

Abstract
Background and Purpose
Despite extensive research in the last decade, the role of serum amyloid A (SAA) in atherogenesis remains highly controversial. The aim of this study was therefore to assess whether SAA is associated with long‐term mortality in patients with subclinical carotid artery disease.
Methods
One thousand sixty‐five patients with neurological asymptomatic carotid atherosclerosis as evaluated by duplex sonography were prospectively followed for cause‐specific mortality.
Results
During a median of 11.8 years, a total of 549 deaths, including 362 cardiovascular deaths, were recorded. Patients who died within the follow‐up period had significantly higher baseline SAA serum levels compared to those who survived (12.9 vs 9.5 mg/dL; P < 0.001). In univariable Cox regression analysis, the risk of all‐cause and cardiovascular mortality significantly increased in patients with elevated serum levels of SAA (crude hazard ratio for cardiovascular mortality per increase of 1 SD of SAA levels was 1.14, 95% CI 1.08‐1.22], P < 0.0001). However, SAA lost its significance after adjusting for high‐sensitivity C‐reactive protein (hsCRP), suggesting that SAA might not be directly associated with atherogenesis, but rather be a mere reflection of the individual patient's inflammatory status.
Conclusions
Serum amyloid A is not independently associated with (cardiovascular) mortality in patients with subclinical carotid atherosclerosis.
Keywords: biomarker, carotid atherosclerosis, risk factor, serum amyloid A
1. INTRODUCTION
Inflammation is recognized as an essential hallmark of atherogenesis.1 In this context, a substantial amount of attention is shifted to the relationship between elevated levels of specific serum proteins in the context of a chronic inflammatory response. These proteins have been studied mainly as markers of cardiovascular risk, but some also have been implicated in a causal role as part of the atherosclerotic process.2
Serum amyloid A (SAA) is a family of apoproteins highly expressed by the liver during an acute‐phase response (APR), a complex systemic early‐defence system activated by infection, traumatic injuries, neoplasia or auto‐inflammatory disease. The SAA family comprises acute‐phase isoforms (SAA1‐ and SAA‐2) and a constitutive isoform (SAA‐4).3 During APR, the plasma level of SAA rises for a brief period of time by up to 1000‐fold.4 The observation that SAA serum levels are chronically increased in patients with diabetes and obesity and are therefore subject to an increased cardiovascular risk 5 have led to extensive experimental research in the last decade, investigating the potential proinflammatory properties of SAA.
SAA exhibits several characteristics that could render it atherogenic. It was shown to enhance monocyte recruitment,6 directly stimulate foam cell formation,7 stimulate chemotaxis of lymphocytes 8 and facilitate the binding of HDL to vascular proteoglycans. Recently, Dong et al overexpressed murine SAA in mice and demonstrated that sustained elevation of SAA leads to increased atherosclerosis.9 In contrast to these findings, De Beer et al showed that the absence of SAA did not affect atherosclerotic lesion formation10 and Tam et al11 demonstrated that administration of SAA might even prevent atherosclerotic lesion development in mice. Taken together, today, experimental studies neither provide enough evidence to designate SAA as a causal factor in atherogenesis nor can it be ruled out definitively. A vast amount of epidemiological data links SAA with cardiovascular diseases (CVDs),12, 13, 14, 15, 16, 17 but, up to now, studies investigating the value of SAA as a biomarker for clinical outcome failed to reach an agreement whether SAA is useful for risk stratification in subjects with or without atherosclerotic diseases. Ridker et al showed in ~28,000 postmenopausal women that SAA is not independently associated with cardiovascular events over a three year follow‐up period.12 Johnson et al on the other hand found a strong and independent association of SAA with cardiovascular events within 3 years after coronary angiography in women with suspected myocardial ischaemia.18 In addition, the group around Eugene Braunwald showed that SAA predicts 14‐day mortality in patients with acute coronary syndrome.13 However, these studies investigated only short‐term outcomes and often used combined endpoints.
To the best of our knowledge, no previous study has examined the predictive value of SAA for long‐term morality in patients with atherosclerosis. Therefore, the aim of this study was to assess whether SAA is associated with cause‐specific mortality in a large prospectively collected cohort of patients with asymptomatic carotid atherosclerosis.
