

Abstract
Regorafenib exposure could potentially be influenced by an interaction with acid‐reducing drugs. In this crossover trial, patients were randomized into two sequence groups consisting of three phases: regorafenib intake alone, regorafenib with concomitant esomeprazole, and regorafenib with esomeprazole 3 hours prior. The primary end point was the relative difference (RD) in geometric means for regorafenib 0–24‐hour area under the concentration‐time curve (AUC 0–24h) and was analyzed by a linear mixed model in 14 patients. AUC 0–24h for regorafenib alone was 55.9 μg·hour/mL (coefficient of variance (CV): 40%), and for regorafenib with concomitant esomeprazole or with esomeprazole 3 hours prior AUC 0–24h was 53.7 μg·hour/mL (CV: 34%) and 53.6 μg·hour/mL (CV: 43%), respectively. No significant differences were identified when regorafenib alone was compared with regorafenib with concomitant esomeprazole (RD: −3.9%; 95% confidence interval (CI): −20.5 to 16.1%; P = 1.0) or regorafenib with esomeprazole 3 hours prior (RD: −4.1%; 95% CI: −22.8 to 19.2%; P = 1.0). These findings indicate that regorafenib and esomeprazole can be safely combined in clinical practice.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑For several kinase inhibitors, reduced absorption and decreased exposure were demonstrated with concurrent use of acid reducing drugs like PPIs. However, there is no knowledge about this potential DDI with regorafenib.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑This is the first randomized pharmacokinetic crossover trial investigating the influence of esomeprazole on regorafenib pharmacokinetics, including a potential time‐dependency of this interaction.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑No significant pharmacokinetic interaction was observed between regorafenib and esomeprazole (used concomitantly or 3 hours prior to regorafenib intake), and, therefore, these two drugs can be combined safely in clinical practice.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑A clinically relevant DDI between regorafenib and esomeprazole was ruled out in a unique study design, which could serve as a template for future studies evaluating the influence of PPIs on exposure of oral (anticancer) drugs.
Regorafenib is an oral multikinase inhibitor that targets angiogenic, stromal, and oncogenic receptor tyrosine kinases (e.g., vascular endothelial growth factor receptor, KIT, B‐type Raf, platelet‐derived growth factor receptor, and fibroblast growth factor receptor).1 It is currently registered for metastatic colorectal cancer (mCRC), gastrointestinal stromal tumor (GIST), and hepatocellular carcinoma (HCC).2, 3, 4 Regorafenib is the first and currently only tyrosine kinase inhibitor (TKI) registered for mCRC, although the median overall survival increase for an unselected group in the third or fourth line of treatment is only 1.4 months compared with placebo.2 For HCC and GIST, regorafenib provides a stronger survival benefit as second and third line TKI‐based therapy.3, 4 For several TKIs, systemic exposure has been demonstrated to influence toxicity and efficacy.5, 6
After oral administration, regorafenib is rapidly absorbed, with a time of maximum concentration (Tmax) reached at 3–4 hours.6, 7 Most TKIs exhibit pH‐dependent solubility.8 For regorafenib, a low basic predicted pK a of around 2 suggests influence of the gastrointestinal pH on the absorption; however, this is not clearly demonstrated.9, 10 Although the physiochemical properties of regorafenib may not predict significant pH‐dependent solubility, regorafenib absorption is multifactorial and may be affected by the concomitant use of acid‐reducing drugs.11 For many TKIs, a pharmacokinetic interaction with an acid‐suppressive agent has already been demonstrated; for example, erlotinib combined with omeprazole resulted in 46% decrease in systemic exposure.8 However, for some TKIs this interaction could be ruled out. To our knowledge, for regorafenib there is no study available yet on a possible drug–drug interaction (DDI) with acid‐reducing drugs.
When the exposure is decreased, the efficacy of TKI treatment could potentially also decrease, as was demonstrated for sorafenib and pazopanib among other TKIs.6 As regorafenib resembles the structure and mechanism of action of sorafenib, an exposure–response relationship could be suspected for regorafenib as well. In a secondary analysis of the phase III RESORCE trial in patients with HCC, median overall survival and time‐to‐progression tended to be longer in patients with higher regorafenib exposure during the first treatment cycle; however, after correction for several covariates, it did not reach statistical significance.12 To our knowledge, this trial is the only available evidence on a possible exposure – response relationship for regorafenib; therefore, more research is necessary on this point.
