

Abstract
Neuropilin 1 (NRP1) is a transmembrane glycoprotein that acts as a co‐receptor for multiple extracellular ligands and typically performs growth‐promoting functions in cancer cells. Accumulating evidence indicates that NRP1 is upregulated, and may be an independent predictor of cancer relapse and poor survival, in many cancer types, including non‐small cell lung cancer (NSCLC). Recent evidence suggests that NRP1 affects tumour cell viability via the epidermal growth factor receptor (EGFR) and Erb‐B2 receptor tyrosine kinase 2 (ErbB2) signalling pathways in venous endothelial cells and in multiple cancer cells. In the present study, we aimed to evaluate the role of NRP1 in NSCLC tumourigenesis and to explore a new post‐transcriptional mechanism of NRP1 regulation via a microRNA that mediates EGFR signalling regulation in lung carcinogenesis. The results showed that miR‐338‐3p is poorly expressed and NRP1 is overexpressed in NSCLC tissues relative to their levels in adjacent noncancerous tissues. Luciferase reporter assays, quantitative real‐time reverse transcription PCR, and Western blot analyses showed that NRP1 is a direct target of miR‐338‐3p. Overexpression of miR‐338‐3p in NSCLC cell lines inhibited cell proliferation in vitro and in vivo. Moreover, cell migration and invasion were inhibited by miR‐338‐3p overexpression. These effects occurred via the EGF signalling pathway. Our data revealed a new post‐transcriptional mechanism by which miR‐338‐3p directly targets NRP1; this mechanism plays a role in enhancing drug sensitivity in EGFR wild‐type patients with NSCLC.
Keywords: epidermal growth factor receptor, EGFR, miR‐338‐3p, Neuropilin 1, non‐small cell lung cancer, NSCLC
1. INTRODUCTION
Lung cancer is the leading cause of cancer‐related death worldwide, and approximately 85% of all lung cancers are non‐small cell lung cancer (NSCLC).1, 2 Despite improvements in therapeutic strategies, the survival, and outcome of patients with NSCLC have not changed dramatically.3 At diagnosis, most patients have advanced stage disease that is past the optimal treatment period. Therefore, understanding the pathogenesis of NSCLC and identifying new treatment targets are important.
Neuropilins (NRPs) are multifunctional proteins involved in development, immunity, and cancer. Neuropilin 1 (NRP1) and its homologue neuropilin 2 (NRP2) are coreceptors that interact with multiple growth factors.4 A study by Kawakami et al demonstrated that the expression level of the NRP1 gene in neoplastic tissue was higher than that in extra neoplastic lung tissue; 55 of 68 NSCLC specimens were positive for NRP1 gene expression (80.9%).5 Another study reported that patients with high NRP1 expression had shorter disease‐free and overall survival times compared with patients with low NRP1 expression.6 In addition, recent evidence suggests that NRP1 affects tumor cell viability via the epidermal growth factor receptor (EGFR) and Erb‐B2 receptor tyrosine kinase 2 (ErbB2) signalling pathways in venous endothelial cells and in multiple cancer cells.7, 8 A molecular biomarker that predicts the efficacy of an EGFR‐tyrosine kinase inhibitor(s) (TKI(s)) in patients with lung cancer with wild‐type EGFR has yet to be established. However, some patients with lung cancer with wild‐type EGFR benefit from EGFR‐TKI therapy,9, 10 possibly because resistance to EGFR‐TKIs can be mediated through multiple signalling pathways that converge upon cap‐dependent translation in NSCLC cells expressing wild‐type EGFR.11, 12 Interestingly, our previous study showed that CD73 affected the efficacy of EGFR‐targeted therapies in NSCLC cells with wild‐type EGFR.13 Thus, based on the literature, we hypothesized that NRP1 plays a role in the EGF signalling pathway and that knockdown of NRP1 expression might sensitize NSCLC cells to therapeutic agents. To determine whether knockdown of NRP1 expression could sensitize NSCLC cells to EGFR‐TKI, we assessed the viability of NRP1‐silenced and control cells exposed to gefitinib. Our data showed that NRP1 inhibition significantly improved the effects of EGFR‐TKIs in vitro.
As the most abundant family of small single‐stranded noncoding RNA gene products, microRNAs (miRNAs) play roles in the maintenance of cellular homeostasis.14 MiRNAs are critical in attenuating the stability and translation of mRNAs via base pairing to partially complementary sites in the 3′ untranslated region (3′‐UTR) of their target genes and are involved in multiple physiological and pathological processes.14, 15 Recently, dysregulation of miRNAs has been shown to be associated with tumour growth, metastasis, diagnosis, and prognosis.16, 17, 18, 19 For example, a comprehensive expression analysis of numerous miRNAs reflected the developmental lineage and differentiation state of human cancers, including lung cancer.20 Studies have shown that the prognosis of cancer is closely related to the altered expression of miRNAs in cancer tissues and in specific expression signatures or panels,21 which can also be used to classify human cancers and distinguish tumour subtypes.22 In particular, accumulating evidence indicates that miRNAs are closely related to the development of human lung cancer.23, 24 We previously used miRNA arrays to identify miRNAs that might affect various cellular pathways and biological processes. Among the identified miRNAs, microRNA‐338‐3p was significantly downregulated in NSCLC tissues.25 In the present study, to identify further novel targets of miR‐338‐3p that might play an important role in NSCLC, we predicted its target mRNAs using computational algorithms. Interestingly, miR‐338‐3p was predicted to bind to the 3′‐UTR of NRP1 mRNA (encoding neuropilin 1), indicating that miR‐338‐3p might be involved in regulating NRP1 and the NRP1‐mediated EGF signalling pathway during lung cancer progression.
In the present study, we evaluated the role of NRP1 in NSCLC tumourigenesis and explored the possible role of miR‐338‐3p in the regulation of NRP1 expression. We found that the regulation of NRP1 by miR‐338‐3p affects EGFR‐TKI‐mediated drug sensitivity in lung carcinogenesis.
2. MATERIAL AND METHODS
2.1. Patients and samples
All participants provided written informed consent for the whole study. Following approval by the Ethics Committee of the First Affiliated Hospital of Soochow University (Suzhou, China), a group of 55 patients diagnosed with NSCLC were recruited consecutively from the First Affiliated Hospital of Soochow University from March 2009 to December 2013. The patients were diagnosed with NSCLC based on their histological and pathological characteristics, according to the Revised International System for Staging Lung Cancer. They had not undergone chemotherapy or radiotherapy before tissue sampling. Tissue samples were snap frozen and stored in a cryofreezer at −80°C.
2.2. Gene expression and survival analysis
The oncomine database (https://www.oncomine.org) was selected to compare NRP1 expression between the NSCLC group and the normal control group (adjusted P < 0.05 and an absolute log2 fold change >2). We also used the Gene Expression Profiling Interactive Analysis (GEPIA) database (http://gepia.cancer-pku.cn/index.html), which performs overall survival (OS) or disease free survival (DFS, also called relapse‐free survival and RFS) analysis based on gene expression. GEPIA uses the log‐rank test (also known as the Mantel–Cox test) for hypothesis testing. Cohort thresholds can be adjusted, and gene‐pairs can be used. GEPIA will generate a survival plot based on user custom input parameters. Using Kaplan–Meier Plotter (http://kmplot.com/), we generated two survival curves to show the association between the expression of NRP1 and the OS of patients with the auto‐select best cut‐off value. The GEO datasets GSE36681 (https://www.ncbi.nlm.nih.gov/gds/) is a public dataset containing 47 paired NSCLC tumors and a normal control group and we extracted the data concerning the expression of miR‐338‐3p between these two groups.