2. METHODS
We prospectively enrolled all consecutive patients who underwent duplex ultrasound investigations of the extracranial carotid arteries from March 2002 until March 2003 who were currently neurologically asymptomatic with respect to carotid stenosis, as described previously.19 A total of 1363 participants were enrolled at the Vienna General Hospital, a university‐affiliated tertiary care centre, and were required to complete a detailed study questionnaire. The acquired data were then reviewed by a physician further assessing the patient's medical history, biometric data, family history, lifestyle factors and medication. Furthermore, all recorded parameters were checked for completeness and accuracy by two independent observers. Cardiovascular risk factors such as hypertension, diabetes mellitus, current smoking habits and lipid disorders were obtained applying the respective guidelines. The follow‐up included a 6‐month revisit, a 3.5‐year clinical follow‐up for major adverse cardiovascular events and a 6‐year and 12‐year search of the national death registry of our patient population, as described previously.15, 19
Cardiovascular and all‐cause mortality were assessed by searching the national death register for the specific cause of death in 2015 (according to the International Statistical Classification of Diseases and Related Health Problems, 10th Revision). Only the specific cause of death (eg stroke) was used to categorize death as either all‐cause, cardiovascular or noncardiovascular death. In 43% the underlying cause was assessed by autopsy. The study protocol complies with the Declaration of Helsinki and was approved by the Ethics Committee of the Medical University of Vienna. The reporting of the study confirms to the STROBE statement and EQUATOR guidelines.20
2.1. Definitions
Definitions of risk factors and comorbidities were published previously.19 Briefly, hypertension was considered present in patients with blood pressure above 140/90 mm Hg or in patients taking antihypertensive medication. Patients with fasting blood glucose levels >126 mg/dL (7.0 mmol/L) or glycohemoglobin A1c levels >6.5% or patients under antidiabetic therapy were considered diabetic. A family history of atherosclerotic disease was considered positive if its presence had been verified in a first‐degree relative. Codes indicating for cardiovascular death (our second study objective) were obtained from the 10th revision of the International Statistical Classification of Diseases and Related Health Problems (ICD), chapter IX, blocks I00 to I99.
2.2. Clinical and laboratory data
Patients underwent a conventional duplex sonographic examination (Acuson 128 XP10) with a 7.5‐MHz linear array probe (Acuson, Malvern, PA) by experienced technical assistants under the supervision of a physician. Duplex grading of carotid stenosis was performed by measurement of the peak systolic and end‐diastolic velocities in the internal carotid artery and the common carotid arteries, as previously described.19
Antecubital venous blood samples were drawn and analysed directly (without freezing) according to local laboratory standard procedures. Serum levels of SAA were determined at admission within 2 to 4 hours after blood collection by N Latex SAA (DADE Behring) with a detection level of 3.8 mg/L and a coefficient of variation of 6.4%.
2.3. Statistical methods
Cox proportional hazards models were applied to assess the potential association between levels of SAA and the occurrence of either all‐cause or cardiovascular death. For this purpose, we constructed four models for multivariable regression analyses:
Model I included age (years) and gender (male/female).
Model II included (in addition to age and gender) data of the individuals patients history and physical examination with respect to cardiovascular risk as well as serum lipid markers: history of myocardial infarction (binary), history of stroke (binary), peripheral arterial disease (binary), body mass index (kg/m2), hypertension (binary), levels of triglycerides (mg/dL), total cholesterol levels (mg/dL), low‐density lipoprotein cholesterol levels (mg/dL) and statin treatment (binary).
In Model III kidney function and metabolic status were added: diabetes mellitus (binary), glycohemoglobin A1 (%) and serum creatinine (mg/dL) as well as the variables of Model II. In model IV, we included hsCRP (mg/dl) in addition to all the variables selected for model III.
In addition, we performed Cox regression analyses in which SAA was modelled as a categorical variable (Quartiles). Results of the Cox models are presented as hazard ratios (HR; 95% confidence interval [CI]). We assessed the overall model fit using Cox–Snell residuals. We also tested the proportional hazard assumption for all covariabes using Schoenfeld residuals (overall test) and the scaled Schoenfeld residuals (variable‐by‐variable testing). Spearman correlation coefficients (rs) were used for univariable analyses, as appropriate.
