

Abstract
Patients with hepatocellular carcinoma (HCC) have a poor prognosis and limited therapeutic options. Alpha‐fetoprotein (AFP) is often expressed at high levels in HCC and is an established clinical biomarker of the disease. Expression of AFP in nonmalignant liver can occur, particularly in a subset of progenitor cells and during chronic inflammation, at levels typically lower than in HCC. This cancer‐specific overexpression indicates that AFP may be a promising target for immunotherapy. We verified expression of AFP in normal and diseased tissue and generated an affinity‐optimized T‐cell receptor (TCR) with specificity to AFP/HLA‐A*02+ tumors. Expression of AFP was investigated using database searches, by qPCR, and by immunohistochemistry (IHC) analysis of a panel of human tissue samples, including normal, diseased, and malignant liver. Using in vitro mutagenesis and screening, we generated a TCR that recognizes the HLA‐A*02‐restricted AFP158‐166 peptide, FMNKFIYEI, with an optimum balance of potency and specificity. These properties were confirmed by an extension of the alanine scan (X‐scan) and testing TCR‐transduced T cells against normal and tumor cells covering a variety of tissues, cell types, and human leukocyte antigen (HLA) alleles. Conclusion: We have used a combination of physicochemical, in silico, and cell biology methods for optimizing a TCR for improved affinity and function, with properties that are expected to allow TCR‐transduced T cells to differentiate between antigen levels on nonmalignant and cancer cells. T cells transduced with this TCR constitute the basis for a trial of HCC adoptive T‐cell immunotherapy.
Abbreviations
- 3D
three‐dimensional
- AFP
alpha fetoprotein
- β2m
β2‐microglobulin
- cDNA
complementary DNA
- EC50
half maximal effective concentration
- GFP
green fluorescent protein
- GTEx
Genotype‐Tissue Expression Portal
- HCC
hepatocellular carcinoma
- HLA
human leukocyte antigen
- IFN
interferon
- IHC
immunohistochemistry
- IL
interleukin
- pHLA
peptide human lymphocyte antigen (complex)
- RPL32
ribosomal protein L32
- TCGA
The Cancer Genome Atlas
- TCR
T‐cell receptor
- wt
wild type
Hepatocellular carcinoma (HCC) is the sixth‐most common cancer and the second‐most frequent cause of cancer‐related death worldwide.1, 2 Current treatment options are limited, and patients suffer from a high rate of recurrence. The 10‐year survival rate is only 7.2%. Most HCC patients cannot tolerate aggressive chemotherapy and therefore are in need of better treatment options.3
Several features make HCC a promising candidate for immunotherapy. Gene products of human leukocyte antigen (HLA)‐class I (HLA‐A, ‐B, and ‐C) are essential for presenting intracellular antigens on the cell surface so that they can be recognized by CD8+ T cells. In contrast to many cancers, HCCs do not generally down‐regulate HLA‐class I expression; HLA‐A is up‐regulated in 54% of patients.4 A recent meta‐analysis confirmed that high levels of HCC tumor infiltration by CD8+ T cells improved clinical outcomes, including overall survival.5 Several groups have isolated class I restricted tumor‐killing T‐cell clones that were generated spontaneously in HCC patients. Most of these clones recognized alpha‐fetoprotein (AFP; see previous works6, 7 and references therein).
AFP is absent from normal adult tissues, except for trace amounts produced by the liver. Fetal AFP synthesis, however, can resume in certain tumors of endodermal origin and to a lesser degree in both liver pathology and regeneration (reviewed in a previous work8). Serum AFP levels are used as a biomarker of the disease, because they correlate inversely with the survival of patients with HCC.9, 10, 11 Moreover, evidence points to active roles of the protein in cell proliferation, transformation, and metastasis, through both autocrine and paracrine actions.8, 12 Because of these properties, AFP has attracted interest as a target for immunotherapy. Several cytotoxic T‐cell epitopes have been identified, including the HLA‐A*02‐restricted AFP158‐166 peptide, FMNKFIYEI.13 Nevertheless, therapeutic interventions based on natural T‐cell receptor (TCR) repertoires for AFP/HLA have been effective in only a minority of treated patients.6, 13, 14
T cells that are naturally generated by thymic selection generally lack high‐affinity receptors for self‐antigens, including tumor‐associated self‐antigens.15 A potentially more successful approach involves improving TCR affinity through protein engineering. This process, however, risks altering specificity and/or generating cytotoxicity against tissues expressing levels of antigen that are too low to be recognized by natural TCRs, but high enough for an affinity‐enhanced mutant TCR.16, 17, 18 Several studies were able to find a “therapeutic window” of TCR affinity within which efficient tumor killing occurred with no or manageable damage to healthy tissue.14, 17 New TCRs recognizing AFP158‐166 /HLA‐A*02:01 have been described recently.19, 20 T cells transduced with these TCRs protected mice against subcutaneous tumor xenografts and displayed cytotoxicity against a limited number of AFP+, but not AFP–, cell lines. However, reactivity against tissues or cells expressing low‐to‐intermediate AFP levels was not tested. Herein, we report on the development of a TCR with optimal affinity and specificity for targeting HCC expressing HLA‐A2 and high levels of AFP.
Materials and Methods
AFP Target Validation
Publicly available data on AFP mRNA expression were obtained from the Genotype‐Tissue Expression Portal (GTEx21; https://gtexportal.org/) and from The Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/) and were reanalyzed using Oncomine (https://www.oncomine.org). qPCR was performed on the TissueScan Cancer cDNA Array obtained from Origene Technologies (catalog no. LVRT101; Rockville, MD) and on complementary DNA (cDNA) prepared from target cell lines (see below). Quantitative RT‐PCR was performed with a commercial Taqman assay (catalog no. 4331182; Thermo Fisher Scientific, Loughborough, UK), using the QuantiTect PCR Master mix with ROX (catalog no. 204345; Qiagen, Manchester, UK). cDNA was added to 25 ng/μL (cell line cDNAs) or 1 ng (Origene). Reactions were set up in duplicate in 96‐ or 384‐well plates covered with MicroAmp Optical Adhesive Film (catalog nos. 4309849 and 4311971; Thermo Fisher Scientific). Amplifications were carried out on a Quantstudio7 Real‐time PCR system (Applied Biosystems, Warrington, UK). Absolute quantification was performed by comparing mean computed tomography values of the test samples to those of a standard (titrations of plasmids with known number of copies of AFP or a housekeeping gene, ribosomal protein L32 [RPL32]). Numbers of AFP transcripts were normalized to 100 ng of total RNA or RPL32.