Acid‐suppressive therapy is frequently used by patients with cancer, both as prophylaxis for gastrointestinal bleeding due to DDIs and as treatment for gastroesophageal reflux disease.13 In 2013, Smelick et al.14 reported that up to 33% of all anticancer patients used any form of acid‐suppressive therapy, most notably a proton pump inhibitor (PPI). TKIs often cause stomach complaints or gastroesophageal reflux disease, which confronts clinicians with a challenge, as the general consensus is to avoid the combination of TKIs and acid‐suppressive agents. Therefore, registration authorities currently recommend investigating this DDI before registration of a new TKI. However, for regorafenib, this potential DDI has not been investigated.
In this study, we assessed the potential pharmacokinetic interaction between esomeprazole and regorafenib. Furthermore, we also assessed the potential influence of timing of esomeprazole intake relative to that of regorafenib (3 hours before regorafenib ingestion or concomitantly).15
Results
Patient characteristics
A total of 31 patients were included, of which 14 patients were evaluable for the primary endpoint analysis. The evaluable patients were equally distributed over the two treatment sequence groups. Patients were not evaluable because of various reasons: screen failures (n = 4), rapid disease progression during treatment (n = 8), and premature treatment interruption (n = 5). Patients who developed progressive disease during the study period were also equally distributed over the two treatment sequences.
Patient characteristics are detailed in Table 1 . All patients were diagnosed with mCRC, were of white origin, and were predominantly men (71%). Median age was 69 years, and most patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 1 (86%). All patients used regorafenib 120 mg at steady‐state on recommendation of the treating physician or due to dosereductions in the first 2 weeks of the trial.
Table 1.
Patient characteristics
| Characteristic | Total |
|---|---|
| Gender | |
| Male | 10 (71%) |
| Female | 4 (29%) |
| Age, years | |
| Median [IQR] | 69 [61–73] |
| ECOG performance status | |
| 0 | 2 (14%) |
| 1 | 12 (86%) |
| Ethnic origin | |
| White | 14 (100%) |
| BMI, kg/m2 | |
| Median [IQR] | 28.6 [24.1–29.9] |
| eGFR, mL/minutea | |
| Median [IQR] | 82 [77–91] |
| Liver function (median [IQR]) | |
| AST | 39 [27–68] |
| ALT | 33 [17–39] |
| Bilirubin | 8 [6–13] |
| Prior therapy | |
| Surgery | 12 (86%) |
| Radiotherapy | 4 (29%) |
| Chemotherapy | 14 (100%) |
| Monoclonal antibodiesb | 9 (64%) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; ECOG, Eastern Cooperative Oncology Group; eGFR, estimated glomerular filtration rate; IQR, interquartile range.
aeGFR was calculated according to the Chronic Kidney Disease Epidemiology Collaboration. bTreatment with monoclonal antibodies included bevacizumab, panitumumab, and cetuximab.
Pharmacokinetics
All obtained pharmacokinetic results are depicted in Table 2 . No statistical difference in geometric means for regorafenib 0–24‐hour area under the concentration‐time curve (AUC0–24h) was found when regorafenib alone was compared with regorafenib and esomeprazole concomitantly (relative difference (RD): −3.9%; 95% confidence interval (CI): −20.5 to 16.1%; P = 1.0) or when compared with regorafenib and esomeprazole 3 hours before regorafenib intake (RD: −4.1%; 95% CI: −22.8 to 19.2%; P = 1.0; Figure 1 ). Furthermore, no differences could be identified in peak plasma concentration (Cmax) or Tmax for regorafenib. For M‐2 and M‐5, no differences could be identified either, although the interindividual variability (expressed as coefficient of variation) was much higher for all these pharmacokinetic parameters compared with regorafenib (Table 2 , Figure S1 ). No sequence or period effects were seen for any of the comparisons of the AUC0–24h and Cmax (results not shown).
Table 2.