A 2‐sided P value of <0.05 was considered significant. All calculations were performed with SPSS (version 20.0, SPSS Inc) for Windows.
3. RESULTS
A total of 1363 patients were enrolled in the study. Ninety‐five (7%) of these patients had missing duplex ultrasound follow‐up data, and 203 patients (16%) were lost to clinical follow‐up, leaving 1065 patients for the final analysis. The 298 patients who had to be excluded did not significantly differ from the patients who were included in terms of baseline and demographic parameters (age, sex, frequency of atherothrombotic risk factors, cardiovascular comorbidities, patients' medical history, family history, results of health assessments and physical examinations and degree of carotid stenosis; data not shown). An overall survival rate of 48.5% was determined for the median follow‐up of 11.8 years (interquartile range [IQR], 6.0‐12.4 years), corresponding to 9871 overall person‐years. A total of 549 (51.5%) deaths were recorded. Of these, 362 (34.0%) patients died of cardiovascular causes, 142 (13.3%) of malignant diseases and 45 (4.2%) of other causes. The patient population comprised of 668 male patients (62.7%); the median age was 69.0 years (interquartile range, 61.2‐76.2 years) at the time of inclusion. Demographic and clinical characteristics of the 1065 patients included are shown in Table 1. The median baseline SAA levels were 6.4 mg/dL (IQR, 3.8‐11.0 mg/dL). We found a significant, but weak association between SAA and hsCRP (P < 0.01; R = 0.56).
Table 1.
Baseline characteristics of study participants
| Variable | All patients |
|---|---|
| Age (y) | 69 (61‐76) |
| Male gender | 668 (62.7) |
| Previous PAD | 456 (42.8) |
| Previous MI | 257 (24.1) |
| Previous stroke | 176 (16.5) |
| Statin | 142 (16.6) |
| BMI | 26.1 (24.0‐28.7) |
| Hypertension | 731 (68.6) |
| Diabetes | 242 (22.7) |
| Current smokers | 287 (26.9) |
| Serum creatinine (mg/dl) | 1.1 (0.9‐1.2) |
| HbA1c% | 6.0 (5.6‐6.6) |
| LDL cholesterol (mg/dL)a | 118 (94‐146) |
| Triglycerides (mg/dL)b | 147 (107‐216) |
| hsCRP (mg/dL) | 0.3 (0.1‐0.6) |
| Serum Amylyoid A | 6.4 (3.9‐11.0) |
Continuous data are presented as the median and the interquartile range. Discrete data are given as counts and percentages.
HbA1c, Glycated haemoglobin A1; hsCRP, High‐sensitivity C‐reactive protein.; LDL, Low‐density lipoprotein; MI, myocardial infarction; PAD, peripheral arterial disease.
Multiply by 0.0259 to convert variable to mmol/L.
Multiply by 0.0113 to convert variable to mmol/L.
3.1. All‐cause mortality
Patients who died within the period of follow‐up had significantly higher mean baseline SAA levels than those who survived (12.9 vs 9.5 mg/dL; P < 0.01). In univariable Cox proportional hazard regression analysis, increased levels of SAA were significantly associated with increased risk of all‐cause mortality (crude HR for an increase of 1 SD of SAA levels was 1.16 [95% CI 1.08‐1.22; P < 0.001]; Table 2). Univariable HRs for the risk of death from any cause for quartiles of SAA levels were 1.03 (95% CI 0.81‐1.32) for the second, 1.20 (95% CI 0.94‐1.53) for the third quartile and 1.40 (95% CI 1.11‐1.77, P < 0.01) for the fourth quartile compared with the lowest quartile, respectively (Table 3, Figure 1). The inclusion of age and gender (Model 1), lipid parameters and history of CV disease (Model 2), as well as kidney function and diabetes (Model 3) did not significantly change the association between SAA and all‐cause mortality. After adjustment for hsCRP (Model 4) SAA lost its significant association with all‐cause mortality (Tables 2 and 3). No statistically significant association was found between serum levels of SAA and noncardiovascular mortality or cancer‐related mortality (data not shown).
Table 2.