For immunohistochemistry (IHC) of AFP protein, tissue microarrays were purchased from US Biomax (LV805a, LV1201, and LV20812; Rockville, MD). We used a polyclonal rabbit antihuman α‐1‐fetoprotein antibody (catalog no. A0008; DAKO, Santa Clara, CA). Tissue sections were pretreated, and staining was performed in a DAKO Autostainer Link 48 with the EnVision Flex detection system and hematoxylin stain (catalog nos. K8002 and K8008; DAKO, Ely, UK), according to protocols supplied by the manufacturer.
Target Cell Lines
The HCC lines, HuH6, JHH‐5, and JHH‐4, were purchased from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan), and Hep3B cells were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig, Germany). Other human nonprimary cell lines were supplied by ATCC. Epstein‐Barr virus (EBV)‐transformed B lymphocytes were supplied by the Immunohistocompatibility Working Group (Seattle, WA). These cell lines were cultured in RPMI 1640 or Iscove's modified Dulbecco's medium supplemented with 10% fetal bovine serum (FBS), 1% PenStrep, and 1% l‐glutamine. Primary cell lines were purchased from Sciencell (Carlsbad, CA) and Promocell (Heidelberg, Germany). In addition, a panel of in vitro differentiated inducible pluripotent stem (iPS) cell‐derived cell lines (iCell) was purchased from Cellular Dynamics International (Madison, WI). These were derived from a single HLA‐A*02:01+ donor and selected to represent all major organs of the human body. Where necessary, cells were stably transduced with lentiviral vector to coexpress human β2‐microglobulin (β2m) with a relevant HLA‐A*02 subtype. Details of primary and iCell lines and the media used for their culture are shown in Supporting Table S1. All cell lines were routinely assessed for mycoplasma contamination (Mycoplasma experience Ltd, Bletchingley, UK), and cell‐line integrity was routinely confirmed by short tandem repeat analysis (LGC Ltd, Teddington, UK).
Protein Expression and Purification
Procedures used for preparation of proteins used in this study have been described.22 AFP158‐166 peptide (FMNKFIYEI, >90% pure) was obtained from Peptide Protein Research Ltd. (Fareham, UK). Codon‐optimized genes for all of the proteins used in this study, including soluble forms of both TCR α and β chains, soluble β2m (residues 21‐119), and a soluble HLA–A*02:01 heavy chain (residues 25‐276), were cloned into the pGMT7 expression vector (Promega, Southampton, UK). Soluble HLA‐A*02:01 heavy chain was expressed with a C–terminal in vitro biotinylation tag and refolded in the presence of both soluble β2m and peptide. After enzymic biotinylation with BirA‐500 (Avidity, Colorado, CA), this refolded complex was purified as soluble HLA‐biotinylated peptide HLA (pHLA) monomers by ion exchange and gel fitration to phosphate‐buffered saline. Refolding of soluble TCR α/β heterodimer was assisted by an artificial disulphide bond introduced by genetic engineering.22
Generation of Affinity‐Enhanced TCR Mutants and Biochemical Characterization
Affinity‐enhanced mutants were engineered by using parental TCR α and β chains as templates for mutagenesis of their complementarity‐determining regions. High‐affinity mutants were selected by phage display panning with pHLA‐coated magnetic beads. Mutations from specific binders were cloned as separate TCR α and β chains and refolded for affinity analysis. Equilibrium dissociation constants (KD) between TCRs and relevant biotinylated pHLA monomers were determined as described using streptavidin–coupled CM5 sensor chips and a BIAcore3000 instrument (GE Healthcare, Little Chalfont, UK).22
T‐Cell Transduction and Culture
Synthetic forms of the gene sequences for full‐length wild‐type (wt) AFP TCR α and β chains were codon‐optimized for maximal expression in human cells (GeneArt; Thermo Fisher Scientific). The genes for each pair of TCR chains were linked together in a single open‐reading frame by a P2A ribosomal skipping sequence.23 These fused genes were cloned into a glycoprotein of the vesicular stomatitis virus–pseudotyped lentiviral gene expression vector.24
Primary T cells expressing wt or affinity‐enhanced TCRs were generated from peripheral blood mononuclear cells (PBMCs) obtained from healthy volunteers. PBMCs were harvested using a Lymphoprep Ficoll gradient, diluted to 1 × 106 cells/mL in serum‐free RPMI 1640 medium, and either subjected to a CD8‐ or CD4‐negative isolation followed by mixing back at a CD4+:CD8+ cell ratio of 1:1, or CD14+ cell depletion. Isolated lymphocytes were incubated overnight with CD3/CD28 antibody‐coated beads (Dynabeads; Thermo Fisher) in RPMI 1640 medium supplemented with 10% FBS (R10), 100 U/mL of recombinant human interleukin (IL)‐2, and anti‐CD3/anti‐CD28. The following day, T cells were transduced to stably express the TCR genes by adding lentiviral vector supernatant at 1‐2× multiplicity of infection. TCR expression on the surface of stably transduced cells was confirmed by flow cytometry, using conjugated antibodies for the TCR β chain (Vβ1‐PE) and CD8. The fraction of CD8+ cells was 60%‐70% in T‐cell cultures transduced with panel 1 and 20%‐30% in cultures transduced with panel 2. For all TCRs except AFPc334 and AFPc335, transduction was 55%‐65% for CD8+ cells and over 70% for CD4+ cells. Similar results were obtained by staining with cognate pHLA tetramers, but sensitivity of the assay was low for some TCRs because of their relatively low affinity (not shown). Additionally, pHLA tetramer staining is poor in CD4+ T cells compared to CD8+ T cells and therefore under‐represents transduced T‐cell populations.
For small‐scale culture, cells were expanded in 48‐ or 24‐well microplates for up to 13 days. Beads were magnetically removed on day 5, and IL‐2‐containing R10 medium was refreshed every other day. For large‐scale “mock clinical” cultures, TCR‐transduced T cells were allowed to expand in TexMACS Good Manufacturing Practice serum‐free culture medium (Miltenyi Biotech, Bisley, UK), T150 culture flasks until day 5, then transferred to WAVE Cellbags and expanded on a WAVE bioreactor (GE Healthcare), until day 12 before harvest and cryopreservation.