2.3. Cell culture
The human NSCLC cell lines A549, HCC827 (lung adenocarcinoma), H226 (lung squamous carcinoma), and the BEAS‐2B cell line (human immortalized normal epithelial cells) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% foetal bovine serum (FBS) (Gibco, Carlsbad, CA, USA) and l‐glutamine (Invitrogen, Carlsbad, CA) at 37°C in a humidified atmosphere containing 5% CO2. The genetic characteristics of the cells were determined by Beijing Microread Genetics Company using a Goldeneye™ 20A Kit and an ABI 3100 instrument. All cell lines were passaged for less than 3 months and tested in Jan 2016.
2.4. RNA extraction and quantitative real‐time reverse transcription PCR analysis
RNA isolation, cDNA synthesis, and quantitative real‐time reverse transcription PCR (qRT‐PCR) analyses were performed as previously described.13 The primer sequences used for NRP1 mRNA detection were 5′‐GAAAAATGCGAATGGCTGAT‐3′ (forward) and 5′‐AATGGCCCTGAAGACACAAC‐3′ (reverse). The bulge‐loop miRNA qRT‐PCR primer sets (one RT primer and a pair of qPCR primers for each set) that were specific for miR‐338‐3p were designed and synthesized by RiboBio (RiboBio Co. Ltd, Guangzhou, Guangdong, China). The cycle threshold (Ct) values for NRP1 mRNA and miR‐338‐3p were equilibrated to those of ACTB (encoding beta‐actin) mRNA and U6, respectively, which were used as internal controls. The △△Ct method was applied to calculate the relative expression levels of these mRNAs.
2.5. Western blotting analysis
Western blotting analysis was performed as previously described.13 The antibodies used in the analysis were anti‐NRP1 (A‐12), anti‐phospho (p)EGFR (Tyr1068) (1H12), anti‐EGFR (A‐10) (all from Santa Cruz, Santa Cruz, CA), anti‐pAKT (Ser473) (D9E), anti‐AKT, anti‐pFAK (Tyr397) (D20B1), anti‐FAK (D2R2E), anti‐Cyclin D1 (92G2), anti‐MMP2 (D8N9Y), anti‐MMP9 (603H), anti‐Snail (C15D) (all from Cell Signaling Technology, Danvers, MA), anti‐N‐cadherin, anti‐Vimentin (RV202) (both from BD Biosciences, San Jose, CA), anti‐β‐actin and anti‐mouse or anti‐rabbit secondary antibodies (all from Cell Signaling Technology). According to the protein loading marker, we cut the whole membranes into small pieces and incubated them with corresponding specific primary antibody overnight at 4°C. On day 2, after washing four times with 1 × TBST (Tris‐buffered saline–Tween 20), we continued to incubate the membrane pieces with the appropriate secondary antibodies. Image J was used to quantify the band density of the immunoreactive proteins. After we opening our immunoblot images in image J, we transformed the image type to 8‐bit to be recognized and then subtracted the background. Finally, we set the associated measurements and selected our target band to obtain integrated density value for further analysis in Excel.
2.6. Plasmid construction, transient transfection, and luciferase assay
To construct a plasmid containing the NRP1 3′‐UTR fused to the 3′‐end of a luciferase reporter for the luciferase assay, a 220‐bp fragment of the NRP1 3′‐UTR containing the miR‐338‐3p target sites (positions 234–240 and 353–359, as predicted by TargetScan) was selected. The wild‐type (WT) (psiCHECK2‐NRP1‐3′‐UTR) and three mutated fragments (psiCHECK2‐NRP1‐3′‐UTR‐mutant1/mutant2/mutant1&2) of the NRP1 3′‐UTR were directly synthesized (Genewiz, Suzhou, China) and fused to the 3′ end of a luciferase reporter, in the psiCHECK2 dual luciferase vector (Promega, Madison, WI). A549 and H226 cells were plated in a 24‐well plate and co‐transfected with the constructed plasmids together with either the miR‐338‐3p mimics or negative control miRNA (miR‐NC) purchased from RiboBio, using Lipofectamine 2000 (Life Technologies, Carlsbad, CA). Forty‐eight hours later, the cells were collected, and the luciferase activity was measured using a Dual Luciferase Reporter Assay kit (Promega). Each experiment was performed in triplicate.
2.7. Establishment of cell lines with stable NRP1 silencing or overexpression
To establish cell lines with stable NRP1 silencing, two DNA fragments (NRP1 shRNA‐1, 5′‐GATCCGCTACGACCGGCTAGAAATCTTTCAAGAGAAGATTTCTAGCCGGTCGTA GCTTTTTTG‐3′ or NRP1 shRNA‐2, 5′‐GATCCGGGAAACTGGCATATCTATGATTCAA GAGATCATAGATATGCCAGTTTCCCTTTTTTG‐3′) were subcloned into the lentiviral vector pGMLV‐SC5 (Genomeditech, Shanghai, China) digested with the endonucleases BamHI and EcoRI. A scrambled sequence of NRP1 short hairpin RNA (shRNA) (sh‐NC, GATCCGTTCTCCGAACGTGTCACGTTCAAGAGACGTGACACGTTCGGAGAACTTTTTTG) served as the negative control. The NRP1‐silencing construct or negative control was co‐transfected with packaging plasmids into human embryonic kidney 293 cells, which were purchased from the Cell Bank of the Chinese Academy of Sciences, using Lipofectamine 2000 (Invitrogen). Forty‐eight hours later, the cells were infected with the packaged lentiviruses, cultured for 2 days, and then selected with 0.4 µg/mL (NRP1‐silenced) or 2 µg/mL (negative control) puromycin (Sigma‐Aldrich, St. Louis, MO).
To establish a cell line in which NRP1 was stably overexpressed, we subcloned the coding sequence of NRP1 into the pLVX‐IRES‐Neo vector using EcoRI and BamHI endonucleases for expression via a Lenti‐X lentiviral expression system (Clontech, Mountain View, CA). The NRP1 expression construct was co‐transfected with packaging plasmids into human embryonic kidney 293 cells using Lipofectamine 2000 (Invitrogen). The empty vector served as the negative control. After incubation, the packaged lentiviruses were collected and used to infect A549 and H226 cells. After 2 days, stable cells were selected with 400 µg/mL G418 (Amresco, Solon, OH).
2.8. Cell proliferation analysis and drug treatment
The cell proliferation analysis and drug treatment were performed as previously described.13 Briefly, according to the manufacturer's instructions, cell proliferation was examined using a Cell Counting Kit‐8 (CCK‐8; Boster, Wuhan, China). Treated cells or the corresponding negative control cells were seeded in 96‐well plates at a density of 2 × 103 cells per well and grown under normal culture conditions for different durations. The experiment was performed in triplicate. We then assessed cell proliferation using a clonogenic assay. Briefly, cells transfected with the miR‐338‐3p mimics and sh‐NRP1 or sh‐NC were diluted in complete culture medium, and 200 cells were reseeded in a 60‐mm plate. After incubation for 14–20 days, depending on the cell growth rate, foci containing at least 50 cells were stained with Giemsa and counted. Cell viability was measured at several time points (24, 48, and 72 h) according to the manufacturer's instructions. Each experiment was performed in triplicate. For drug treatment, stable NRP1‐knockdown cells were plated in 96‐well plates, and gefitinib and TAE226 (Selleck Chemicals, Houston, TX) were added to the cultures. Cell viability was assessed 72 h after drug treatment.