Results of univariable and multivariable Cox Regression analyses for continuous measurement of serum amyloid A
| Variable | All‐cause mortality | Cardiovascular mortality | ||||
|---|---|---|---|---|---|---|
| Hazard ratio | CI | P‐value | Hazard ratio | CI | P‐value | |
| Univariable | ||||||
| SAA | 1.14 | 1.06‐1.22 | <0.001 | 1.14 | 1.08‐1.22 | <0.001 |
| Multivariable | ||||||
| Model 1 | 1.14 | 1.08‐1.22 | <0.001 | 1.18 | 1.10‐1.26 | <0.001 |
| Model 2 | 1.14 | 1.08‐1.22 | <0.001 | 1.16 | 1.08‐1.26 | <0.001 |
| Model 3 | 1.16 | 1.08‐1.22 | <0.001 | 1.18 | 1.10‐1.26 | <0.001 |
| Model 4 | 1.03 | 0.95‐1.13 | 0.52 | 1.06 | 0.95‐1.18 | 0.25 |
Model 1: Adjusted for age and sex. Model 2: Adjusted for Model 1 + body mass index, hypertension smoking, history of peripheral artery disease, history of stroke history of myocardial infarction, low‐density lipoprotein cholesterol levels, triglyceride levels, statin treatment. Model 3: Adjusted for Model 2 + glycohemoglobin A1 level, diabetes mellitus, serum creatinine. Model 4: Adjusted for Model 3 + hsCRP. HRs refer to an increase of 1 mg/dL of serum amyloid A.
CI, confidence interval; HR, hazard ratio; hsCRP, high‐sensitivity C‐reactive protein.
Table 3.
Results of univariable and multivariable cox regression analyses for quartiles of serum amyloid A
| Variable | All‐cause mortality | Cardiovascular mortality | ||||
|---|---|---|---|---|---|---|
| Hazard ratio | CI | P‐value | Hazard ratio | CI | P‐value | |
| Univariate | ||||||
| SAA Quartile 2 | 1.03 | 0.81‐1.32 | 0.80 | 1.19 | 0.88‐1.61 | 0.25 |
| SAA Quartile 3 | 1.20 | 0.94‐1.53 | 0.13 | 1.27 | 0.93‐1.71 | 0.13 |
| SAA Quartile 4 | 1.40 | 1.11‐1.77 | <0.01 | 1.67 | 1.25‐2.23 | <0.01 |
| Multivariate | ||||||
| Model1 | ||||||
| SAA Qartile 2 | 1.05 | 0.83‐1.34 | 0.68 | 1.22 | 0.90‐1.65 | 0.21 |
| SAA Qartile 3 | 1.23 | 0.97‐1.57 | 0.09 | 1.29 | 0.95‐1.75 | 0.10 |
| SAA Qartile 4 | 1.45 | 1.15‐1.83 | <0.01 | 1.73 | 1.29‐2.31 | <0.01 |
| Model2 | ||||||
| SAA Qartile 2 | 1.05 | 0.82‐1.34 | 0.72 | 1.19 | 0.87‐1.61 | 0.28 |
| SAA Qartile 3 | 1.15 | 0.90‐1.47 | 0.26 | 1.19 | 0.87‐1.63 | 0.27 |
| SAA Qartile 4 | 1.34 | 1.06‐1.70 | 0.02 | 1.60 | 1.19‐2.15 | <0.01 |
| Model3 | ||||||
| SAA Qartile 2 | 0.99 | 0.78‐1.27 | 0.96 | 1.12 | 0.83‐1.53 | 0.46 |
| SAA Qartile 3 | 1.14 | 0.89‐1.46 | 0.29 | 1.18 | 0.86‐1.65 | 0.29 |
| SAA Qartile 4 | 1.30 | 1.02‐1.65 | 0.03 | 1.56 | 1.16‐2.10 | <0.01 |
| Model4 | ||||||
| SAA Qartile 2 | 0.97 | 0.76‐1.25 | 0.83 | 1.10 | 0.81‐1.50 | 0.54 |
| SAA Qartile 3 | 1.05 | 0.82‐1.34 | 0.70 | 1.10 | 0.80‐1.51 | 0.54 |
| SAA Qartile 4 | 0.96 | 0.74‐1.25 | 0.78 | 1.20 | 0.87‐1.66 | 0.26 |
Model 1: Adjusted for age and sex. Model 2: Adjusted for Model 1 + body mass index, hypertension, smoking, history of peripheral artery disease, history of stroke history of myocardial infarction, blood pressure, low‐density lipoprotein cholesterol levels, triglyceride levels, statin treatment. Model 3: Adjusted for Model 2 + glycohemoglobin A1 level, diabetes mellitus, serum creatinine. Model 4: Adjusted for Model 3 + hsCRP. Quartile 1 served as the reference category.
