1 overexpression on expression of AKT and p‐AKT in both HepG2 and PMH cells was detected by Western blot analysis. The effect of NF‐κB inhibitor PDTC and TREM‐1 overexpression on expression of p65 and p‐p65 in both HepG2 and PMH cells was detected by Western blot analysis. ***P < 0.001 and ### P < 0.001. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; NF‐κB, nucleear factor‐κB; PDTC, pyrrolidine dithiocarbamate; PMH, primary mouse hepatocytes; TREM‐1, triggering receptor expressed on myeloid cells‐1
3.4. Knockdown of TREM‐1 alleviates inflammation in HFD‐fed mice via regulating NF‐κB
In vitro cell model has confirmed the proinflammatory function of TREM‐1 through NF‐κB signal pathway, we then decided to detect the in vivo effect of TREM‐1 on inflammation of NAFLD. The HFD‐fed mice were injected with supernatant in cell culture of shRNAs of TREM‐1 (shTREMs). By the means of qRT‐PCR (Figure 4A) and Western blot analysis (Figure 4B), the efficiency of shTREMs was detected, and suggested significant downregulation of TREM in Lv‐shTREM‐1, Lv‐shTREM‐2 and Lv‐shTREM‐3 compared with the Lv‐shRNA group (P < 0.001). With the more efficiency, Lv‐shTREM‐2 and Lv‐shTREM‐3 were chosen for the next experiments. Then, the expression of p‐p65 was detected for the Lv‐shTREM‐2 and Lv‐shTREM‐3 treatment, which showed a dramatically decrease compared with the Lv‐shRNA (P < 0.001) (Figure 4C), consistent with intraperitoneal injection of NF‐κB inhibitor PDTC (Figure 4C), suggesting that knockdown of TREM‐1 could reduce activation of NF‐κB as its inhibitor. Proinflammatory cytokines IL‐1β, IL‐6, TNF‐α, IFN‐γ, MCP‐1, and MIP‐1α secretion of liver tissues were also decreased in both Lv‐shTREM‐2/Lv‐shTREM‐3 and PDTC (Figure 4D). Histological examination of liver tissues through H&E staining showed structural abnormalities and fatty degeneration in Lv‐shRNA group, while treatment with Lv‐shTREM‐2/Lv‐shTREM‐3 and PDTC showed marked improvement in tissue morphology and architecture compared with Lv‐shRNA (Figure 4E), especially in Lv‐shTREM‐2. Taken together, knockdown of TREM‐1 could alleviate inflammation in NAFLD via regulating NF‐κB.
Figure 4.
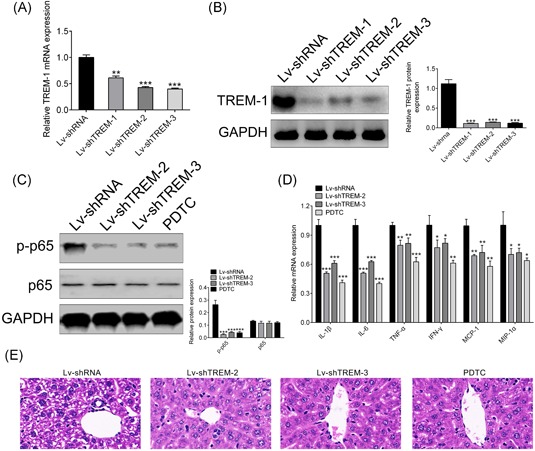
Knockdown of TREM‐1 alleviates inflammation in HFD‐fed mice via regulating NF‐κB. qRT‐PCR analysis of TREM‐1 mRNA in HFD‐fed mice injected with supernatant in cell culture of shTREMs (Lv‐shTREMs) that knockdown of TREM‐1. Western blot analysis of TREM‐1 protein in HFD‐fed mice injected with supernatant in cell culture of shTREMs (Lv‐shTREMs) that knockdown of TREM‐1. The effect of NF‐κB inhibitor PDTC and Lv‐shTREMs on expression of p65 and p‐p65 in HFD‐fed mice was detected by Western blot analysis. qRT‐PCR analysis of IL‐1β, IL‐6, TNF‐α, IFN‐γ, MCP‐1 and MIP‐1α mRNA in HFD‐fed mice injected with Lv‐shTREMs that knockdown of TREM‐1. H&E staining analysis of the effect of NF‐κB inhibitor PDTC and Lv‐shTREMs on tissue morphology and architecture in HFD‐fed mice. *P < 0.05, **P < 0.01, and ***P < 0.001. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HFD, high‐fat diet; H&E, hematoxylin and eosin; IFN‐γ, interferon‐γ; IL, interleukin; Lv, lentiviral vector; MCP‐1, monocyte chemoattractant protein‐1; MIP‐1α, macrophage inflammatory protein‐1α; mRNA, messenger RNA; PDTC, pyrrolidine dithiocarbamate; PMH, primary mouse hepatocytes; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; shTREM, short hairpin RNAs of TREM‐1; TNF‐α, tumor necrosis factor‐α; TREM‐1, triggering receptor expressed on myeloid cells‐1