2.9. Wound healing, migration, and invasion assays
Cell motility was analyzed as previously described.13 Briefly, for the wound‐healing assay, cells were seeded in six‐well plates 48 h after transfection to form a monolayer. The cell monolayer was then scratched across the centre of the well with a fresh 10‐μL pipette tip, washed gently twice with phosphate‐buffered saline (PBS) and replenished with fresh medium. The cells were grown for an additional 24 h, and images were acquired under a microscope (IX73, Olympus). For the cell migration assay, according to the instructions from the manufacturer, 5 × 104 cells in medium containing 1% FBS were seeded into the upper chamber of a Transwell insert, and 800 µL of medium containing 10% FBS was placed into the lower chamber, followed by incubation at 37°C for 24 h. For the invasion assay, the inserts were coated with Matrigel matrix (BD Biosciences, Sparks, MD) diluted in serum‐free medium and incubated at 37°C for 2 h. Finally, the cells were photographed and counted.
2.10. Cell cycle analysis
According to the instructions of the Cell Cycle Analysis kit (Beyotime, Shanghai, China), cells were cultured in six‐well plates. The cells were collected, washed with cold PBS, fixed in 70% ethanol at 4°C for 24 h, washed with cold PBS again, and stained with a propidium iodide (PI)/RNase A mixture. The cells were then incubated in the dark at 37°C for 30 min and analysed using a fluorescence‐activated cell sorting (FACS) FACSCalibur system (Beckman Coulter, Brea, CA).
2.11. Animal experiments
2.11.1. Agomir treatment
Female BALB/c athymic nude mice (4‐6 weeks old and weighing 16‐20 g) were purchased from the Experimental Animal Centre of Soochow University and bred under pathogen‐free conditions. All animal experiments were carried out in accordance with the Soochow University Guide for the Care and Use of Experimental Animals. An miR‐338‐3p agomir and a NC agomir (RiboBio) were directly injected into the tumour, which was formed from implanted A549 cells, at a dose of 2 nmol (in 20 μL of PBS) per mouse every 4 days. After seven treatments, chemically stabilized miRNAs may have markedly improved the pharmacological properties, as described previously.13 The tumor volume (V) was determined by measuring the tumour length (L) and width (W) using Vernier callipers and applying the formula V = (L × W2) × 0.5.
2.11.2. Tumour metastasis model
In the experimental lung metastasis model, cells were resuspended in PBS (1 × 106 cells/100 µL PBS/mouse) before being injected into each mouse (BALB/c, 6 weeks old) via the tail vein on day 0. Eight weeks after tail vein injection, all mice were sacrificed, and the numbers of lung nodules were counted under a microscope after the appropriate tissues were stained with haematoxylin and eosin (H&E).
2.12. Statistical analysis
Differences in NRP1 and miR‐338‐3p expression between NSCLC tissues (T) and adjacent noncancerous lung tissues (N) were analyzed using a paired t‐test (two‐tailed). The clinicopathological characteristics of the patients and the expression levels of mRNA and miRNA in the NSCLC samples were compared using nonparametric tests (the Mann–Whitney U test for comparisons between two groups, and the Kruskal–Wallis test for comparisons among three or more groups). Two‐way analysis of variance was used to determine the difference in cell growth between two groups. In all analyses, P < 0.05 was considered to indicate a statistically significant difference. All statistical analyses were performed using GraphPad Prism 5.02 (GraphPad, San Diego, CA) and SPSS 16.0 (IBM Corp., Armonk, NY) software.
3. RESULTS
3.1. NRP1 is upregulated in NSCLC tissues and cell lines
First, we found that NRP1 mRNA expression was significantly upregulated in lung carcinoma tissues relative to that in normal lung tissues in the public data deposited in the Oncomine database (http://www.oncomine.org, Figures 1A and 1B). Furthermore, using data in the Gene GEPIA (http://gepia.cancer-pku.cn/index.html) database, we found that high NRP1 expression in lung squamous cell carcinoma tissues was significantly associated with poor prognosis (P < 0.05, Figure 1C). To further verify the prognostic role of NRP1 in NSCLC, Kaplan–Meier analyses and log–rank tests were performed. As shown in Figure 1D, high expression of NRP1 was significantly associated with poor survival in patients with NSCLC. Moreover, we detected NRP1 mRNA expression in 55‐paired NSCLC tissues and adjacent noncancerous lung tissues. The results indicated that the NRP1 mRNA levels were significantly higher in NSCLC tissues than in adjacent noncancerous lung tissues (P < 0.05, Figure 1A and Supplemental Table S1). Next, we assessed the NRP1 mRNA and protein levels in three NSCLC cell lines and a bronchial epithelial cell line, BEAS‐2B, using qRT‐PCR and Western blotting (Figure 1D). The results showed that the NRP1 mRNA and protein levels were higher in the NSCLC cell lines than in the BEAS‐2B cell line. Collectively, our data show that NRP1 is upregulated in NSCLC tissues and cell lines.
Figure 1.
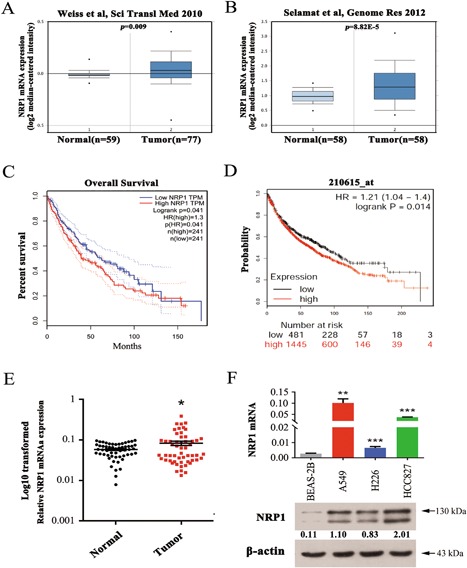
NRP1 is upregulated in NSCLC tissues and cell lines. A and B, Data on NRP1 mRNA expression in lung carcinoma and normal lung tissues from several study groups deposited in the Oncomine database (http://www.oncomine.org). C, Survival analysis of 241 NSCLC patients from the GEPIA database stratified by NRP1 expression. (P = 0.041). D, The effect of the NRP1 expression level on overall survival in 1926 lung cancer patients was analysed, and Kaplan–Meier plots were generated using a Kaplan–Meier plotter (http://www.kmplot.com, Affy ID: 210615_at). E, NRP1 mRNA levels in 55 NSCLC tissues and paired noncancerous lung tissues. F, The level of NRP1 mRNA and protein in human NSCLC cells was measured using qRT‐PCR and western blotting, respectively. N, paired noncancerous lung tissues, T, non‐small cell lung cancer tissues. *P < 0.05; *P < 0.01; ***P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
3.2. The functional role of NRP1 in NSCLC cells
To elucidate the role of NRP1 in NSCLC, we first established A549 and H226 cell lines with stable knockdown of NRP1. The expression of NRP1 mRNA and protein was significantly reduced after the stable transfection of A549 and H226 cells with either of two NRP1 shRNAs (Figure 2A). The CCK‐8 assay showed that the growth of cells with stable knockdown of NRP1 was significantly inhibited compared with that of control cells at 24, 48, and 72 h (Figure 2B). Next, we confirmed these findings via a clonogenic assay (Figure 2C). Moreover, the flow cytometry results for NRP1‐silenced A549 and H226 cells demonstrated a high proportion of G0/G1 phase cells and a low proportion of S and G2/M phase cells compared with these proportions in the control cells (P < 0.05, Figure 2D). These results indicated that NRP1 inhibits cell proliferation in NSCLC cells via its effects on the cell cycle.