CI, confidence interval; HR, hazard ratio; hsCRP, high‐sensitivity C‐reactive protein.
Figure 1.
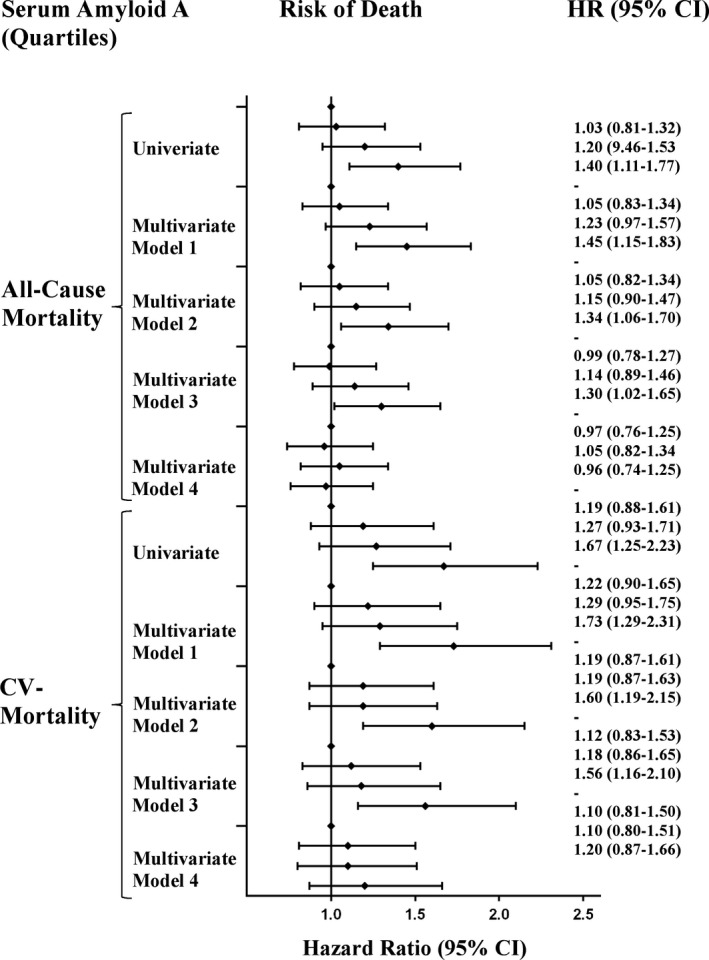
Risk of all‐cause and cardiovascular mortality according to quartiles of SAA. Diamonds indicate HR The first quartile as the reference category for each model. Model 1: Adjusted for age and sex. Model 2: Adjusted for Model 1 + body mass index, hypertension, smoking, history of peripheral artery disease, history of stroke history of myocardial infarction, blood pressure, low‐density lipoprotein cholesterol levels, triglyceride levels, statin treatment. Model 3: Adjusted for Model 2 + glycohemoglobin A1 level, diabetes mellitus, serum creatinine. Model 4: Adjusted for Model 3 + hsCRP. CI, confidence interval; HR, hazard ratio; hsCRP, high‐sensitivity C‐reactive protein; SAA, Serum Amyloid A
3.2. Cardiovascular mortality
Patients who died of cardiovascular diseases had significantly higher mean baseline SAA levels than those who survived (13.3 mg/dL versus 9.5 mg/dL; P < 001). Increased levels of SAA were significantly associated with increased risk of cardiovascular death in univariable analysis (crude HR for an increase of 1 mg/dL of SAA levels 1.14 [95% CI 1.08‐1.22; P < 0.001]). Univariable HRs for the risk of death from CV causes for quartiles of SAA levels were 1.19 (95% CI 0.88‐1.61) for the second, 1.27 (95% CI 0.93‐1.71) for the third quartile and 1.67 (95% CI 1.25‐2.23, P < 0.01) for the fourth quartile compared with the lowest quartile, respectively (Table 3, Figur1). The inclusion of age and gender (Model 1), lipid parameters and history of CV disease (Model 2), as well as kidney function and diabetes (Model 3) did not significantly change the association between SAA and all‐cause mortality. After adjustment for hsCRP (Model 4), SAA lost its significant association with CV mortality (Tables 2 and 3).