Regorafenib pharmacokinetics
| PK parameters | Regorafenib (phase A) | Regorafenib + Esomeprazole concomitant (phase B) | Regorafenib + Esomeprazole 3 hours prior (phase C) | Relative difference B vs. A (95% CI) | P value | Relative difference C vs. A (95% CI) | P value |
|---|---|---|---|---|---|---|---|
| Regorafenib | |||||||
| AUC0–24h (μg·hour/mL (CV)) | 55.9 (40.3) | 53.7 (33.5) | 53.6 (42.6) | −3.9% (−20.5 to 16.1%) | 1.00 | −4.1% (−22.8 to 19.2%) | 1.00 |
| Cmax (μg/mL (CV)) | 5.3 (28.6) | 4.4 (24.2) | 4.7 (25.5) | −16.5% (−34.9 to 7.0%) | 0.18 | −12.1% (−32.0 to 13.8%) | 0.45 |
| Tmax (median hours (IQR)) | 2.5 (2.0–3.0) | 2.5 (2.0–3.0) | 3.0 (2.5–3.1) | 1.00 | 0.83 | ||
| M‐2 | |||||||
| AUC0–24h (μg·hour/mL (CV)) | 36.6 (71.4) | 35.1 (66.2) | 35.0 (64.5) | −4.0% (−28.6 to 29.2%) | 1.00 | −4.3% (−30.1 to 31.0%) | 1.00 |
| Cmax (μg/mL (CV)) | 2.9 (72.0) | 2.6 (60.9) | 2.6 (44.2) | −11.0% (−38.7 to 29.1%) | 0.88 | −9.3% (−38.1 to 32.9%) | 1.00 |
| Tmax (median hours (IQR)) | 3.3 (2.0–6.0) | 2.6 (2.1–3.5) | 3.5 (2.5–6.0) | 1.00 | 1.00 | ||
| M‐5 | |||||||
| AUC0–24h (μg·hour/mL (CV)) | 21.9 (103.4) | 21.6 (125.7) | 20.0 (128.9) | −1.4% (−22.5 to 25.4%) | 1.00 | −8.9% (−40.4 to 39.1%) | 1.00 |
| Cmax (μg/mL (CV)) | 1.6 (118.8) | 1.4 (132.4) | 1.4 (107.6) | −10.4% (−34.6 to 22.8%) | 0.78 | −9.1% (−43.2 to 45.5%) | 1.00 |
| Tmax (median hours (IQR)) | 2.6 (1.5–4.0) | 2.3 (1.5–8.0) | 3.5 (2.5–6.0) | 1.00 | 0.76 | ||
AUC0–24h, 0–24‐hour area under the concentration‐time curve (expressed as geomean μg·hour/mL (CV)); CI, confidence interval; Cmax, peak plasma concentration (expressed as geomean μg/mL (CV)); CV, coefficient of variation expressed in %; IQR, interquartile range; PK, pharmacokinetic; Tmax, time until maximum concentration (expressed as median hours (IQR)).
Figure 1.
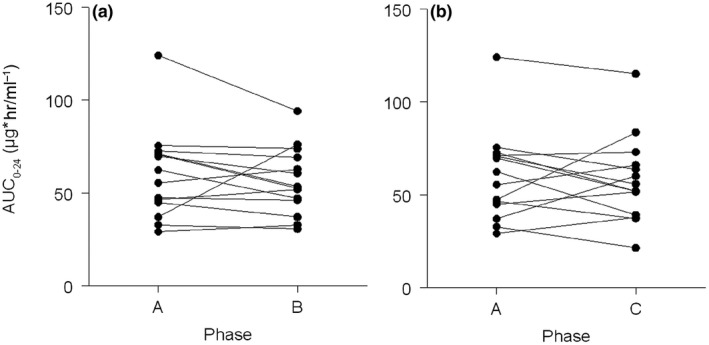
Regorafenib area under the curve. Regorafenib exposure compared (a) between phase A (regorafenib alone) and phase B (regorafenib concomitantly with esomeprazole), and (b) between phases A and C (regorafenib with esomeprazole 3 hours prior). AUC 0–24, 0–24‐hour area under the concentration‐time curve; hr, hour.