3.5. Knockdown of TREM‐1 alleviates lipid accumulation in HFD‐fed mice via regulating PI3K/AKT
In vitro cell model also has confirmed the positive regulator function of TREM‐1 in lipogenesis through PI3K/AKT signal pathway, we then decided to detect the in vivo effect of TREM‐1 on lipid accumulation of NAFLD. The expression of p‐AKT was detected for the Lv‐shTREM‐2 and Lv‐shTREM‐3, which showed a dramatically decrease compared with Lv‐shRNA (P < 0.001) (Figure 5A), consistent with intraperitoneal injection of AKT inhibitor MK‐2206 (Figure 5A), suggesting that knockdown of TREM‐1 could reduce activation of PI3K/AKT just as its inhibitor. Genes involved in uptake of fatty acid MSR1 and LDLR, with cholesterol transporter NPC1 and STARD4, were downregulated in both Lv‐shTREM‐2/Lv‐shTREM‐3 and MK‐2206 (Figure 5B), cholesterol and phospholipids exporters ABCA1 and ABCG1 with NPC2 were upregulated in both Lv‐shTREM‐2/Lv‐shTREM‐3 and MK‐2206 (Figure 5B). Oil Red O staining showed less lipid accumulation in Lv‐shTREM‐2/Lv‐shTREM‐3 and MK‐2206 (Figure 5C). Therefore, all these results suggested that knockdown of TREM‐1 could alleviate lipid accumulation in NAFLD by regulating PI3K/AKT.
Figure 5.
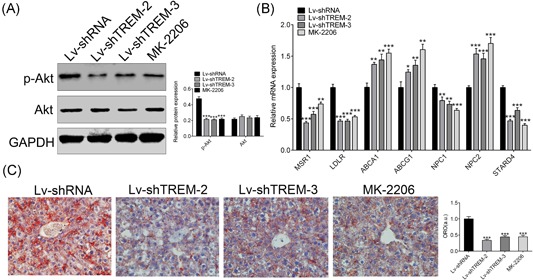
Knockdown of TREM‐1 alleviates lipid accumulation in HFD‐fed mice via regulating PI3K/AKT. The effect of AKT inhibitor MK‐2206 and Lv‐shTREMs on expression of AKT and p‐AKT in HFD‐fed mice was detected by Western blot analysis. qRT‐PCR analysis of MSR1, LDLR, ABCA1, ABCG1, NPC1/2 and STARD4 mRNA in HFD‐fed mice injected with Lv‐shTREMs that knockdown of TREM‐1.Oil Red O staining of liver tissues of HFD‐fed mice treated with MK‐2206 and Lv‐shTREMs. *P < 0.05, **P < 0.01, and ***P < 0.001. ABCA1, ATP‐binding cassette transporter‐1; ABCG1, ATP‐binding cassette sub‐family G member 1; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HFD, high‐fat diet; LDLR, low‐density lipoprotein receptor; Lv, lentiviral vector; mRNA, messenger RNA; MSR1, macrophage scavenger receptor 1; NPC1, Niemann‐Pick disease, type C1; NPC2, NPC intracellular cholesterol transporter 2; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; shTREM, short hairpin RNAs of TREM‐1; STARD4, StAR‐related lipid transfer protein 4; TREM‐1, triggering receptor expressed on myeloid cells‐1
4. DISCUSSION
With changes in people's diet and lifestyle, NAFLD is gradually becoming a worldwide health problem, with up to 80% of obese people have this disease and up to 20% normal‐weight people might develop it.27 Unfortunately, there are no fully proven medications for NAFLD for now and liver transplantation is the only treatment option for endstage NAFLD.28 Thus, there is an urgent need to find effective drugs for NAFLD. In the present study, we demonstrated that inhibition of TREM‐1 attenuated HFD diet‐induced NAFLD by suppressing key regulators of inflammation and lipid accumulation.