Cytokine Secretion
Interferon (IFN)‐γ ELISpot plate assays were performed with kits from Becton Dickinson (catalog numbers 551849 and 552572; Oxford, UK). TCR‐transduced T cells were incubated overnight with the appropriate targets at a density of 50,000 cells each per well. Plates were read using an Immunospot analyzer (version 5.0; Cellular Technology Limited, Cleveland, OH).
Lymphokine secretion by fresh autologous whole‐blood cells was assessed following the addition of AFPc332T cells expanded in microplate culture. The Luminex panel (catalog number LHC0009M; Thermo Fisher Scientific) included granulocyte macrophage colony‐stimulating factor, IL‐6, eotaxin, IFN‐α, IL‐7, interferon γ‐induced protein 10, IFN‐γ, IL‐8, monocyte chemoattractant protein‐1, IL‐1β, IL‐10, monokine inducible by IFN‐γ, IL‐1RA, IL‐12, macrophage inflammatory protein 1 (MIP‐1)α, IL‐2, IL‐13, MIP‐1β, IL‐2R, IL‐15, regulated on activation normally T‐cell expressed secreted, IL‐4, IL‐17, IL‐5, and tumor necrosis factor α. Reactions were set up in triplicate at 50,000 and 250,000 cells per well in 96‐well plates. Included were three positive controls: cells stimulated with anti‐CD3/CD28–coated beads, targets pulsed with 5 μg/mL of AFP158‐166, and HepG2 liver cancer cells. Plate values were read with a Luminex MAGPIX reader.
Measurement of T‐Cell–Mediated Cytotoxicity
Cytotoxicity assays were performed using the IncuCyte ZOOM platform (Essen Bioscience, Welwyn Garden City, UK), using time‐lapse microscopy and detection of apoptotic cells through cleavage of a fluorogenic caspase‐3/7 substrate (CellPlayer 96‐Well Kinetic Caspase‐3/7 reagent; Essen Bioscience). For this, target cells were seeded in 96‐well plates the day before T‐cell addition, followed by addition of AFPc332T or nontransduced T cells with 5 μM of fluorogenic reagent. Images were taken of the wells at 3‐ or 4‐hour intervals for >60 hours, and fluorescent objects were enumerated using IncuCyte software (Essen Bioscience). A size‐exclusion gate was applied to remove dead T cells from further analysis.
Killing of Native Antigen‐Expressing Three‐Dimensional Microtissues
Three‐dimensional (3D) microtissue killing assays were performed using the IncuCyte ZOOM, using time‐lapse microscopy to measure the cytotoxic activity of AFPc332T cells toward green fluorescent protein (GFP)‐labeled 3D cancer cell‐line microtissues. For the generation of 3D microtissues, ultra‐low‐adhesion plates were used, facilitating the adhesion of cells to one another to form 3D microtissue. Target cells were seeded approximately 9 days before T‐cell addition at various cell densities (350‐1,000 cells/well). Images were acquired at 4‐hour intervals at 4× magnification. Microtissue formation was confirmed in each well before the addition of AFPc332T or nontransduced T cells (200,000 cells/well) from 2 separate donors. Killing of target GFP‐positive cancer lines by AFPc332T or nontransduced T cells was assessed by quantification of total GFP fluorescence area of the central microtissue core. Image analysis was performed using custom Zeiss Axiovision software.
Results
AFP Expression is Weak or Undetectable in Normal Tissues but High in a Significant Fraction of Liver Cancers
For a preliminary assessment of target safety and therapeutic potential, we analyzed AFP expression profiles in public RNA sequencing databases. TCGA allowed us to compare the abundance of AFP sequence reads in HCC samples (n = 371) and in nonmalignant controls adjacent to diverse tumors (n = 741, representing 23 different tissues). In addition, the GTEx Portal provided information on AFP mRNA expression in 51 normal tissues (n = 8,153 samples). Distribution of AFP transcript levels in both data sets is shown grouped by organ system and for selected tissues with the highest expression (Fig. 1). In both databases, AFP was most abundant in kidney, biliary, breast, and liver. Most HCC samples also contained AFP transcript numbers in a similar range, but ~30% of HCC expressed levels substantially above those found in any of the nonmalignant controls (note the log scale).
Figure 1.
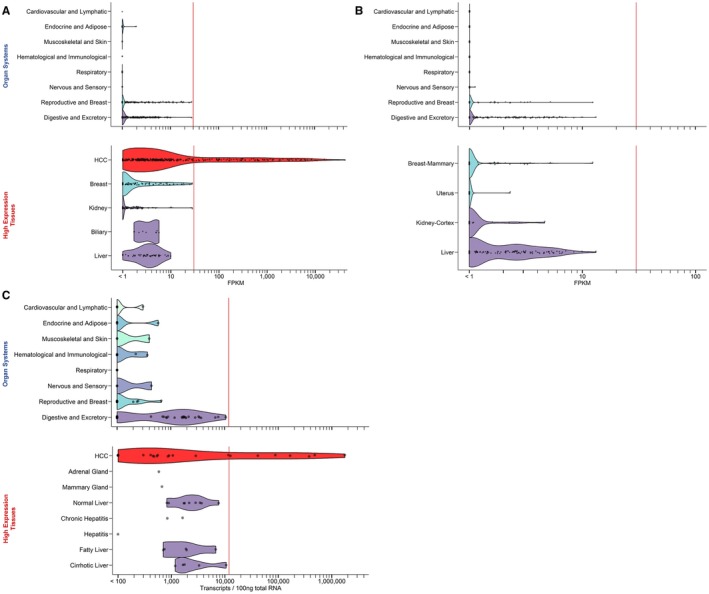
Expression of AFP mRNA in normal tissues and cancer, according to public databases. (A) Liver cancer and nonmalignant tissues adjacent to tumors, samples in TCGA. (B) Tissues from healthy individuals in GTEx. Data were obtained as fragments per kilobase million (FPKM) using the Oncomine platform. Samples were grouped by organ system (top panels) or tissue types in which samples with the highest expression levels were found (lower panels). The width of the ribbons shows the smoothed distribution of samples within a category as a function of AFP expression. Categories belonging to the same organ system are shown in identical colors in all four panels. (C) Analysis of AFP mRNA levels in selected tissues by RT‐qPCR. Presented as in (A) and (B).