4. DISCUSSION
We previously demonstrated that SAA is significantly and independently associated with progression of atherosclerotic carotid stenosis within a 6‐ to 9‐month follow‐up19 and we recently showed in our patient population that hsCRP is significantly associated with long‐term cardiovascular mortality, independent of known cardiovascular risk factors.15 We now aimed to evaluate the clinical value of SAA as a maker for survival in these patients. Patients within the fourth quartile of serum levels of SAA are subject to a significantly higher risk for cardiovascular‐related mortality within the 12‐year follow‐up period in univariable analysis. However, the inclusion of cardiovascular risk factors based on four predefined models in multivariable Cox regression analysis diminished this observed association. Even though age, gender, lipid parameters, history of CV disease, kidney function and diabetes did not significantly change the association between SAA and (cardiovascular) mortality, after adjustment for hsCRP the association fell from a highly significant to a nonsignificant level.
We are aware that our prognostic model is not applicable for the evaluation of causality. Still, regression analysis could be helpful to falsify a potential causal relationship. The group around Altman et al recently stated that even though not every predictor is a true cause, every causal factor is a predictor.21 The converse argument implies that SAA would have to be significantly associated with cardiovascular outcome, if it were to be causally involved in atherosclerosis. In appraisal of this knowledge, the results of our study do not support the hypothesis that SAA plays a causal role in the long‐term development of atherosclerosis. Nevertheless, although we did not detect a statistically significant independent association between SAA and cardiovascular mortality, we believe that it cannot be entirely ruled out that SAA is directly involved in atherogenesis. As mentioned above, we previously showed in this patient population that SAA is significantly and independently of hsCRP associated with 6 months progression of carotid stenosis.19 One explanation for the discrepancy of our results might be that atherosclerosis is considered a chronic inflammatory disease and usually becomes clinically significant after years or decades of disease progression. Therefore, after only 6 months follow‐up, the effect of SAA might have yet not been attenuated by hsCRP. However, at this point, our study design does not allow any conclusion why serum amyloid A predicts progression of carotid atherosclerosis, but not cardiovascular mortality in patients with asymptomatic carotid artery disease. Further, (experimental) research will be necessary to elucidate the dissonance of our findings.
In the last decades or so, a plethora of observational clinical studies aimed to investigate the role of SAA in conditions which are generally associated with a chronic inflammatory response such as metabolic syndrome,16 diabetes,22 rheumatoid arthritis,14 renal disease 23 or vasculitis.24 Furthermore elevated serum levels of SAA were found to be associated with an increased body mass index 25 and diet‐induced weight loss led to a profound reduction in SAA in obese subjects.26 These findings lend substantial support to the assumption that SAA might be also involved in the development and progression of atherosclerosis. In this context, the usefulness of SAA as a biomarker of carotid atherosclerotic disease has been repeatedly evaluated. A recent study of patients with rheumatoid arthritis demonstrated that SAA levels are markedly higher in patients with increased CIMT (=carotid intima thickness).27 Furthermore, Eren et al found that women with gestational diabetes (confirmed by oral glucose tolerance tests [OGTT]) are subject to a significant clinical correlation between SAA and CIMT.17 In contrast, Jylhävä et al's “Cardiovascular Risk in Young Finns Study” (n = 2280) effectively establishes that SAA lost its significant association with CIMT as well as with carotid artery compliance after adjustment for BMI and serum lipids.28 Wohlin et al analysed cytokine‐mediated inflammatory biomarkers in a population‐based cohort of 234 elderly men without anti‐inflammatory medications and found no significant association between SAA and CIMT.29 Taken together, the currently available data do not provide enough evidence to designate SAA as clinically useful for patients with carotid atherosclerosis.