Figure 2.
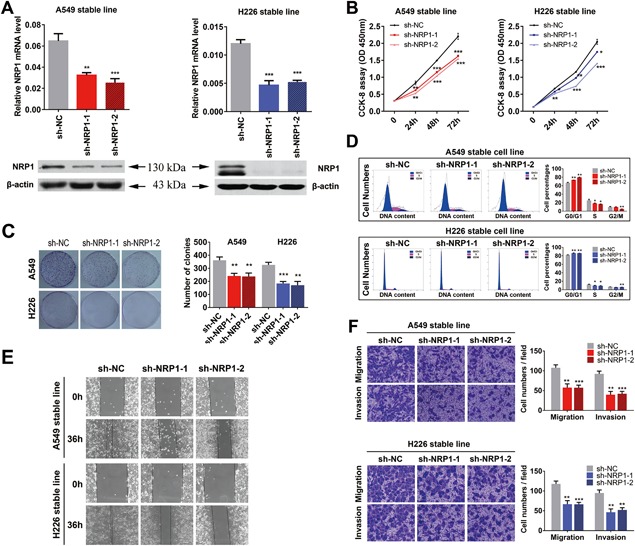
Silencing of NRP1 inhibits NSCLC cell proliferation and motility. A, NRP1 mRNA and protein levels in stably transfected A549 and H226 cells. B, CCK‐8 assay for cell viability in NSCLC cell lines; cell viability was determined at 24, 48, and 72 h. C, Representative images of the clonogenic cell proliferation assay in NSCLC cells. Bar charts showing the clonogenic growth of the cells. D, Flow cytometric analysis of NSCLC cell lines (NRP1‐silenced cells vs. NC cells). Cells were harvested at 72 h after transfection and stained with propidium iodide (PI). The percentage of cells in each cell cycle phase is shown in the inset of each panel; the values are the means ± SDs of three measurements. E, A wound‐healing assay was performed to investigate the effect of NRP1 silencing on cells; the migration rate of cells towards the wounded area was slower for sh‐NRP1‐transfected cells than for the control cells. F, NRP1 silencing inhibited the invasion and migration of NSCLC cells. NRP1‐silenced NSCLC cells were allowed to migrate through an 8‐µM pore size Transwell insert. The cells that migrated were stained and counted in at least three microscopic fields (magnification, ×100). Then, cells were treated as above and allowed to invade through the Matrigel‐coated membrane in the Transwell inserts. Invaded cells were stained and counted under a light microscope. *P < 0.05; **P < 0.01; ***P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
A wound‐healing assay was performed to observe the effects of sh‐NRP1 transfection on the migration of NSCLC cells. sh‐NRP1‐transfected A549 and H226 cells migrated toward the scratch more slowly than the control cells (Figure 2E). A Transwell assay with the stable NSCLC cell lines further indicated that the loss of NRP1 expression considerably suppressed the migratory and invasive abilities of NSCLC cells (Figure 2F).
To further investigate the function of NRP1 in NSCLC cells, we established A549 and H226 cell lines with stable overexpression of NRP1 (Figure 3A). High levels of NRP1 mRNA and protein were consistently observed in cells overexpressing NRP1. The CCK‐8 and clonogenic assays showed that the growth of cells overexpressing NRP1 was significantly increased compared with that of the control cells (Figure 3B‐D). Moreover, the wound‐healing and Transwell assays showed that overexpression of NRP1 enhanced the migratory and invasive abilities of A549 and H226 cells (Figure 3E‐G).
Figure 3.
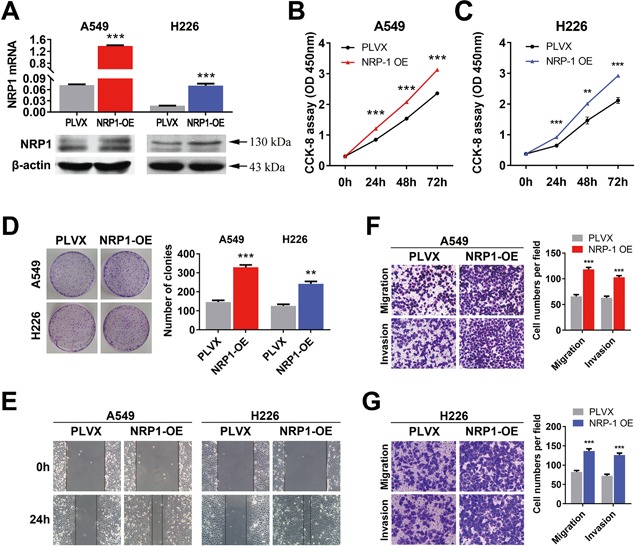
Increasing the level of NRP1 promotes NSCLC cell proliferation and motility. A, NRP1 mRNA and protein levels in stable cell lines overexpressing NRP1 (NRP1‐OE). B and C, CCK‐8 assay for NSCLC cell viability at 24, 48, and 72 h. C, Representative images of the clonogenic cell proliferation assay in NRP1‐OE and control cells. Bar charts showing the clonogenic growth of the cells. D, Representative images of the clonogenic cell proliferation assay in NSCLC cells. Bar charts showing the clonogenic growth of the cells. E, A wound‐healing assay was performed to investigate the effect of NRP1 overexpression (NRP1‐OE) on cells; the migration rate of the cells towards the wounded area was faster for NRP1‐OE cells than for control cells. F and G, Overexpressing NRP1 promotes the invasion and migration of NSCLC cells. NRP1‐OE NSCLC cells were allowed to migrate through an 8‐µM pore size Transwell insert. The cells that migrated were stained and counted in at least three microscopic fields. Then, the cells were treated as above and allowed to invade through the Matrigel‐coated membrane in the Transwell inserts. Invaded cells were stained and counted under a light microscope. The values are the means ± SEs of three measurements. *P < 0.05; **P < 0.01; ***P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
3.3. NRP1 promotes NSCLC metastasis in vivo
We next sought to clarify the mechanisms underlying NRP1‐mediated tumor metastasis. NRP1‐silenced and control A549 cells were injected into the tail vein of nude mice. As shown in Supplemental Figure S1, mice injected with NRP1‐silenced A549 cells developed fewer pulmonary metastatic nodules than those injected with the control cells.
3.4. miR‐338‐3p targets NRP1 in NSCLC cells by binding to its 3′‐UTR
MiRNAs are critical for attenuating the stability and translation of mRNAs via base pairing to partially complementary sites in the 3′‐UTR of their target genes and are involved in multiple physiological and pathological processes. First, by searching publicly available databases (TargetScan Human: http://www.targetscan.org/), we identified the 3′‐UTR of NRP1 as having a sequence complementary to miR‐338‐3p (Figure 4A). To validate this prediction, the NRP1 wild‐type (WT) 3′‐UTR (containing the miR‐338‐3p complementary sequence) and NRP1 MUT 3′‐UTR (miR‐338‐3p mutated sequence) constructs were synthesized, and dual luciferase reporter assays were performed in A549 and H226 cells. MiR‐338‐3p significantly inhibited luciferase activity in cells transfected with the wild‐type NRP1 3′‐UTR but had no effect in cells transfected with the mutant1&2 construct (Figure 4B).
Figure 4.