To confirm these findings, we performed RT‐qPCR on mRNA isolated from human tissues and cell lines (Fig. 1C). Our RT‐qPCR primers, from a validated commercial assay, spanned exons 14 and 15 of the AFP gene. No variant isoforms of human AFP have been described that carried this region without the AFP158‐166 sequence in exons 4‐5. Conversely, variants of human AFP lacking the AFP158‐166 sequence but containing the probe region exist, but these are expressed at extremely low levels compared to wt AFP.25 Normal tissues, including healthy and inflamed liver, expressed <7,500 AFP transcripts per 100 ng of RNA. The only exception was a sample of cirrhotic tissue adjacent to a liver tumor (1.1 × 104 transcripts/100 ng of RNA). In agreement with the TCGA data, ~35% of HCC samples had AFP expression levels above those found in any of the nonmalignant tissues. Similar results were obtained when transcripts were normalized by RPL32 as a reference gene (not shown).
Tissue microarrays were used to evaluate AFP protein expression in HCC and liver disease (Fig. 2). Based on a semiquantitative scale of staining, 26% (15 of 57) of HCC samples had high and 16% (9 of 57) low‐to‐medium expression. Expression in the HCC line, HepG2, showed some variability; however, it was always equal to or less than expression in HCC patient samples. Only 5 of 224 nonmalignant liver samples had high‐intensity AFP staining. These tissues were from patients with cirrhosis or hepatitis, and 4 of 5 also had a diagnosis of HCC (no data were available for the fifth patient). In summary, AFP mRNA and protein is overexpressed in approximately one third of HCC with RNA transcript numbers typically 100‐ to 1,000‐fold above levels found in noncancerous tissue. This obsvervation formed the basis for developing a TCR with optimal sensitivity for AFP for adoptive T‐cell therapy of HCC.
Figure 2.
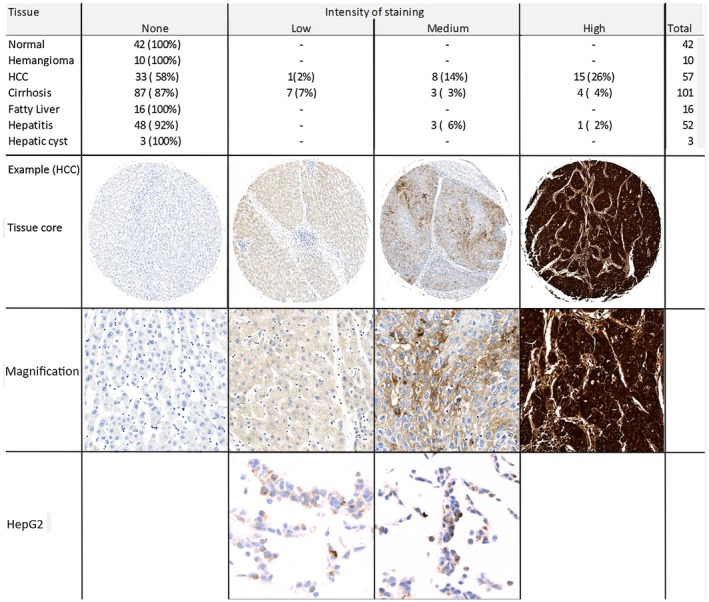
Detection of AFP protein in tissue samples by IHC staining of tissue microarrays. Shown are numbers and percentages of samples of normal and diseased liver. Staining intensity was scored visually, as shown in the example images. Representative samples of the HCC cell line, HepG2, are shown for comparison.
Generation and Selection of a TCR with Optimal Affinity and Specificity for AFP‐Expressing Cells
The wt parental T‐cell receptor AFPc239 was isolated from a T cell recognizing AFP158‐166 (FMNKFIYEI) presented by HLA*A02:01. When expressed in Escherichia coli and refolded, soluble AFPc293 demonstrated a specific, but weak, binding affinity for the cognate pHLA (KD 754 µM). We recloned both TCR chains into a phage display vector and applied random mutagenesis to the complementarity‐determining regions. The resulting library of mutant TCRs showed affinities across a broad range, increasing up to ~2,500‐fold compared to the parent TCR control (Table 1). wt and selected affinity‐enhanced mutant TCRs were recloned as full‐length codon‐optimized single open‐reading frame genes into lentiviral vectors.
Table 1.
Initial Panel of Soluble Mutant TCRs Engineered From the wt AFPc293 TCR
| TCR No. | T½ [s] | KD [µM] | Fold wt KD |
|---|---|---|---|
| c293 (wt) | NA | 754 | 1.0 |
| c327* | NA | 489 | 1.5 |
| c329* | NA | 356 | 2.1 |
| c330* | NA | 178 | 4.2 |
| c331 | 0.4 | 80 | 9.4 |
| c328 | NA | 37 | 20 |
| c332 | 0.7 | 11 | 71 |
| c326 | 1.5 | 1.5 | 503 |
| c333 | 3.8 | 0.71 | 1,062 |
| c334 | 4.3 | 0.52 | 1,450 |
| c335 | 14 | 0.31 | 2,432 |
Shown are the binding half‐life and biochemical affinity, as determined by BIAcore analysis, and the fold change in affinity over the wt TCR.
Low affinity: not tested further.
Abbreviation: NA, not available.
IFN‐γ secretion demonstrated that TCR AFPc332 (KD 10.6 µM) had the best ability to discriminate between AFP‐positive and –negative cells (Fig. 3A). Lower affinity resulted in loss of potency, whereas TCRs with higher affinity were cross‐reactive. To identify the optimal affinity with higher resolution, we generated a second panel of mutant AFP TCRs with KD values in the range bracketed by the two TCRs closest to AFPc332 in panel 1 (Table 2; Fig. 3B). As in the low‐resolution panel 1, IFN‐γ secretion by TCR‐transduced T cells increased with target affinity, reaching a plateau when stimulated by levels of AFP expressed by HepG2 liver cancer cells. Off‐target cross‐reactivity with AFP– cells emerged for KD values lower than 4.5 µM. Similar response data were obtained both for granzyme B secretion and for the direct killing of target cells by TCR‐transduced T cells from 3 different donors (not shown). We concluded that AFPc350, AFPc332, and AFPc351 TCRs occupied a window of optimal TCR affinity, characterized by enhanced potency and full retention of specificity. From these, TCR AFPc332 was selected for further investigation. AFPc350 and AFPc351 would have been pursued further if AFPc332 failed in specificity or potency testing.