In obese individuals, adipocytes turned into hypertrophy and secreted large amount of FFAs.29 On the other hand, the excessive production of FFAs may cause hepatotoxicity and induce NAFLD via several mechanisms beyond direct cytotoxicity.30, 31 In patients with NAFLD, the upregulation of circulating FFAs was confirmed, higher level correlated with disease severity.32, 33 In the present study, we successfully constructed NAFLD the in vivo mice model fed with HFD and in vitro cell model incubated with 5 mmol/L OA, according to obvious lipid accumulation showed by the liver morphology and Oil Red O staining. Analysis of Gene Expression Omnibus (GEO) database found that the expression of TREM‐1 in the liver tissues of NAFLD patients was high, which may be related to the development of NAFLD. Consistent with GEO analysis, significantly higher expression of TREM‐1 in liver biopsies of HFD diet‐induced NAFLD mice and OA‐treated HepG2/PMH cells were found. The elevation of TREM‐1 in liver with steatosis or steatohepatitis could at least partly due to the excessive liver influx of FAs. Previous studies reported that TREM‐1 could be a common stress response of hepatocytes to various insults, such as inflammation and lipid accumulation.13, 19, 22 Therefore, we speculated that TREM‐1 may attribute to inflammation and lipid accumulation during NAFLD development.
“Initial hit” of NAFLD pathogenesis was related to lipid accumulation in livers, which sensitized hepatocytes to the following pathogenic factors, including inflammatory cytokines that promoted hepatocellular damage,34 which resulted from the impairment of cholesterol metabolism and enhancement of FA synthesis.35, 36 We applied well‐accepted NAFLD mice and cell models, and showed that TREM‐1 increased liver lipid droplets and augmented relative gene expression involved in lipid metabolism, and the gene expression were reversed by knocking down of TREM‐1. MSR1 and LDLR for the amplified uptake of FA were upregulated due to TREM‐1 overexpression and downregulated by knocking down of TREM‐1 in our study, consistent with previous study that Ldlr −/− mice with lacking Msr1 significantly reduced hepatic inflammation and lipid oxidation.37 Additionally, our data showed that genes involved in cholesterol metabolism were also modulated by TREM‐1. Cholesterol exporter ABCA1 and ABCG1 were downregulated with TREM‐1 overexpression and upregulated with TREM‐1 knockdown, also fit with the previous study.26 As the previous study, administration of antibody specific for NPC1‐like 1 could ameliorate NAFLD38 and increased StAR expression in steatosis and NASH patients enhanced mitochondrial free cholesterol accumulation and toxicity,39 overexpression of TREM‐1 increased NPC1 and STARD4 thus aggravating lipid accumulation in NAFLD. NPC2 deficiency leads to fatty liver, obesity, and metabolic syndrome,40 in accordance with our result that overexpression of TREM‐1 decreased NPC2 in NAFLD. Overall, TREM‐1 has the ability of upregulating lipogenesis related genes and dysregulating genes in cholesterol metabolism. In addition, in vivo oil Red O staining showed less lipid accumulation with knockdown of TREM‐1 by Lv‐shTREM treatment, suggesting that knocking down of TREM‐1 likely ameliorates hepatic steatosis of NAFLD through regulating key regulators of lipid and cholesterol metabolism.