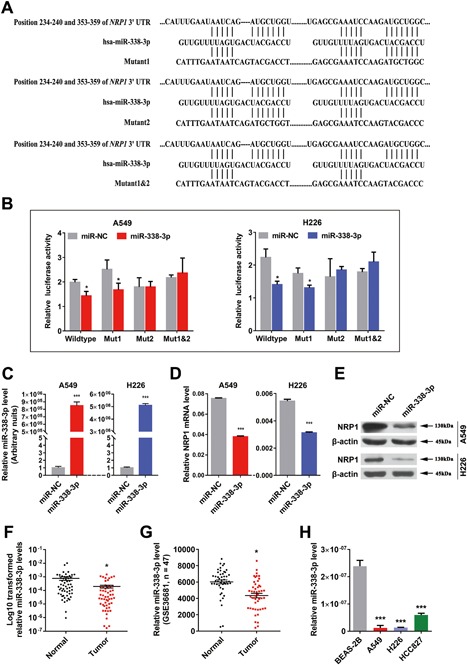
NRP1 is a direct target gene of miR‐338‐3p, an miRNA whose expression is decreased in NSCLC tissues and cell lines. A, Schematic diagram showing the subcloning of the predicted miR‐338‐3p binding site at two positions (234‐240 and 353‐359) in the NRP1 3′‐UTR into the psiCHECK‐2 luciferase vector. The predicted duplex formation between miR‐338‐3p and the wild‐type or mutant miR‐338‐3p binding site on NRP1 is indicated. B, Luciferase activity of the construct containing the wild‐type or mutant NRP1 3′‐UTR reporter gene in A549 and H226 cells co‐transfected with negative control (NC) or miR‐338‐3p. Scrambled sequences were used as the NC. The relative Renilla luciferase activity was determined and normalized to the firefly luciferase activity. C‐E, Expression of miR‐338‐3p and NRP1 in NSCLC cells transfected with the miR‐338‐3p mimics or inhibitor was detected by qRT‐PCR and Western blotting, respectively. F, Relative miR‐338‐3p levels in 55 NSCLC tissues (T) and paired noncancerous lung tissues (N). G, Scatter plot showing the relative miR‐338‐3p expression levels in NSCLC tumor and adjacent normal lung tissues from a public dataset (GSE36681). H, QRT‐PCR analysis of relative miR‐338‐3p expression in human NSCLC cell lines and a human bronchial epithelial cell line. [Color figure can be viewed at wileyonlinelibrary.com]
In addition, we measured NRP1 expression in A549 and H226 cells transfected with the miR‐338‐3p mimics and in control cells transfected with miR‐NC. The expression of miR‐338‐3p was higher in NSCLC cells transfected with the miR‐338‐3p mimics than in cells transfected with miR‐NC (Figure 4C). Consistent with the expression of miR‐338‐3p, the expression of NRP1 was downregulated in cells transfected with the miR‐338‐3p mimics, as determined via qRT‐PCR (Figure 4D) and Western blotting (Figure 4E). Collectively, our data show that NRP1 is a target of miR‐338‐3p in NSCLC cells.
3.5. miR‐338‐3p expression is downregulated in NSCLC tissues and cell lines
Our previous study showed that miR‐338‐3p expression is lower in lung cancer tissues than in noncancerous tissues (Supplemental Figure S2A). Considering that dysregulation of microRNAs is associated with multiple biological processes, we performed Gene Ontology and pathway analyses and found that miR‐338‐3p was predicted to play a significant role in the pathogenesis of cancer (Supplemental Figure S2B). To verify the miRNA array results, we examined the expression of miR‐338‐3p in 55‐paired NSCLC tissues via qRT‐PCR and found that its expression was significantly lower in tumor tissues than in paired noncancerous tissues (Figure 4F and Supplemental Table S1). Furthermore, a public Gene Expression Omnibus dataset (GSE36681) containing 47 NSCLC tissues and 47 normal lung tissues showed that miR‐338‐3p expression was downregulated in human NSCLC tissues (Figure 4G). We then examined miR‐338‐3p expression in three NSCLC cell lines and found that miR‐338‐3p levels were significantly lower in NSCLC cell lines than in BEAS‐2B cells (Figure 4H).
3.6. MiR‐338‐3p overexpression can inhibit NSCLC cell growth, cell cycle progression, and metastasis in vitro
To determine the function of miR‐338‐3p in NSCLC, we induced miR‐338‐3p overexpression using miR‐338‐3p mimics and then evaluated the effect of miR‐338‐3p on cell growth. CCK‐8 assays showed that NSCLC cells overexpressing miR‐338‐3p had significantly lower proliferation abilities than the control cells (Figures 5A and 5B). These results were confirmed using a clonogenic assay (Figure 5C), suggesting that miR‐338‐3p inhibits NSCLC cell proliferation.
Figure 5.
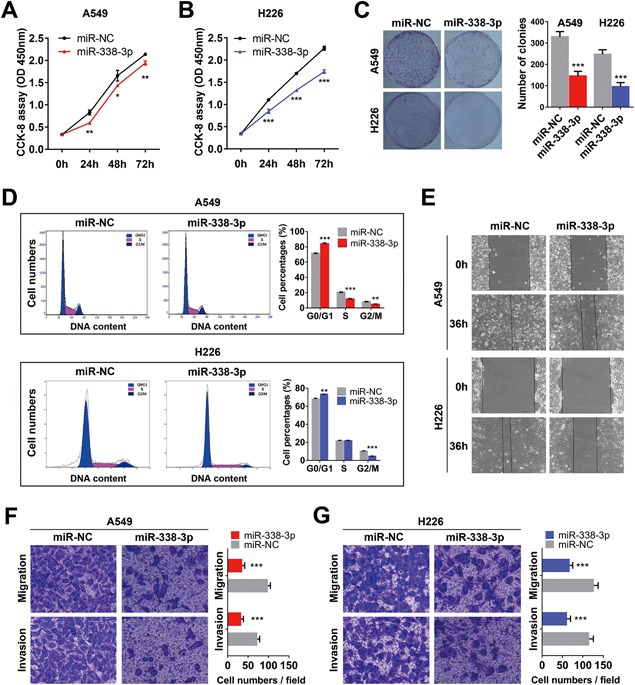
Overexpression of miR‐338‐3p inhibits NSCLC cell proliferation and motility. A and B, CCK‐8 assay of cell viability in NSCLC cell lines transfected with the miR‐338‐3p mimics at 24, 48, and 72 h. C, Representative images of the clonogenic cell proliferation assay in NSCLC cells. Bar charts showing the clonogenic growth of the cells. D, Flow cytometric analysis of NSCLC cell lines (miR‐338‐3p‐ vs miR‐NC‐transfected cells). Cells were harvested at 72 h after transfection and stained with propidium iodide (PI). The percentage of cells in each cell cycle phase is shown in the inset of each panel; the values are the means ± SDs of three measurements. E, A wound‐healing assay was performed to investigate the effect of miR‐338‐3p transfection on cells. The migration rate of the cells toward the wounded area was slower for the cells transfected with the miR‐338‐3p mimics than for the control cells. F and G, Overexpression of miR‐338‐3p inhibited the invasion and migration of NSCLC cells. The A549 and H226 cell lines were transfected with the miR‐338‐3p mimics and allowed to migrate through 8‐µM pore size Transwell inserts. The cells that migrated were stained and counted in at least three microscopic fields (magnification, ×100). Then, cells were treated as previously described and allowed to invade through the Matrigel‐coated membrane in the Transwell inserts. Invaded cells were stained and counted under a light microscope. *P < 0.05; **P < 0.01; ***P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
To determine the mechanism by which miR‐338‐3p suppresses the proliferation of NSCLC cells, we examined the distribution of cell cycle phases using flow cytometry and found that a higher numbers of NSCLC cells overexpressing miR‐338‐3p than negative control cells were arrested in G0/G1 phase (P < 0.05, Figure 5D). These results indicated that miR‐338‐3p inhibits cell proliferation in NSCLC cells via its effects on the cell cycle.