Figure 3.
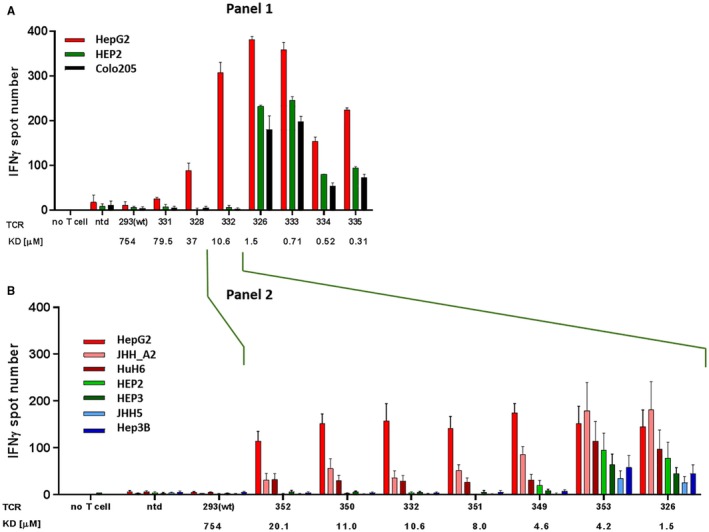
Functional analysis of AFP TCR mutant panels by IFN‐γ ELISpot. Bars represent mean numbers of responding T cells in at least 6 donors, each tested in triplicate, with the standard error for interdonor variation. Red: HLA‐A*02:01+AFP+ HCC cell lines (HepG2, JHH5.A2, and HuH6). Blue: HLA‐A*02‐AFP+ HCC cell lines (JHH5, Hep3B). Green: normal primary hepatocytes (HEP2, HEP3). TCRs are arranged by increasing affinity from left to right. (A) wt TCR AFPc239 and initial panel of mutants. Data are representative of 2 donors, showing mean ± SEM in triplicate wells. (B) Refined panel of enhanced affinity TCRs against HCC cell lines and normal liver cells. Abbreviation: ntd, nontransduced T cells.
Table 2.
Panel 2 Mutant TCRs Following High‐Resolution Engineering
| TCR No. | T½ [s] | KD [µM] | Fold wt KD |
|---|---|---|---|
| c352 | 0.41 | 20.1 | 38 |
| c350 | 0.79 | 11.0 | 68 |
| c332 | 0.70 | 10.6 | 71 |
| c351 | 0.77 | 8.0 | 94 |
| c349 | 1.82 | 4.6 | 166 |
| c353 | 1.53 | 4.2 | 181 |
Shown are the binding half‐lives and biochemical affinities, as determined by BIAcore analysis (repeated for c332), and the fold change in affinity over the wt TCR.
Safety and Potency Testing
To further assess the safety and potency of AFPc332T cells, we investigated how their responses correlated with levels of AFP mRNA in a variety of target cell lines naturally expressing AFP. In addition, Colo205 cells (for IFN‐γ assays) or HCT116 cells (for cytotoxicity assays) were genetically modified to generate batches producing a range of AFP levels (unpublished work) that were verified by qPCR. A consistent trend was apparent, indicating that 1‐2 × 106 transcripts per 100 ng of RNA are required for triggering IFN‐γ secretion or cytotoxicity in a detectable number of AFPc332T cells in vitro (Fig. 4). Potency of AFPc332T cells was further investigated by quantifying their ability to kill antigen‐positive 3D microtissues. Addition of AFPc332T cells to HepG2‐GFP microtissues resulted in an almost complete loss of GFP fluoresence with time, indicating the rapid and complete destruction of microtissues (Fig. 5, representative data). Together, these results confirmed that AFPc332T cells are specific for high levels of AFP mRNA and protein that distinguish a subset of HCC from other human tissues. AFPc332T cells also recognized AFP158‐166 presented by HLA‐A*02:02 and A*02:07 with functional avidities that were similar to A*02:01 (Supporting Fig. S1). Responses toward A*02:05 and A*02:06 were slightly increased compared to A*02:01.
Figure 4.
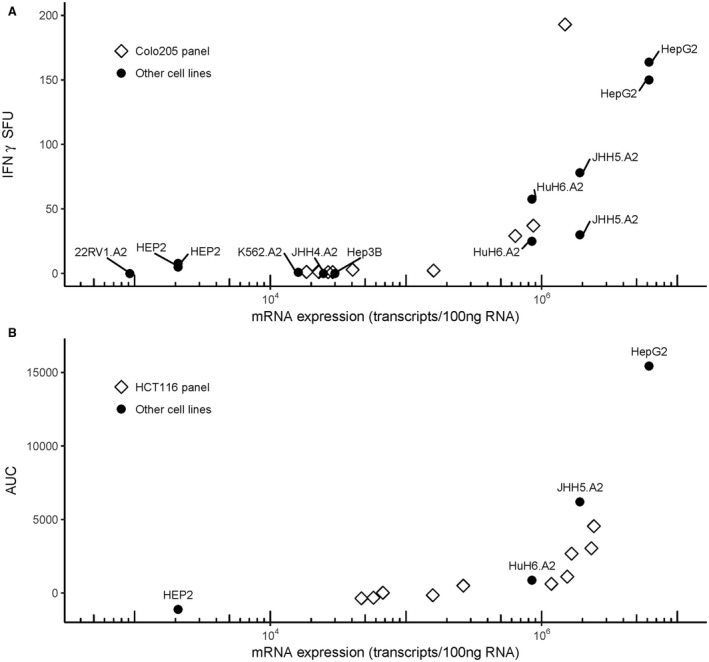
Correlation between AFP mRNA expression in target cells and responses by AFPc332T cells. Numbers of IFN‐γ secreting AFPc332T cells (ELISpot count; A) or T‐cell–mediated cytotoxicity (B) were plotted against AFP trancripts in target cells. The suffix “.A2” indicates HLA‐A*02:01 transduced lines. AFP transcript levels shown for JHH4.A2 are those measured in the parental JHH4 HCC line before transduction with HLA‐A*02. T‐cell–mediated cytotoxicity was quantified from event counts (spot‐forming units, SFU) as the area under the curve (AUC) from 1 to 36 hrs, after subtraction of cytotoxicity observed in the presence of nontransduced T cells.