Several limitations of this study should be noted. Various comorbidities, previous diseases, as well as environmental and time‐dependent factors which were not included in the multivariable model may influence the relationship between SAA carotid atherosclerosis and mortality. In addition, overadjustment in the multivariable model could also distort the relation between SAA and outcome. However, hsCRP, the only variable which affected the association between SAA and outcome in the multivariable model, displayed only a weak correlation with SAA (rs = 0.5).
These are the first results of a prospective cohort study investigating the usefulness of SAA as a biomarker for long‐term (cardiovascular) outcome in patients with subclinical atherosclerosis. Our findings suggest that SAA might be a reflection of the subliminal chronic inflammation in atherosclerosis, but our data do not support the hypothesis that SAA is causally involved in atherosclerosis. The question whether SAA has a verifiably causal relationship with atherosclerosis or purely mirrors the inflammatory response of the underlying disease remains ambiguous. In addition, the results of our study speak against the clinical usefulness of SAA as a biomarker of cardiovascular outcome in patients with asymptomatic carotid artery disease. Whereas SAA lost its association with mortality after including hsCRP, the association between hsCRP and outcome remained highly significant for both all‐cause as well as CV mortality.15 Further prospective studies of patients with (symptomatic) atherosclerotic diseases are warranted to determine the clinical value of measurements of SAA in patients at risk for adverse outcome.
CONFLICT OF INTEREST
None of the authors has any personal or financial relationships that could inappropriately influence his or her actions or manuscript. No financial or other potential conflicts of interest exist (includes involvement with any organization with a direct financial or other interest) regarding the manuscript.
Mayer FJ, Binder CJ, Krychtiuk KA, Schillinger M, Minar E, Hoke M. The prognostic value of serum amyloid A for long‐term mortality among patients with subclinical carotid atherosclerosis. Eur J Clin Invest. 2019;49:e13095 10.1111/eci.13095
REFERENCES
- 1. Libby P, Ridker PM, Hansson GK. Leducq Transatlantic Network on A. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54(23):2129‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ridker PM. From C‐reactive protein to Interleukin‐6 to Interleukin‐1: moving upstream to identify novel targets for atheroprotection. Circ Res. 2016;118(1):145‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ye RD, Sun L. Emerging functions of serum amyloid A in inflammation. J Leukoc Biol. 2015;98(6):923‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gabay C, Kushner I. Acute‐phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340(6):448‐454. [DOI] [PubMed] [Google Scholar]
- 5. Zhao H, Guan J, Lee HM, et al. Up‐regulated pancreatic tissue microRNA‐375 associates with human type 2 diabetes through beta‐cell deficit and islet amyloid deposition. Pancreas. 2010;39(6):843‐846. [DOI] [PubMed] [Google Scholar]
- 6. Lee Hy, Kim Sd, Shim Jw, et al. Serum amyloid A induces CCL2 production via formyl peptide receptor‐like 1‐mediated signaling in human monocytes. J Immunol. 2008;181(6):4332‐4339. [DOI] [PubMed] [Google Scholar]
- 7. Lee HY, Kim SD, Baek S‐H, et al. Serum amyloid A stimulates macrophage foam cell formation via lectin‐like oxidized low‐density lipoprotein receptor 1 upregulation. Biochem Biophys Res Commun. 2013;433(1):18‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Badolato R, Wang JM, Murphy WJ, et al. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med. 1994;180(1):203‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong Z, Wu T, Qin W, et al. Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E‐deficient mice. Mol Med. 2011;17(11–12):1357‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Beer MC, Wroblewski JM, Noffsinger VP, et al. Deficiency of endogenous acute phase serum amyloid A does not affect atherosclerotic lesions in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol. 2014;34(2):255‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tam SP, Ancsin JB, Tan R, Kisilevsky R. Peptides derived from serum amyloid A prevent, and reverse, aortic lipid lesions in apoE‐/‐ mice. J Lipid Res. 2005;46(10):2091‐2101. [DOI] [PubMed] [Google Scholar]