A wound‐healing assay was performed to determine the role of miR‐338‐3p transfection in regulating the migration of A549 and H226 cells. As shown in Figure 5E, the migration rate of the cells transfected with the miR‐338‐3p mimics towards the wounded area was slower than that of the control cells. A Transwell assay using A549 and H226 cells further indicated that overexpression of miR‐338‐3p considerably suppressed the migration ability of A549 (Figure 5F) and H226 cells (Figure 5G). Taken together, these observations suggest that miR‐338‐3p might function as a tumor suppressor in NSCLC.
A rescue experiment was performed to confirm that NRP1 is a functional target of miR‐338‐3p in A549 and H226 cells. First, cell lines stably overexpressing NRP1 were transfected with the miR‐338‐3p mimics or miR‐NC. As shown in Supplemental Figure S3A and B, NRP1 overexpression partially reversed the suppression of cell proliferation, migration and, invasion induced by miR‐338‐3p. Next, we investigated whether the NRP1 gene links miR‐338‐3p with EGFR signalling. The results indicated that pLVX‐transfected cells overexpressing miR‐338‐3p exhibited significantly lower levels of p‐EGFR than miR‐NC‐transfected cells and that the restoration of NRP1 expression in miR‐338‐3p mimic‐transfected NSCLC cells recovered the inhibitory effect on p‐EGFR expression induced by miR‐338‐3p overexpression (Supplemental Figure S3C and D).
3.7. NRP1 and its associated pathways may be useful therapeutic targets
Recent evidence suggests that NRP1 affects tumor cell viability via the EGFR and ErbB2 signalling pathways in venous endothelial cells and multiple cancer cells.7, 8 We thus hypothesized that NRP1 plays a role in the EGF signalling pathway and that knockdown of NRP1 expression might sensitize NSCLC cells to therapeutic agents. First, we assessed the levels of p‐EGFR and its downstream signalling molecules. As shown in Figure 6A, the p‐EGFR, p‐FAK, and p‐AKT levels were significantly lower in NRP1‐silenced cells than in the control cells. Correspondingly, the p‐EGFR, p‐FAK, and p‐AKT levels were significantly higher in NRP1‐overexpressing cells (Figure 6C). Furthermore, in the cell lines with stable NRP1 knockdown, the EGF‐induced increase in p‐EGFR expression was inhibited (Figure 6B). Moreover, our data showed that the p‐EGFR, p‐FAK, and p‐AKT levels were significantly lower in the cell lines transfected with the miR‐338‐3p mimics than in the control cells (Figure 6D).
Figure 6.
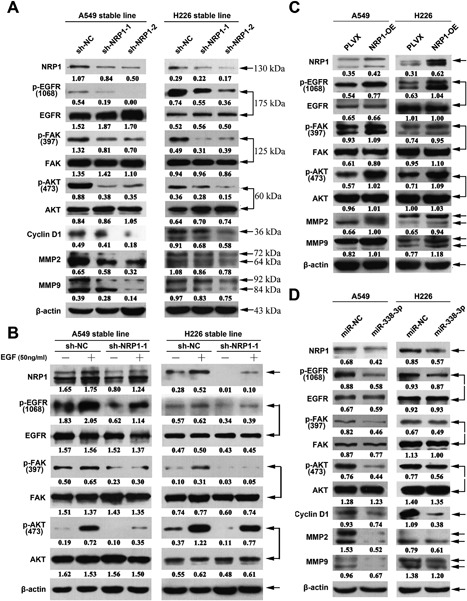
Western blotting analysis of proteins from related signalling pathways in the indicated cells. A, The expression levels of proteins in the NRP1 and EGFR signaling pathways were assessed in stably transformed A549 and H226 cell lines. The levels of p‐EGFR, p‐FAK, p‐AKT, CyclinD1, MMP2, and MMP9 were significantly lower in NRP1‐silenced cell lines than in control cell lines. B, After serum starvation for 24 h, NRP1‐silenced A549 and H226 cells were treated with or without EGF (50 ng/mL) for 30 min. The levels of NRP1, p‐EGFR, EGFR, p‐FAK, FAK p‐AKT, and AKT were analysed using western blotting. C, In contrast to the results for NRP1‐silenced cell lines, the levels of p‐EGFR, p‐FAK, p‐AKT, CyclinD1, MMP2, and MMP9 were significantly higher in NRP1‐overexpressing cell lines than in control cell lines. D, The p‐EGFR, p‐FAK, and p‐AKT levels were significantly lower in the cell lines transfected with the miR‐338‐3p mimics than in the control cell lines. [Color figure can be viewed at wileyonlinelibrary.com]
EGFR‐TKI therapy significantly improves treatment outcomes of patients with lung cancers that harbour EGFR mutations; however, some patients with lung cancer with wild‐type EGFR also benefit from EGFR‐TKI therapy.9, 10 Therefore, we performed the following experiment: A549 and H226 cells (Mock), stable NRP1‐silenced cells, and negative control cells (sh‐NC) were treated with gefitinib or TAE226, an inhibitor of active FAK, which is characterized by phosphorylation at the Y397 residue.26 Cell viability was assessed at 72 h after drug treatment. As shown in Supplemental Figure S4C and D, the viability of untreated A549 cells and sh‐NC treated A549 cells decreased by 50% when exposed to gefitinib. Similar effects were observed when the cells were treated with TAE226 (P < 0.001, Figure 7D). However, no significant changes were observed in control NSCLC cells exposed to either gefitinib or TAE226 (Supplemental Figure S4A and B). Our data seemed to indicate that NRP1 inhibition augments the effects of EGFR‐TKIs.
Figure 7.

MiR‐338‐3p inhibits tumor growth by targeting NRP1 in vivo. A, At the experimental endpoint, tumors treated with the miR‐338‐3p agomir were dissected and photographed as indicated. Growth of A549 xenograft tumors in nude mice treated with the miR‐338‐3p agomir and NC agomir (n = 7). B, The graph shows the tumor growth curves at the time of sacrifice with respect to the baseline measurements and after the administration of 2 nmol miR‐338‐3p agomir or NC agomir per mouse seven times every 4 days. C and D, qRT‐PCR analysis of miR‐338‐3p levels and NRP1 mRNA expression in excised tumours transfected with the miR‐338‐3p agomir or the NC agomir; U6 and β‐actin were used as the internal controls, respectively. E, A working model of the mechanistic interaction of miR‐338‐3p and NRP1 in controlling the EGFR signalling pathway: NRP1 targeted by miR‐338‐3p modulates EGFR signalling in NSCLC. *P < 0.05; **P < 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
3.8. MiR‐338‐3p overexpression inhibits tumour growth in nude mice by targeting NRP1
MiR‐338‐3p is significantly downregulated in NSCLC; therefore, a miR‐338‐3p agomir was used for replacement therapy. As shown in Figures 7A and 7B, tumors treated with the miR‐338‐3p agomir were smaller and weighed less than the control tumors. Moreover, miR‐338‐3p agomir treatment resulted in a significant increase in the level of miR‐338‐3p and a significant decrease in the level of NRP1 (Figures 7C and 7D). These findings indicated that overexpression of miR‐338‐3p inhibits lung cancer cell growth in vivo through NRP1; however, these findings need to be confirmed with a larger sample size.