Figure 5.
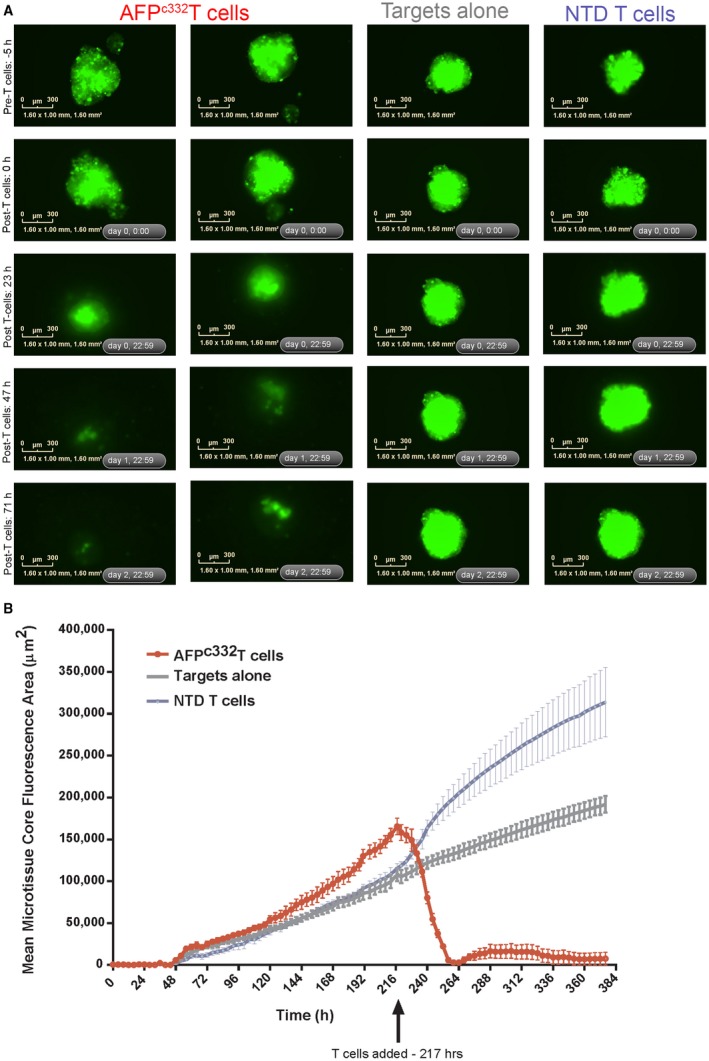
Cytotoxic activity of AFPc332T cells toward small AFP expressing HepG2‐GFP 3D microtissues. HepG2‐GFP cells seeded at 350 cells/well resulted in microtissues of ~450‐500 μm estimated diameter at the time of T‐cell addition. (A) Images show the GFP fluoresence of microtissues during the course of the assay. Time points shown refer to the approximate times after initial HepG2‐GFP target cell seeding: pre‐T cells –5 hours = 212 hours posttarget seeding, 0 hours post‐T cells = 217 hours, 23 hours post‐T cells = 240 hours, 47 hours post‐T cells = 264 hours, 71 hours post‐T cells = 288 hours. Scale bars = 300 μm. (B) The GFP fluorescent area of the microtissue in each well was measured over time for each treatment. Black arrow indicates T‐cell addition. Data are expressed as mean microtissue fluorescence area ± SEM of all three wells for each condition. Abbreviation: NTD, nontransduced T cells.
Cross‐reactivity for nontarget pHLA can occur in adoptive therapy with TCR engineered T cells16, 26 and chimeric antigen receptor T cells.27 To mitigate this risk, we used two complemenatry approaches: testing a large panel of AFP– cell types from a variety of tissues, and a combined in vitro/in silico approach to scan the human proteome for potentially cross‐reactive peptides.
To test whether AFPc332T cells might produce a systemic inflammatory response (cytokine release syndrome), autologous transduced T cells generated from 3 different donors were incubated with matched whole blood at T‐cell concentrations similar to those in patients after infusion. None of the 25 serum cytokines and chemokines tested (see Materials and Methods) were induced, unless either anti‐CD3/CD28 antibodies or AFP158‐166 peptide were added as positive stimulatory controls (data not shown).
We next evaluated the off‐target recognition, shown by IFN‐γ production by AFPc332T cells, using a panel of 123 human primary cells and iPS‐derived cells representing 10 tissues and 67 cell types (Supporting Table S1). Where necessary, cell lines were transduced with lentiviral vector to coexpress HLA‐A*02 subtypes and β2m. Weak, but consistent, responses by AFPc332T cells were found toward one of eight brain vascular pericyte cell lines and one of two thyroid fibroblast lines (Supporting Fig. S2). In both cases, no AFP mRNA was detectable by RT‐qPCR, ruling out reactivity attributed to AFP expression or contamination with AFP‐producing cells (data not shown). Because both cell lines naturally expressed the HLA‐A*02:02 subtype, we investigated the possibility of alloreactivity. Several HLA‐A*02– brain vascular pericyte lines generated responses when they were transduced with HLA‐A*02:02/β2m, but not with HLA‐A*02:01/β2m (Supporting Fig. S2B). Similar results were obtained for cytotoxic killing (data not shown). Two other A*02:02+ lines, HEF2 (esophageal fibroblasts) and HCPEpiC3 (choroid plexus epithelial cells), did not generate a response. Further HLA‐alloreactivity testing was conducted against an extensive panel of EBV‐transformed human B‐cell lines naturally expressing 129 of the most common HLA alleles (Supporting Table S2). Two of these lines, FH42 and 230699, induced IFN‐γ release. Their only shared HLA allele, HLA‐A*24:02, did not generate a response in several other cell lines. Each cell line also carried a unique allele, HLA‐C*04:04 in FH42 and HLA‐B*51:03 in 230699. Because these alleles are extremely rare, it was not viable to confirm their alloreactivity in alternate lines. None of the cell lines shown in Fig. 4 have been reported to carry these alleles.