HFD and FFA‐promoted hepatic lipid accumulation can facilitate the development of hepatic inflammation and provide a suitable advanced NAFLD mice model for researchers to study the mechanisms driving NAFLD‐related inflammation.41, 42, 43, 44, 45 As the central participator in the development of NAFLD, proinflammatory cytokines and chemokines recruited inflammatory cells and kill hepatocytes.46 In our study, TREM‐1 could induce production of proinflammatory cytokines IL‐1β, IL‐6, TNF‐α, IFN‐γ, MCP‐1, and MIP‐1α gene expression and secretion from hepatocytes. Otherwise, these “dangerous” signals were downregulated with the knockdown of TREM‐1. This should be taken that TREM‐1 was an important mediator of the lipotoxicity strengthening for production of hepatocyte‐derived proinflammatory cytokines, thus facilitating progression of the possible ensuing inflammation, which promoted the development of NAFLD. These results also supported the proposition that the therapeutic effects of knockdown of TREM‐1 on NAFLD were also associated with downregulating proinflammatory cytokines in HFD mice model, consistent with the in vivo histological examination of liver tissues showed marked improvement in tissue morphology and architecture by Lv‐shTREM treatment.
NF‐κB‐mediated proinflammatory effects47 and PI3K/AKT‐mediated lipid accumulation48 had been considered as the major concern in the development of steatosis progression to steatohepatitis, thus leading to the occurrence of NAFLD. In the present study, as a downstream positive regulator, TREM‐1 induced the activation of the NF‐κB pathway and promoted recruitment of inflammation, thus mediating hepatic lesion function. Together with the evidence that knocking down of TREM‐1 alleviated the inflammation via decreased p‐p65, thus weakening the NF‐κB activation, consistent with the effect of NF‐κB inhibitor PDTC, our results supported previous findings and newly demonstrated that the regulatory effects of TREM‐1 on FFAs‐induced expression of proinflammatory cytokines from hepatocyte were probably through modulation of NF‐κB activation. Through the inhibition of NF‐kB and proinflammatory factors, knocking down of TREM‐1 alleviated hepatic inflammation in HFD‐induced steatohepatitis of NAFLD. On the other hand, in our study, we found p‐AKT expression was elevated under TREM‐1 overexpression. In line with it, we demonstrated that knockdown of TREM‐1 significantly downregulated the expression level of p‐AKT, the same effect as AKT inhibitor MK‐2206. Thus, knocking down of TREM‐1 mediated amelioration of lipid accumulation in hepatocytes probably through downregulation of genes involved in FA metabolism and modulation of PI3K/AKT activation.
To the best of our knowledge, this is the first study determining TREM‐1 regulates lipid accumulation and inflammation in NAFLD via specifically targeting the NF‐κB and PI3K/AKT signal pathway. In summary, in this study, we report that TREM‐1 could be a physiological factor to FFA stimulation in the pathogenesis of NAFLD, and also acted as an important prolipotoxic and proinflammatory mediator through promotion of lipid accumulation and proinflammatory cytokines release. Understanding the regulatory mechanism of TREM‐1 in NAFLD could lead to the identification of useful clinical biomarker or indicator. In addition, a potential link between TREM‐1 and NAFLD suggested that TREM‐1 might be a potential therapeutic target for developing a novel approach to treat NAFLD with more in vivo studies to confirm the therapeutic potential of TREM‐1 in future.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
We declare that this study was done by the researchers listed in this study. All liabilities related with the content of this study will be borne by the authors. SR and JH designed all the experiments and revised the paper. CX and MZ formed the experiments, and XL wrote the paper.
Rao S, Huang J, Shen Z, Xiang C, Zhang M, Lu X. Inhibition of TREM‐1 attenuates inflammation and lipid accumulation in diet‐induced nonalcoholic fatty liver disease. J Cell Biochem. 2019;120:11867‐11877. 10.1002/jcb.28468
Contributor Information
Jingsong Huang, Email: JingsongHuangasd@163.com.