4. DISCUSSION
Although the survival rate for lung cancer has improved incrementally over the last several decades, the improvements in survival seen in other common malignancies have not been realized in lung cancer, which remains the leading cause of cancer mortality worldwide, including in China.2 The current 5‐year survival rate for lung cancer is a discouraging 15%. Therefore, understanding the molecular mechanisms of cancer development is important to develop effective therapies.
Recently, several studies have directly profiled miRNA expression in lung cancers, and unique groups of miRNAs that either characterize neoplastic tissues or identify patients with poor prognosis have been distinguished.20, 21, 22, 23, 24, 25 Our previous study indicated that miR‐338‐3p expression is significantly decreased in NSCLC,25 consistent with the results of a study by Tan et al.21 Previous research has suggested that miR‐338‐3p functions as a tumor suppressor and is downregulated in breast cancer,27 nasopharyngeal carcinoma,28 hepatocellular carcinoma,29 gastric cancer,30 and lung cancer.31, 32 Recently, attention has focused on the expression of miR‐338‐3p and its target gene; however, few studies have addressed the mechanism of action of miR‐338‐3p, especially its associated signalling transduction.30 In the present study, we first confirmed the significantly decreased expression of miR‐338‐3p in NSCLC reportedly previously. At the cellular level, upregulation of miR‐338‐3p inhibited the proliferation, colony formation, invasion, and migration of NSCLC cells. Moreover, overexpressing miR‐338‐3p in NSCLC cells reduced tumorigenesis in vivo; however, these results need to be confirmed in a larger sample in the future. Taken together, our results suggested a tumor suppressor role for miR‐338‐3p in NSCLC.
NRP1 is a transmembrane glycoprotein that acts as a co‐receptor for numerous extracellular ligands, including class III/IV semaphorins,33 certain isoforms of vascular endothelial growth factor (VEGF),34 transforming growth factor beta (TGF‐β),35 and platelet‐derived growth factor.36 NRP1 is upregulated, and may be an independent predictor of cancer relapse and poor survival, in patients with NSCLC.6 In addition, the synergistic effect of ErbB2 and NRP1 has been observed in venous endothelium.7 Rizzolio et al demonstrated that NRP1‐blocking antibodies and NRP1 silencing could counteract ligand‐induced EGFR activation in cancer cells.8 However, the above studies were unable to demonstrate whether transcriptional regulation events are involved in NRP1 inhibition and whether NRP1 inactivation enhances EGFR‐TKI sensitivity. In the current study, using bioinformatic analysis and luciferase assays, we found that NRP1 is a target gene of miR‐338‐3p, as has been confirmed in different types of cancer.30, 37 Downregulation of NRP1 inhibited the proliferation, colony formation, invasion, and migration of NSCLC cells in vitro. In contrast, upregulation of NRP1 promoted the proliferation, colony formation, invasion, and migration of NSCLC cells.
Although our findings revealed a new post‐transcriptional mechanism of NRP1 regulation via miR‐338‐3p in lung cancer, we cannot exclude the possibility that other target genes of miR‐338‐3p are involved in modulating EGF signalling. Our study focused on the role of the miR‐338‐3p/NRP1/EGFR axis in NSCLC; however, determining whether this complex plays functional roles in other types of cancer would be interesting. Moreover, we observed that the loss of NRP1 considerably suppressed the migratory ability of NSCLC cells. Epithelial‐mesenchymal transition (EMT) is vital for morphogenesis during embryonic development and the conversion of early‐stage tumors into invasive malignancies,38, 39 and accumulating evidence indicates that TGF‐β signalling is a potent inducer of EMT in various cancers, including NSCLC.40, 41, 42 Glinka et al demonstrated that NRP1 is a co‐receptor for TGF‐β1 and augments responses to latent and active TGF‐β in breast cancer.35 Therefore, whether NRP1 can affect TGF‐β signalling in NSCLC requires further verification.
In summary, this study is the first to report that miR‐338‐3p expression is downregulated in NSCLC and is correlated with an increase in NRP1 expression. Furthermore, we found that miR‐338‐3p inhibits NRP1 expression by directly targeting the NRP1 3′‐UTR, thereby repressing NSCLC cell proliferation and mobility. Thus, our findings revealed the mechanistic interaction between miR‐338‐3p and NRP1 in NSCLC carcinogenesis. Importantly, our data seemed to indicate that miR‐338‐3p/NRP1 inhibition could augment the effects of EGFR‐TKIs. Thus, miR‐338‐3p‐mediated downregulation of NRP1 might lead to new therapeutic strategies for NSCLC (Figure 7E).
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Supporting Table S1.
Supporting Figure S1.
Supporting Figure S2.
Supporting Figure S3.
Supporting Figure S4.
ACKNOWLEDGMENTS
ZLD, JJZ, YYZ, WWD, YZ, HCT, YLZ, and HLQ participated in the data collection and analysis. ZLD, JAH, and ZYL participated in the design of the study. ZYL participated in the writing of the manuscript and the interpretation of the data. All authors read and approved the final manuscript. We thank all patients who participated in this study. This work was supported by grants from the National Natural Science Foundation of China (No. 81201575), the Science and Technology Plan Project of Suzhou (No. SYS201612), the Jiangsu Provincial Medical Youth Talent (No. QNRC2016746), the Societal and Developmental Project of Suzhou (No. SS201630), the Foundation of Health Care Rejuvenation by Science and Education (No. KJXW2015002), the Huai'an City Science and Technology Support Program (No. hAS2015013‐4), the Suzhou Key Laboratory for Respiratory Medicine (No. SZS201617), the Clinical Medical Center of Suzhou (No. Szzx201502), the Jiangsu Provincial Key Medical Discipline (No. ZDXKB2016007), and the Clinical Key Specialty Project of China.
Ding Z, Zhu J, Zeng Y, et al. The regulation of Neuropilin 1 expression by miR‐338‐3p promotes non‐small cell lung cancer via changes in EGFR signaling. Molecular Carcinogenesis. 2019;58:1019–1032. 10.1002/mc.22990
Zongli Ding, Jianjie Zhu, Yuanyuan Zeng and Wenwen Du have contributed equally to this work.
Contributor Information
Zeyi Liu, Email: liuzeyisuda@163.com.
Jian‐an Huang, Email: huang_jian_an@163.com.