Cell panels and animal models might be insufficient to assay the full range of human self‐peptides presented by HLA.26 We therefore performed a comprehensive mutational analysis to identify single amino acid substitutions in the AFP158‐166 peptide that were tolerated by the AFPc332 TCR (“X‐scan”).28 Matching sequences were synthesized as synthetic peptides, and those which generated a response were tested as naturally processed and presented antigen. No evidence for recognition of antigens other than AFP by the AFPc332 TCR was found (Supporting Table S3).
Discussion
Although T cells transduced with affinity‐enhanced TCRs can show superior antitumor activity in vitro and in vivo, this strategy can also produce reactivity toward normal tissues. Off‐target cross‐reactivity has been observed in clinical trials toward homologous peptides (such as from other proteins of the same family16), but also in absence of any recognizable sequence homology.26 The X‐scan was designed to screen for the latter and complement testing in cell lines where these might not fully represent all proteins expressed in the human body.28 Novel specificities that might arise from mispairing between transduced and endogenous TCR chains are much harder to predict. Autoimmunity caused by this scenario has so far been observed only in vitro and in a mouse model,29 but not in clinical trials. Strategies to promote the selective pairing of the transduced TCR chains exist (reviewed in a previous work30), but can cause problems of their own.31 We used a lentiviral vector optimized for TCR expression,24 codon optimization,32 and a 2A peptide‐linked retroviral vector23 to favor the expression of transduced TCRs over endogenous and mispaired TCR α and β chains.
On‐target/off‐tumor reactions against healthy tissues are a specific risk when targeting genes that are expressed preferentially, but not exclusively, by tumors. In most cases, adverse effects were either absent or manageable in mice33, 34 and humans (17, 18; also reviewed in another work27). However, cases of fatal toxicity arose from recognition of peptide epitopes shared by normal brain16 and lung tissue.35 We therefore aimed to generate a TCR with an optimal affinity, sufficient to trigger responses against high densities of AFP158‐166 HLA‐A*02:01 complexes on tumor cells, but not against the lower densities that might exist on nonmalignant cells. Unfortunately, neither the pHLA densities on individual cells in patient tissues nor the activation threshold of AFP‐specific T cells in a clinical setting are easily determined. We used AFP mRNA levels and in vitro T‐cell responses as surrogate makers. Through bioinformatic searches, RT‐qPCR, and immunohistology, we confirmed and extended previous observations that a sizeable fraction of hepatocellular tumors express AFP at levels exceeding those in any nonmalignant tissue, including inflamed liver. In fact, the highest levels of AFP in samples not classified as tumor were from 5 of 135 patients with cirrhosis or hepatitis, of which 4 also had a diagnosis of HCC (no data were available for the fifth patient). In these, we theorize that AFP overexpression might reflect early stages of transformation,36 driven by viral proteins.12
A major source of AFP in nonmalignant liver is a type of cells resembling fetal hepatoblasts.37 These cells constitute less than 0.1% of all cells in the adult organ, but can expand in liver disease and regeneration. Hepatoblasts arise through differentiation of hepatic stem cells, but in contrast to the latter produce high amounts of AFP. AFP expression is lost again with further differentiation of hepatoblasts into bilary and hepatic cells.37 Therefore, off‐tumor cytoxicity against hepatoblasts might delay liver regeneration, but without affecting the critical stem cell population or fully differentiated tissue. An autoimmune hepatitis caused by AFP vaccination of partially hepatectomized mice resolved spontaneously when the organ recovered.38 Less evidence exists for low or intermediate AFP expression at the cellular level in situ in nonmalignant liver or other tissues. In normal rat liver, partial hepatectomy induced a weak AFP mRNA hybridization signal in all hepatocytes.39 In humans, variable fluorescence immunostaining for AFP outside the tumor area was observed in combined hepatocellular cholangiocarcinoma and was associated with hepatocyte dedifferentiation and proliferation.40 In our in vitro experiments, however, primary hepatocytes (and other cells expressing similar levels of AFP mRNA) did not generate IFN‐γ or cytotoxic responses to AFPc332 transduced cells. Moreover, we found only low levels of HLA‐A, ‐B, and ‐C in normal liver, compared to HCC (unpublished observations), which could further enhance treatment safety.
Protection of mice against subcutaneous xenografts of tumor cell lines has been used to assess the potency of TCRs in vivo 19, 20; however, these models involve immune‐compromised hosts. Syngeneic grafts in wt animals tend to elicit stronger responses than naturally evolved tumors.41 In addition, species differences exist regarding antigen expression, presentation, and immune systems. Taken together, it seems unlikely that current animal models can provide additional information on the risk‐benefit profile of new TCRs, beyond what can be demonstrated from a carefully chosen combination of in vitro assays.
In general, affinity dissociation constants (KDs) of 1‐10 μM seem to produce the strongest and greatest number of effector functions for HLA class I–restricted TCRs (reviewed in a previous work42). The 11‐μM KD affinity of AFPc332 is just below this range, but we anticipate that it will be sufficient, considering that the target density is much higher than that of NY‐ESO‐1 presented by HLA‐A2.43 A better correlate of TCR potency is functional avidity (half maximal effective concentration; EC50), which is the concentration of antigenic peptide triggering half‐maximal responses such as cytokine secretion or target cell killing. AFPc332 recognized AFP158‐166 presented by several HLA‐A2 subtypes with an EC50 of ~1 nM. This functional avidity is within the range of those of the NY‐ESO‐1–specific TCRs that have produced encouraging clinical results.17, 44, 45 Moreover, 2 HCC patients who generated T‐cell clones with an EC50 of ≤10 nM in response to AFP peptide vaccination had better and longer‐lasting tumor responses. No autoimmunity was observed.14
AFPc332 lacks alloreactivity with common HLA‐class I alleles; however, the rare subtypes HLA‐C*04:04 and HLA‐B*51:03 were identified as potentially alloreactive. In addition, reactivity was observed to HLA‐A*02:02+ brain pericytes and thyroid fibroblasts, but no other HLA‐A*02:02+ cell lines. This observation suggests the involvement of a peptide or peptides specific for these cell lineages and demonstrates the importance of testing alloreactivity in a large panel of cell types. Based on current data, a clinical trial with AFPc332 should exclude patients who are carriers of the above alleles.