Zhijun Shen, Email: ZhijunShenasd@163.com.
References
REFERENCES
- 1. Käräjämäki AJ, Hukkanen J, Ukkola O. The association of non‐alcoholic fatty liver disease and atrial fibrillation: a review. Ann Med. 2018;50(5):371‐380. 10.1080/07853890.2018.1492147 [DOI] [PubMed] [Google Scholar]
- 2. Haas JT, Francque S, Staels B. Pathophysiology and mechanisms of nonalcoholic fatty liver disease. Annu Rev Physiol. 2016;78:181‐205. 10.1146/annurev-physiol-021115-105331 [DOI] [PubMed] [Google Scholar]
- 3. Tomeno W, Kawashima K, Yoneda M, et al. Non‐alcoholic fatty liver disease comorbid with major depressive disorder: The pathological features and poor therapeutic efficacy. J Gastroenterol Hepatol. 2015;30(6):1009‐1014. 10.1111/jgh.12897 [DOI] [PubMed] [Google Scholar]
- 4. Yeh MM, Brunt EM. Pathological features of fatty liver disease. Gastroenterology. 2014;147(4):754‐764. 10.1053/j.gastro.2014.07.056 [DOI] [PubMed] [Google Scholar]
- 5. French SW, Takahashi H, Wong K, Mendenhall CL. Ito cell activation induced by chronic ethanol feeding in the presence of different dietary fats. Alcohol Alcohol Suppl. 1991;1:357‐361. [PubMed] [Google Scholar]
- 6. Nanji AA. Role of different dietary fatty acids in the pathogenesis of experimental alcoholic liver disease. Alcohol. 2004;34(1):21‐25. 10.1016/j.alcohol.2004.08.005 [DOI] [PubMed] [Google Scholar]
- 7. Li P, Robertson TA, Thorling CA, et al. Hepatic pharmacokinetics of cationic drugs in a high‐fat emulsion‐induced rat model of nonalcoholic steatohepatitis. Drug Metab Dispos. 2011;39(4):571‐579. 10.1124/dmd.110.036806 [DOI] [PubMed] [Google Scholar]
- 8. Lieber CS, Leo MA, Mak KM, et al. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79(3):502‐509. 10.1093/ajcn/79.3.502 [DOI] [PubMed] [Google Scholar]
- 9. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836‐1846. 10.1002/hep.24001 [DOI] [PubMed] [Google Scholar]
- 10. Ratziu V, Goodman Z, Sanyal A. Current efforts and trends in the treatment of NASH. J Hepatol. 2015;62(suppl 1):S65‐S75. 10.1016/j.jhep.2015.02.041 [DOI] [PubMed] [Google Scholar]
- 11. Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol. 2018;53(3):362‐376. 10.1007/s00535-017-1415-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nathan C, Ding A. TREM‐1: a new regulator of innate immunity in sepsis syndrome. Nat Med. 2001;7(5):530‐532. 10.1038/87846 [DOI] [PubMed] [Google Scholar]
- 13. Bleharski JR, Kiessler V, Buonsanti C, et al. A role for triggering receptor expressed on myeloid cells‐1 in host defense during the early‐induced and adaptive phases of the immune response. J Immunol. 2003;170(7):3812‐3818. [DOI] [PubMed] [Google Scholar]
- 14. Haselmayer P, Grosse‐Hovest L, von Landenberg P, Schild H, Radsak MP. TREM‐1 ligand expression on platelets enhances neutrophil activation. Blood. 2007;110(3):1029‐1035. 10.1182/blood-2007-01-069195 [DOI] [PubMed] [Google Scholar]
- 15. Ormsby T, Schlecker E, Ferdin J, et al. Btk is a positive regulator in the TREM‐1/DAP12 signaling pathway. Blood. 2011;118(4):936‐945. 10.1182/blood-2010-11-317016 [DOI] [PubMed] [Google Scholar]
- 16. Wang YS, Li XJ, Zhao WO. TREM‐1 is a positive regulator of TNF‐alpha and IL‐8 production in U937 foam cells. Bosn J Basic Med Sci. 2012;12(2):94‐101. 10.17305/bjbms.2012.2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fortin CF, Lesur O, Fulop T, Jr . Effects of TREM‐1 activation in human neutrophils: activation of signaling pathways, recruitment into lipid rafts and association with TLR4. Int Immunol. 2007;19(1):41‐50. 10.1093/intimm/dxl119 [DOI] [PubMed] [Google Scholar]