Toxicity
Most common adverse events during the whole study period were hoarseness (79%), anorexia (71%), hypertension (71%), hand/foot skin reaction (64%), fatigue (71%), stomatitis (57%), and nausea (50%). In addition, most common blood value disorders included transaminase increase (79%), bilirubin increase (50%), and hypophosphatemia (29%). The majority of adverse events were of low grade; the incidence of toxicity ≥ grade 3 occurred mainly as hypertension (64%), anorexia (14%), and hand/foot skin reaction (14%). The incidence of adverse events seems comparable between different phases. Two patients developed major cardiac events, possibly related to regorafenib treatment: myocardial infarction and atrial fibrillation. One patient developed hypertrichosis, although this rare side effect is seen more often with other TKIs, such as erlotinib16; to our knowledge, it has not been described for regorafenib. All observed adverse events are described in Table S1 .
Discussion
This randomized, three‐phase, crossover clinical trial did not reveal a significant pharmacokinetic interaction between esomeprazole and regorafenib at the two timepoints studied. Therefore, we can conclude that esomeprazole can be combined with regorafenib safely, in contrast with other TKIs.
In this study, esomeprazole was used because it exhibits the strongest pH‐reducing effect of all acid‐reducing drugs currently available.8, 17 In addition, esomeprazole does not influence other enzymes or transporters, such as P‐glycoprotein (ABCB1), that could potentially influence the pharmacokinetics of regorafenib's active metabolites M‐2 and M‐5.18 Therefore, our findings cannot be extrapolated to other PPIs, such as pantoprazole, which is known to influence P‐glycoprotein. We examined two timepoints regarding the intake time of esomeprazole (i.e., concomitantly or 3 hours prior regorafenib intake), because PPIs are assumed to have their maximum acid‐reducing effect 3 hours after intake and a possible interaction would be the strongest at this timepoint.15 However, even at this timepoint we did not demonstrate an influence of esomeprazole on the pharmacokinetics of regorafenib, M‐2, and M‐5.
Regorafenib exhibits low solubility, which is mainly caused by its chemical structure as no strong basic or acidic group is attached (regorafenib: 4‐[4‐({[4‐chloro‐3‐(trifluoromethyl)phenyl]carbamoyl} amino)‐3‐fluorophenoxy]‐N‐methylpyridine‐2‐carboxamide).19 Furthermore, to improve the solubility, regorafenib is formulated as a solid dispersion consisting of small powder particles in which the drug and excipient are integrated.20 Despite this formulation, regorafenib exhibits low solubility compared with other TKIs. As a result, regorafenib absorption is, in theory, less affected by intragastric pH alterations, and the results of this study were not totally unexpected. However, because TKI absorption is multifactorial, a DDI with PPIs cannot always be fully ruled out based on modeling and physiochemical properties alone.11 Therefore, a drug interaction should always be verified in an in vivo setting, as was done in this study for regorafenib.
In order to reach the required sample size of 14 evaluable patients, a total of 31 patients had to be included in the study, due to the fact that many patients were not able to complete three cycles of regorafenib at 160 or 120 mg due to treatment‐related adverse events or progression of disease. In addition, we aimed to include both patients with mCRC and GIST, but mainly patients with mCRC were included, which resulted in a possible selection bias. In general, patients with mCRC are in a worse condition and more heavily pretreated compared with patients with GIST, which could have resulted in more adverse events and a higher dropout rate. However, we do not think it influenced the pharmacokinetic end points. In addition, the CORRECT trial demonstrated a median overall survival increase of 1.4 months compared with placebo in patients with mCRC.2 Therefore, it was not completely surprising that quite some patients developed early disease progression during study treatment, hampering prolonged study participation. In addition, all patients eventually used 120 mg at steady‐state instead of 160 mg, due to known severe treatment‐related adverse events (e.g., hypertension), which also occurred in up to 50% of patients in the registration studies.2, 3, 4 Furthermore, because this study was designed as a pharmacokinetic crossover study, we could not compare toxicity between different cycles. However, because we found no differences in regorafenib pharmacokinetics, a difference in exposure‐related toxicity seems unlikely.
This study was designed to demonstrate a difference based on two primary comparisons on regorafenib exposure depending on esomeprazole intake time (concomitantly or 3 hours prior). Because of the assumption of a difference between those cycles, we did not include a bioequivalence analysis. However, the boundaries of the adjusted 90% CI of the RDs of the regorafenib AUC found in this study almost fit the limits for bioequivalence (B vs. A, RD: −3.9%; 90% CI: −18.2 to 12.9%; and C vs. A, RD: −4.1%; 90% CI: −20.3 to 15.4%),21 which supports the interpretation of our results.