- 12. Ridker PM, Hennekens CH, Buring JE, Rifai N. C‐reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342(12):836‐843. [DOI] [PubMed] [Google Scholar]
- 13. Morrow DA, Rifai N, Antman EM, et al. Serum amyloid A predicts early mortality in acute coronary syndromes: A TIMI 11A substudy. J Am Coll Cardiol. 2000;35(2):358‐362. [DOI] [PubMed] [Google Scholar]
- 14. Shen C, Sun X‐G, Liu Na, et al. Increased serum amyloid A and its association with autoantibodies, acute phase reactants and disease activity in patients with rheumatoid arthritis. Mol Med Rep. 2015;11(2):1528‐1534. [DOI] [PubMed] [Google Scholar]
- 15. Mayer FJ, Binder CJ, Wagner OF, et al. combined effects of inflammatory status and carotid atherosclerosis: a 12‐year follow‐up study. Stroke. 2016;47(12):2952‐2958. [DOI] [PubMed] [Google Scholar]
- 16. Zhao Y, He X, Shi X, et al. Association between serum amyloid A and obesity: a meta‐analysis and systematic review. Inflamm Res. 2010;59(5):323‐334. [DOI] [PubMed] [Google Scholar]
- 17. Eren MA, Vural M, Cece H, et al. Association of serum amyloid A with subclinical atherosclerosis in women with gestational diabetes. Gynecol Endocrinol. 2012;28(12):1010‐1013. [DOI] [PubMed] [Google Scholar]
- 18. Johnson BD, Kip KE, Marroquin OC, et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the National Heart, Lung, and Blood Institute‐Sponsored Women's Ischemia Syndrome Evaluation (WISE). Circulation. 2004;109(6):726‐732. [DOI] [PubMed] [Google Scholar]
- 19. Schillinger M, Exner M, Mlekusch W, et al. Inflammation and carotid artery‐risk for atherosclerosis study (ICARAS). Circulation. 2005;111(17):2203‐2209. [DOI] [PubMed] [Google Scholar]
- 20. Simera I, Moher D, Hoey J, Schulz KF, Altman DG. A catalogue of reporting guidelines for health research. Eur J Clin Invest. 2010;40(1):35‐53. [DOI] [PubMed] [Google Scholar]
- 21. Moons KG, Royston P, Vergouwe Y, Grobbee DE, Altman DG. Prognosis and prognostic research: what, why, and how? BMJ. 2009;338:b375. [DOI] [PubMed] [Google Scholar]
- 22. Marzi C, Huth C, Herder C, et al. Acute‐phase serum amyloid A protein and its implication in the development of type 2 diabetes in the KORA S4/F4 study. Diabetes Care. 2013;36(5):1321‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weichhart T, Kopecky C, Kubicek M, et al. Serum amyloid A in uremic HDL promotes inflammation. J Am Soc Nephrol. 2012;23(5):934‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mitani Y, Sawada H, Hayakawa H, et al. Elevated levels of high‐sensitivity C‐reactive protein and serum amyloid‐A late after Kawasaki disease: association between inflammation and late coronary sequelae in Kawasaki disease. Circulation. 2005;111(1):38‐43. [DOI] [PubMed] [Google Scholar]
- 25. Panagiotakos DB, Pitsavos C, Yannakoulia M, Chrysohoou C, Stefanadis C. The implication of obesity and central fat on markers of chronic inflammation: The ATTICA study. Atherosclerosis. 2005;183(2):308‐315. [DOI] [PubMed] [Google Scholar]
- 26. O'Brien KD, Brehm BJ, Seeley RJ, et al. Diet‐induced weight loss is associated with decreases in plasma serum amyloid A and C‐reactive protein independent of dietary macronutrient composition in obese subjects. J Clin Endocrinol Metab. 2005;90(4):2244‐2249. [DOI] [PubMed] [Google Scholar]
- 27. Targonska‐Stepniak B, Majdan M. Serum amyloid A as a marker of persistent inflammation and an indicator of cardiovascular and renal involvement in patients with rheumatoid arthritis. Mediators Inflamm. 2014;2014:793628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jylhävä J, Haarala A, Eklund C, et al. Serum amyloid A is independently associated with metabolic risk factors but not with early atherosclerosis: the cardiovascular risk in young Finns study. J Intern Med. 2009;266(3):286‐295. [DOI] [PubMed] [Google Scholar]
- 29. Wohlin M, Helmersson J, Sundström J, et al. Both cyclooxygenase‐ and cytokine‐mediated inflammation are associated with carotid intima‐media thickness. Cytokine. 2007;38(3):130‐136. [DOI] [PubMed] [Google Scholar]