- 18. Boufenzer A, Lemarié J, Simon T, et al. TREM‐1 mediates inflammatory injury and cardiac remodeling following myocardial infarction. Circ Res. 2015;116(11):1772‐1782. 10.1161/CIRCRESAHA.116.305628 [DOI] [PubMed] [Google Scholar]
- 19. Subramanian S, Pallati PK, Sharma P, Agrawal DK, Nandipati KC. TREM‐1 associated macrophage polarization plays a significant role in inducing insulin resistance in obese population. J Transl Med. 2017;15(1):85 10.1186/s12967-017-1187-7 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20. Zhou J, Chai F, Lu G, et al. TREM‐1 inhibition attenuates inflammation and tumor within the colon. Int Immunopharmacol. 2013;17(2):155‐161. 10.1016/j.intimp.2013.06.009 [DOI] [PubMed] [Google Scholar]
- 21. Tang J, Dong Q. Knockdown of TREM‐1 suppresses IL‐1beta‐induced chondrocyte injury via inhibiting the NF‐kappaB pathway. Biochem Biophys Res Commun. 2017;482(4):1240‐1245. 10.1016/j.bbrc.2016.12.019 [DOI] [PubMed] [Google Scholar]
- 22. Zysset D, Weber B, Rihs S, et al. TREM‐1 links dyslipidemia to inflammation and lipid deposition in atherosclerosis. Nat Commun. 2016;7:13151 10.1038/ncomms13151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Severgnini M, Sherman J, Sehgal A, et al. A rapid two‐step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology. 2012;64(2):187‐195. 10.1007/s10616-011-9407-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Al‐Khami AA, Rodriguez PC, Ochoa AC. Metabolic reprogramming of myeloid‐derived suppressor cells (MDSC) in cancer. Oncoimmunology. 2016;5(8):e1200771 10.1080/2162402X.2016.1200771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Demers A, Samami S, Lauzier B, et al. PCSK9 induces CD36 degradation and affects long‐chain fatty acid uptake and triglyceride metabolism in adipocytes and in mouse liver. Arterioscler Thromb Vasc Biol. 2015;35(12):2517‐2525. 10.1161/ATVBAHA.115.306032 [DOI] [PubMed] [Google Scholar]
- 26. Min HK, Kapoor A, Fuchs M, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15(5):665‐674. 10.1016/j.cmet.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11‐20. 10.1038/nrgastro.2017.109 [DOI] [PubMed] [Google Scholar]
- 28. Musso G, Cassader M, Gambino R. Non‐alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov. 2016;15(4):249‐274. 10.1038/nrd.2015.3 [DOI] [PubMed] [Google Scholar]
- 29. Estadella D, da Penha Oller do Nascimento CM, Oyama LM, Ribeiro EB, Dâmaso AR, de Piano A. Lipotoxicity: effects of dietary saturated and transfatty acids. Mediators Inflamm. 2013;2013:10. 10.1155/2013/137579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Joshi‐Barve S, Barve SS, Amancherla K, et al. Palmitic acid induces production of proinflammatory cytokine interleukin‐8 from hepatocytes. Hepatology. 2007;46(3):823‐830. 10.1002/hep.21752 [DOI] [PubMed] [Google Scholar]
- 31. Meli R, Mattace Raso G, Calignano A. Role of innate immune response in non‐alcoholic fatty liver disease: metabolic complications and therapeutic tools. Front Immunol. 2014;5:177 10.3389/fimmu.2014.00177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tavares de Almeida I, Cortez‐Pinto H, Fidalgo G, Rodrigues D, Camilo ME. Plasma total and free fatty acids composition in human non‐alcoholic steatohepatitis. Clin Nutr. 2002;21(3):219‐223. [DOI] [PubMed] [Google Scholar]