In conclusion, we have shown that esomeprazole did not influence regorafenib exposure on two different intake timepoints, and that these drugs can be combined in clinical practice without the appearance of a significant pharmacokinetic interaction.
Methods
This study was a randomized, two‐armed, three‐phase, crossover clinical trial in patients using regorafenib. Between May 2016 and February 2018, the study was performed at the Erasmus Medical Center, Rotterdam, the Netherlands. Approval of the medical ethics committee and the board of directors from the Erasmus University Medical Center and the competent authorities were obtained. The study was registered at the European Clinical Trials Database (EudraCT 2015‐005784‐17) and www.clinicaltrials.gov (NCT02800330).
Patients
Patients were included if they were 18 years or older, had a pathological confirmed diagnosis of mCRC or GIST, ECOG performance status ≤ 1, with adequate kidney and liver function. Patients were excluded if they could not abstain from dietary supplements or medication, which could interact with regorafenib or esomeprazole, if they could not interrupt acid‐suppressive therapy, or if they had a known impaired drug absorption or serious illness that could interfere with study conduct (e.g., infection, bleeding diathesis or hemorrhage, arterial or venous thrombotic or embolic events, uncontrolled hypertension despite optimal medical management, human immunodeficiency virus, hepatitis, organ transplants, or kidney, cardiac, and respiratory diseases). All patients provided written informed consent before any study‐related procedure was pursued.
Study design
The main objectives of this study were to compare the AUC of regorafenib alone to regorafenib concomitantly used with esomeprazole, and to regorafenib used with esomeprazole 3 hours prior, in patients with mCRC or GIST. Patients started with regorafenib on 120 or 160 mg once daily during a loading phase of 14 consecutive days (Figure 2 ). Regorafenib dose adjustments were only allowed during these first 2 weeks of the trial. However, because of (reversible) toxicity, the study was allowed to be temporarily interrupted for a maximum of one full regorafenib dosing cycle (i.e., 28 days). After reaching steady state, patients either used regorafenib alone (phase A) or with esomeprazole (40 mg once daily) for 5 consecutive days (phase B and C). During phase B of the study, regorafenib was administered concomitantly with esomeprazole, whereas during phase C regorafenib was administered 3 hours after esomeprazole intake, presuming a maximally elevated intragastric pH at the time of regorafenib ingesture.17 Subjects were randomized into two sequence groups (i.e., A‐B‐C or C‐B‐A) to rule out sequence and time effects (Figure 2 ).
Figure 2.
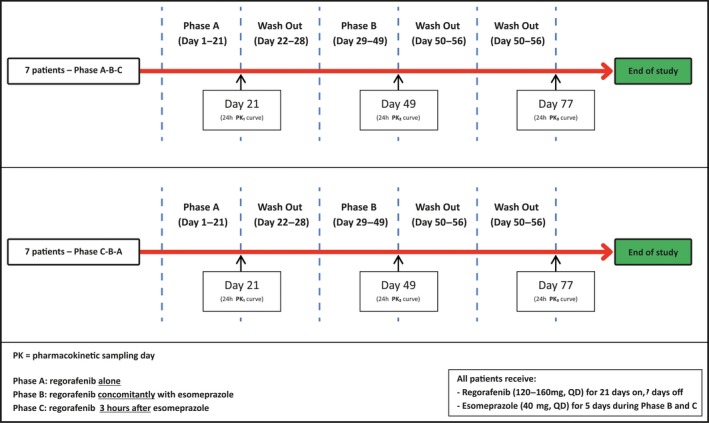
Study procedures. QD, every day. [Colour figure can be viewed at wileyonlinelibrary.com]
Pharmacokinetics
Patients were admitted to the hospital on the 21st, the 49th, and the 77th day of the trial for pharmacokinetic blood sampling. Blood samples were collected before regorafenib administration and at the 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 12, and 24‐hour timepoints after regorafenib administration (at 10:00 am). Blood samples were collected in 4‐mL lithium heparin blood collection tubes, processed into plasma within 10 minutes by centrifugation for 10 minutes at 2,500 g (at 4°C), and stored at T<−70°C until analysis. Regorafenib, M‐2, and M‐5 plasma concentrations were measured using a validated ultraperformance liquid chromatography–tandem mass spectrometry method (detailed description in Methods S1 ). Pharmacokinetic parameters were calculated by using Phoenix WinNonlin version 7.0 (Certara, Princeton, NJ), and included exposure expressed as dose corrected AUC0–24h, maximum observed concentration (Cmax), and time until maximum observed concentration (Tmax).