REFERENCES
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. [DOI] [PubMed] [Google Scholar]
- 2. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. [DOI] [PubMed] [Google Scholar]
- 3. Mulshine JL, Sullivan DC. Clinical practice. Lung cancer screening. N Engl J Med. 2005;352:2714–2720. [DOI] [PubMed] [Google Scholar]
- 4. Prud'homme GJ, Glinka Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget. 2012;3:921–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kawakami T, Tokunaga T, Hatanaka H, et al. Neuropilin 1 and neuropilin 2 co‐expression is significantly correlated with increased vascularity and poor prognosis in nonsmall cell lung carcinoma. Cancer. 2002;10:2196–2201. [DOI] [PubMed] [Google Scholar]
- 6. Hong TM, Chen YL, Wu YY, et al. Targeting neuropilin 1 as an antitumor strategy in lung cancer. Clin Cancer Res. 2007;13:4759–4768. [DOI] [PubMed] [Google Scholar]
- 7. Aghajanian H, Cho YK, Manderfield LJ, et al. Coronary vasculature patterning requires a novel endothelial ErbB2 holoreceptor. Nat Commun. 2016;7:12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rizzolio S, Rabinowicz N, Rainero E, et al. Neuropilin‐1‐dependent regulation of EGF‐receptor signaling. Cancer Res. 2012;72:5801–5811. [DOI] [PubMed] [Google Scholar]
- 9. Cappuzzo F, Ciuleanu T, Stelmakh L, et al. Erlotinib as maintenance treatment in advanced non‐small‐cell lung cancer: a multicentre, randomised, placebo‐controlled phase 3 study. Lancet Oncol. 2010;11:521–529. [DOI] [PubMed] [Google Scholar]
- 10. Zhu CQ, Santos G, Ding K, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in national cancer institute of Canada clinical trials group study BR.21. J Clin Oncol. 2008;26:4268–4275. [DOI] [PubMed] [Google Scholar]
- 11. Patel MR, Jay‐Dixon J, Sadiq AA, et al. Resistance to EGFR‐TKI can be mediated through multiple signaling pathways converging upon cap‐dependent translation in EGFR‐wild type NSCLC. J Thorac Oncol. 2013;9:1142–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raimbourg J, Joalland MP, Cabart M, et al. Sensitization of EGFR wild‐Type non‐Small cell lung cancer cells to EGFR‐Tyrosine kinase inhibitor erlotinib. Mol Cancer Ther. 2017;8:1634–1644. [DOI] [PubMed] [Google Scholar]
- 13. Zhu J, Zeng Y, Li W, et al. CD73/NT5E is a target of miR‐30a‐5p and plays an important role in the pathogenesis of non‐small cell lung cancer. Mol Cancer. 2017;16:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. [DOI] [PubMed] [Google Scholar]
- 15. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bueno MJ, Perez de Castro I, Malumbres M. Control of cell proliferation pathways by microRNAs. Cell Cycle. 2008;7:3143–3148. [DOI] [PubMed] [Google Scholar]
- 17. Jovanovic M, Hengartner MO. MiRNAs and apoptosis: RNAs to die for. Oncogene. 2006;25:6176–6187. [DOI] [PubMed] [Google Scholar]
- 18. Gregory PA, Bert AG, Paterson EL, et al. The miR‐200 family and miR‐205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. [DOI] [PubMed] [Google Scholar]
- 19. Chao CH, Chang CC, Wu MJ, et al. MicroRNA‐205 signaling regulates mammary stem cell fate and tumorigenesis. J Clin Invest. 2014;124:3093–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198. [DOI] [PubMed] [Google Scholar]
- 21. Tan X, Qin W, Zhang L, et al. A 5‐microRNA signature for lung squamous cell carcinoma diagnosis and hsa‐miR‐31 for prognosis. Clin Cancer Res. 2011;17:6802–6811. [DOI] [PubMed] [Google Scholar]
- 22. Lebanony D, Benjamin H, Gilad S, et al. Diagnostic assay based on hsa‐miR‐205 expression distinguishes squamous from nonsquamous non‐small‐cell lung carcinoma. J Clin Oncol. 2009;27:2030–2037. [DOI] [PubMed] [Google Scholar]
- 23. Cai J, Fang L, Huang Y, et al. MiR‐205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non‐small cell lung cancer. Cancer Res. 2013;73:5402–5415. [DOI] [PubMed] [Google Scholar]
- 24. Wang J, Tian X, Han R, et al. Downregulation of miR‐486‐5p contributes to tumor progression and metastasis by targeting protumorigenic ARHGAP5 in lung cancer. Oncogene. 2014;33:1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu J, Zeng Y, Xu C, et al. Expression profile analysis of microRNAs and downregulated miR‐486‐5p and miR‐30a‐5p in non‐small cell lung cancer. Oncol Rep. 2015;34:1779–1786. [DOI] [PubMed] [Google Scholar]
- 26. Liu TJ, LaFortune T, Honda T, et al. Inhibition of both focal adhesion kinase and insulin‐like growth factor‐I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol Cancer Ther. 2007;6:1357–1367. [DOI] [PubMed] [Google Scholar]
- 27. Jin Y, Zhao M, Xie Q, Zhang H, Wang Q, Ma Q. MicroRNA‐338‐3p functions as tumor suppressor in breast cancer by targeting SOX4. Int J Oncol. 2015;47:1594–1602. [DOI] [PubMed] [Google Scholar]
- 28. Shan Y, Li X, You B, Shi S, Zhang Q, You Y. MicroRNA‐338 inhibits migration and proliferation by targeting hypoxia‐induced factor 1 alpha in nasopharyngeal carcinoma. Oncol Rep. 2015;34:1943–1952. [DOI] [PubMed] [Google Scholar]
- 29. Wang G, Sun Y, He Y, Ji C, Hu B. MicroRNA‐338‐3p inhibits cell proliferation in hepatocellular carcinoma by target forkhead box P4 (FOXP4). Int J Clin Exp Pathol. 2015;8:337–344. [PMC free article] [PubMed] [Google Scholar]
- 30. Peng Y, Liu YM, Li LC, Wang LL, Wu XL. MicroRNA‐338 inhibits growth, invasion and metastasis of gastric cancer by targeting NRP1 expression. PLoS ONE 2014;9:e94422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang P, Shao G, Lin X, Liu Y, Yang Z. MiR‐338‐3p inhibits the growth and invasion of non‐small cell lung cancer cells by targeting I RS2. Am J Cancer Res. 2017;7:53–63. [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang G, Zheng H, Cheng R, Lu C, Guo Y, Zhao G. MicroRNA‐338‐3p suppresses cell proliferation and induces apoptosis of non‐small‐cell lung cancer by targeting sphingosine kinase 2. Cancer Cell Int. 2017;17:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Janssen BJ, Malinauskas T, Weir GA, Cader MZ, Siebold C, Jones EY. Neuropilins lock secreted semaphorins onto plexins in a ternary signaling complex. Nat Struct Mol Biol. 2012;19:1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koch S, van Meeteren LA, Morin E, et al. NRP1 presented in trans to the endothelium arrests VEGFR2 endocytosis, preventing angiogenic signaling and tumor initiation. Dev Cell. 2014;28:633–646. [DOI] [PubMed] [Google Scholar]
- 35. Glinka Y, Stoilova S, Mohammed N, Prud'homme GJ. Neuropilin‐1 exerts co‐receptor function for TGF‐beta‐1 on the membrane of cancer cells and enhances responses to both latent and active TGF‐beta. Carcinogenesis. 2011;32:613–621. [DOI] [PubMed] [Google Scholar]
- 36. Muhl L, Folestad EB, Gladh H, et al. Neuropilin 1 binds PDGF‐D and is a co‐receptor in PDGF‐D‐PDGFRbeta signaling. J Cell Sci. 2017;130:1365–1378. [DOI] [PubMed] [Google Scholar]
- 37. Liu C, Wang Z, Wang Y, Gu W. MiR‐338 suppresses the growth and metastasis of OSCC cells by targeting NRP1. Mol Cell Biochem. 2015;398:115–122. [DOI] [PubMed] [Google Scholar]
- 38. Kang Y, Massague J. Epithelial‐mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–279. [DOI] [PubMed] [Google Scholar]
- 39. Thiery JP. Epithelial‐mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. [DOI] [PubMed] [Google Scholar]
- 40. Massague J. TGFbeta in cancer. Cell. 2008;134:215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gregory PA, Bracken CP, Smith E, et al. An autocrine TGF‐beta/ZEB/miR‐200 signaling network regulates establishment and maintenance of epithelial‐mesenchymal transition. Mol Biol Cell. 2011;22:1686–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zeng Y, Zhu J, Shen D, et al. Repression of Smad4 by miR205 moderates TGF‐beta‐induced epithelial‐mesenchymal transition in A549 cell lines. Int J Oncol. 2016;49:700–708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Supporting Table S1.
Supporting Figure S1.
Supporting Figure S2.
Supporting Figure S3.
Supporting Figure S4.