- 33. Nehra V, Angulo P, Buchman AL, Lindor KD. Nutritional and metabolic considerations in the etiology of nonalcoholic steatohepatitis. Dig Dis Sci. 2001;46(11):2347‐2352. [DOI] [PubMed] [Google Scholar]
- 34. Day CP, James OFW. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114(4):842‐845. [DOI] [PubMed] [Google Scholar]
- 35. Banini BA, Sanyal AJ. Nonalcoholic fatty liver disease: epidemiology, pathogenesis, natural history, diagnosis, and current treatment options. Clin Med Insights Ther. 2016;8:75‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benedict M, Zhang X. Non‐alcoholic fatty liver disease: an expanded review. World J Hepatol. 2017;9(16):715‐732. 10.4254/wjh.v9.i16.715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bieghs V, Wouters K, van Gorp PJ, et al. Role of scavenger receptor A and CD36 in diet‐induced nonalcoholic steatohepatitis in hyperlipidemic mice. Gastroenterology. 2010;138(7):2477‐2486. 2486 e2471‐2473. 10.1053/j.gastro.2010.02.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bae JS, Park JM, Lee J, et al. Amelioration of non‐alcoholic fatty liver disease with NPC1L1‐targeted IgY or n‐3 polyunsaturated fatty acids in mice. Metabolism. 2017;66:32‐44. 10.1016/j.metabol.2016.10.002 [DOI] [PubMed] [Google Scholar]
- 39. Caballero F, Fernández A, De Lacy AM, Fernández‐Checa JC, Caballería J, García‐Ruiz C. Enhanced free cholesterol, SREBP‐2 and StAR expression in human NASH. J Hepatol. 2009;50(4):789‐796. 10.1016/j.jhep.2008.12.016 [DOI] [PubMed] [Google Scholar]
- 40. Jelinek D, Millward V, Birdi A, Trouard TP, Heidenreich RA, Garver WS. Npc1 haploinsufficiency promotes weight gain and metabolic features associated with insulin resistance. Hum Mol Genet. 2011;20(2):312‐321. 10.1093/hmg/ddq466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen R, Wang Q, Song S, Liu F, He B, Gao X. Protective role of autophagy in methionine‐choline deficient diet‐induced advanced nonalcoholic steatohepatitis in mice. Eur J Pharmacol. 2016;770:126‐133. 10.1016/j.ejphar.2015.11.012 [DOI] [PubMed] [Google Scholar]
- 42. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54(1):133‐144. 10.1002/hep.24341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Imajo K, Yoneda M, Kessoku T, et al. Rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Int J Mol Sci. 2013;14(11):21833‐21857. 10.3390/ijms141121833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Willebrords J, Pereira IVA, Maes M, et al. Strategies, models and biomarkers in experimental non‐alcoholic fatty liver disease research. Prog Lipid Res. 2015;59:106‐125. 10.1016/j.plipres.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bian Z, Peng Y, You Z, et al. CCN1 expression in hepatocytes contributes to macrophage infiltration in nonalcoholic fatty liver disease in mice. J Lipid Res. 2013;54(1):44‐54. 10.1194/jlr.M026013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145‐171. 10.1146/annurev-pathol-121808-102132 [DOI] [PubMed] [Google Scholar]
- 47. Li L, Hai J, Li Z, et al. Resveratrol modulates autophagy and NF‐kappaB activity in a murine model for treating non‐alcoholic fatty liver disease. Food Chem Toxicol. 2014;63:166‐173. 10.1016/j.fct.2013.08.036 [DOI] [PubMed] [Google Scholar]
- 48. Zhao S, Zhu L, Duan H, et al. PI3K/Akt pathway mediates high glucose‐induced lipid accumulation in human renal proximal tubular cells via spliced XBP‐1. J Cell Biochem. 2012;113(10):3288‐3298. 10.1002/jcb.24207 [DOI] [PubMed] [Google Scholar]