Statistical analysis
A difference in systemic exposure to regorafenib of 30% was determined to be clinically relevant. Because two primary comparisons were to be made (i.e., regorafenib with esomeprazole concomitant or 3 hours prior compared with regorafenib alone), a Bonferroni correction was applied. The Bonferroni correction was implemented by multiplying the obtained P values by 2 and calculation of 97.5% CIs, which correspond to the alpha of 0.025 with the interpretation of Bonferroni corrected 95% CIs. It was assumed that the within‐patient SD in regorafenib pharmacokinetics was 30%. Given a power of 80%, the sample size calculation resulted in a required number of 14 evaluable patients.22 Patients were considered evaluable when they completed all three phases, including all required blood samples.
Analyses of the AUC0–24h and Cmax were performed on log‐transformed observations because they were assumed to follow a log‐normal distribution.21 Estimates for the mean differences in (log) AUCs and Cmax of regorafenib, M‐2, and M‐5 were obtained for the two comparisons separately using a linear mixed effect model with treatment, sequence, and phase as fixed effects and subject within sequence as a random effect.23 Variance components were estimated based on restricted maximum likelihood methods, and the Kenward‐Roger method of computing the denominator degrees of freedom was used. The mean differences and CIs for the differences were exponentiated to provide point estimates of the ratio of geometric means and CIs for these ratios, which can be interpreted as RDs in percentages. The Tmax was analyzed by means of the Wilcoxon signed rank test and described with medians and interquartile ranges.
Toxicity was described as the incidence of toxicity per phase and was corrected for baseline toxicity by describing only new or worsened toxicity compared with baseline. This study was not powered to detect a difference in toxicity between treatment phases; therefore, these results only have a descriptive character.
Funding
This study was supported by an unrestricted grant from Bayer.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
F.M.dM., K.G.A.M.H., F.A.L.M.E., T.vG., R.W.F.vL., and R.H.J.M. wrote the manuscript. F.M.dM., T.vG., R.W.F.vL., and R.H.J.M. designed the research. F.M.dM., K.G.A.M.H., M.dW., H.K.vH., N.C.H.P.vdB.‐dG., F.A.L.M.E., and R.H.J.M. performed the research. F.M.dM., K.G.A.M.H., E.O.‐dH., and P.dB. analyzed the data. P.dB. contributed new reagents/analytical tools.
Supporting information
Figure S1. M‐2 and M‐5 AUC.
Table S1. Toxicity.
Methods S1. Detailed description assay regorafenib, M‐2, and M‐5.
Acknowledgments
This work has been presented at the 2018 ESMO Annual Meeting, #473 (Munich, Germany), October 2018.
References
- 1. Wilhelm, S.M. et al Regorafenib (BAY 73‐4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 129, 245–255 (2011). [DOI] [PubMed] [Google Scholar]
- 2. Grothey, A. et al Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet 381, 303–312 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Demetri, G.D. et al Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet 381, 295–302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bruix, J. et al Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 389, 56–66 (2017). [DOI] [PubMed] [Google Scholar]
- 5. Yu, H. , Steeghs, N. , Nijenhuis, C.M. , Schellens, J.H.M. , Beijnen, J.H. & Huitema, A.D.R. Practical guidelines for therapeutic drug monitoring of anticancer tyrosine kinase inhibitors: focus on the pharmacokinetic targets. Clin. Pharmacokinet. 53, 305–325 (2014). [DOI] [PubMed] [Google Scholar]
- 6. de Wit, D. , Guchelaar, H.J. , den Hartigh, J. , Gelderblom, H. & van Erp, N.P. Individualized dosing of tyrosine kinase inhibitors: are we there yet? Drug Discov. Today 20, 18–36 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Tlemsani, C. et al Effect of glucuronidation on transport and tissue accumulation of tyrosine kinase inhibitors: consequences for the clinical management of sorafenib and regorafenib. Expert Opin. Drug Metab. Toxicol. 11, 785–794 (2015). [DOI] [PubMed] [Google Scholar]
- 8. van Leeuwen, R.W. , van Gelder, T. , Mathijssen, R.H. & Jansman, F.G. Drug‐drug interactions with tyrosine‐kinase inhibitors: a clinical perspective. Lancet Oncol. 15, e315–e326 (2014). [DOI] [PubMed] [Google Scholar]
- 9. DrugBank . Regorafenib <https://www.drugbank.ca/drugs/DB08896>.
- 10. European Bioinformatics Institute . CHEMBL1946170 <https://www.ebi.ac.uk/chembl/compound/inspect/CHEMBL1946170>.
- 11. Herbrink, M. , Nuijen, B. , Schellens, J.H. & Beijnen, J.H. Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treat. Rev. 41, 412–422 (2015). [DOI] [PubMed] [Google Scholar]
- 12. Solms, A. et al Exposure‐response relationship of regorafenib efficacy in patients with hepatocellular carcinoma. Eur. J. Pharm. Sci. 109S, S149–S153 (2017). [DOI] [PubMed] [Google Scholar]
- 13. van Leeuwen, R.W. et al Drug‐drug interactions in patients treated for cancer: a prospective study on clinical interventions. Ann. Oncol. 26, 992–997 (2015). [DOI] [PubMed] [Google Scholar]
- 14. Smelick, G.S. et al Prevalence of acid‐reducing agents (ARA) in cancer populations and ARA drug‐drug interaction potential for molecular targeted agents in clinical development. Mol. Pharm. 10, 4055–4062 (2013). [DOI] [PubMed] [Google Scholar]
- 15. van Leeuwen, R.W.F. et al Tyrosine kinase inhibitors and proton pump inhibitors: an evaluation of treatment options. Clin. Pharmacokinet. 56, 683–688 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vergou, T. et al Facial hypertrichosis and trichomegaly developing in patients treated with the epidermal growth factor receptor inhibitor erlotinib. J. Am. Acad. Dermatol. 63, e56–e58 (2010). [DOI] [PubMed] [Google Scholar]
- 17. Hunfeld, N.G. , Touw, D.J. , Mathot, R.A. , van Schaik, R.H. & Kuipers, E.J. A comparison of the acid‐inhibitory effects of esomeprazole and rabeprazole in relation to pharmacokinetics and CYP2C19 polymorphism. Aliment. Pharmacol. Ther. 35, 810–818 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Oostendorp, R.L. , Buckle, T. , Beijnen, J.H. , van Tellingen, O. & Schellens, J.H. The effect of P‐gp (Mdr1a/1b), BCRP (Bcrp1) and P‐gp/BCRP inhibitors on the in vivo absorption, distribution, metabolism and excretion of imatinib. Invest. New Drugs 27, 31–40 (2009). [DOI] [PubMed] [Google Scholar]
- 19. European Medicines Agency . Assessment report: stivarga (European Medicines Agency, London, 2013). [Google Scholar]
- 20. Sawicki, E. , Schellens, J.H. , Beijnen, J.H. & Nuijen, B. Inventory of oral anticancer agents: pharmaceutical formulation aspects with focus on the solid dispersion technique. Cancer Treat. Rev. 50, 247–263 (2016). [DOI] [PubMed] [Google Scholar]
- 21. Committee for Medicinal Products for Human Use (CHMP), European Medicines Agency . Guideline on the investigation of bioequivalence <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf> (January 2010).
- 22. Statistical considerations for a cross‐over study where the outcome is a measurement . <http://hedwig.mgh.harvard.edu/sample_size/js/js_crossover_quant.html>.
- 23. Jones, B. & Kenward, M.G. Design and Analysis of Cross‐Over Trials 2nd edn (Chapman & Hall/CRC, Raton, FL, 2003). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. M‐2 and M‐5 AUC.
Table S1. Toxicity.
Methods S1. Detailed description assay regorafenib, M‐2, and M‐5.