AFP is an attractive target for adoptive T‐cell therapy of HCC, particularly in aggressive forms for which limited treatment options exist. Using TCR engineering and rigorous testing (Fig. 6), we have developed a TCR with the potential to benefit HLA‐A2+ individuals with AFP‐overexpressing HCC. In an ongoing clinical trial (NCT03132792), we are examining biopsies for AFP expression and T‐cell infiltration while closely monitoring liver chemistries. These observations will increase our understanding of the role of TCR affinity and T‐cell avidity in vivo and help to enable safe and effective targeting of AFP and other cancer‐associated antigens.
Figure 6.
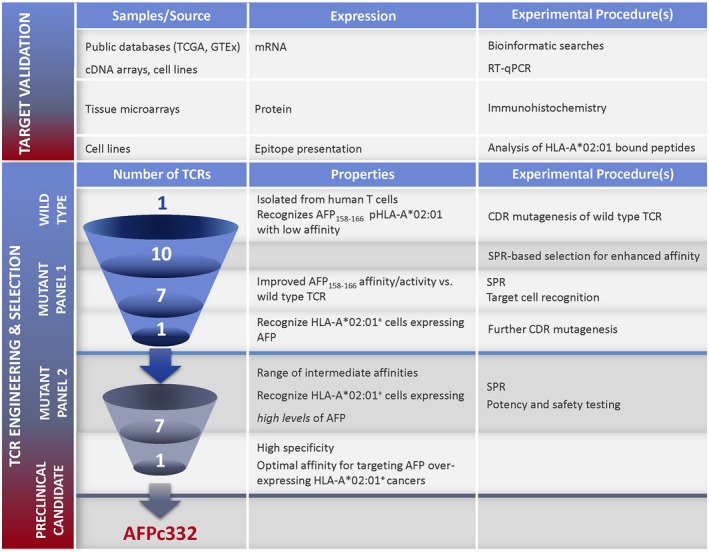
Summary of the experimental strategy for generating a TCR with high specificity and optimal affinity for targeting AFP‐overexpressing HLA‐A*02:01+ cancers. Abbreviations: CDR, complementarity determining region; SPR, surface plasmon resonance.
Supporting information
Acknowledgment
We thank our colleagues who contributed to this work: Bent Jakobsen (conceptualizing and refining research ideas), Qian Gao (database searches), Adriano Quattrini and David Rigby (cell culture), Adele Richmond (lentiviral vector preparation), Joana Senra and Karen Howe (RT‐qPCR data), Darragh Crowley (T‐cell potency assays), Barbara Tavano and Tina Ahmed (T‐cell phenotyping and proliferation), Rachel Abbott, Terri Cornforth, and Louise Rice (T‐cell function assays), and Peter Molloy, Immunocore Ltd (phage display).
Supported by Adaptimmune.
Potential conflict of interest: Dr. Wiederman is employed by and owns stock in Adaptimmune. Dr. Saini is employed by and owns stock in Adaptimmune. Dr. Anderson is employed by and owns stock in Adaptimmune. Dr. Bennett is employed by and owns stock in Adaptimmune. Dr. Ferjentsik is employed by and owns stock in Adaptimmune. Dr. Gerry is employed by and owns stock in Adaptimmune. Dr. Norry is employed by and owns stock in Adaptimmune. Dr. Pumphrey is employed by and owns stock in Adaptimmune. Dr. Sanderson is employed by and owns stock in Adaptimmune. Dr. Weissensteiner is employed by and owns stock in Adaptimmune. Dr. Maroto is employed by and owns stock in Adaptimmune. Dr. Ferronha is employed by and owns stock in Adaptimmune.
References
Author names in bold designate shared co‐first authorship.
- 1. Flores A, Marrero JA. Emerging trends in hepatocellular carcinoma: focus on diagnosis and therapeutics. Clin Med Insights Oncol 2014;8:71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 3. Gluer AM, Cocco N, Laurence JM, Johnston ES, Hollands MJ, Pleass HC, et al. Systematic review of actual 10‐year survival following resection for hepatocellular carcinoma. HPB (Oxford) 2012;14:285‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shen Y, Xia M, Zhang J, Xu L, Yang J, Chen A, et al. IRF‐1 and p65 mediate upregulation of constitutive HLA‐A antigen expression by hepatocellular carcinoma cells. Mol Immunol 2009;46:2045‐2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yao W, He JC, Yang Y, Wang JM, Qian YW, Yang T, Ji L. The prognostic value of tumor‐infiltrating lymphocytes in hepatocellular carcinoma: a systematic review and meta‐analysis. Sci Rep 2017;7:7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, et al. Immunodominance and functional alterations of tumor‐associated antigen‐specific CD8+ T‐cell responses in hepatocellular carcinoma. Hepatology 2014;59:1415‐1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mizukoshi E, Yamashita T, Arai K, Sunagozaka H, Ueda T, Arihara F, et al. Enhancement of tumor‐associated antigen‐specific T cell responses by radiofrequency ablation of hepatocellular carcinoma. Hepatology 2013;57:1448‐1457. [DOI] [PubMed] [Google Scholar]
- 8. Mizejewski GJ. Does alpha‐fetoprotein contribute to the mortality and morbidity of human hepatocellular carcinoma? A commentary. J Hepatocell Carcinoma 2016;3:37‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamashita T, Forgues M, Wang W, Kim JW, Ye Q, Jia H, et al. EpCAM and alpha‐fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res 2008;68:1451‐1461. [DOI] [PubMed] [Google Scholar]
- 10. Agopian VG, Harlander‐Locke MP, Markovic D, Zarrinpar A, Kaldas FM, Cheng EY, et al. Evaluation of patients with hepatocellular carcinomas that do not produce alpha‐fetoprotein. JAMA Surg 2017;152:55‐64. [DOI] [PubMed] [Google Scholar]
- 11. Kimura H, Ohkawa K, Miyazaki M, Sakakibara M, Imanaka K, Tamura T, et al. Subclassification of patients with intermediate‐stage (Barcelona Clinic Liver Cancer stage‐B) hepatocellular carcinoma using the up‐to‐seven criteria and serum tumor markers. Hepatol Int 2017;11:105‐114. [DOI] [PubMed] [Google Scholar]
- 12. Zhu M, Li W, Lu Y, Dong X, Lin B, Chen Y, et al. HBx drives alpha fetoprotein expression to promote initiation of liver cancer stem cells through activating PI3K/AKT signal pathway. Int J Cancer 2017;140:1346‐1355. [DOI] [PubMed] [Google Scholar]
- 13. Butterfield LH, Ribas A, Dissette VB, Lee Y, Yang JQ, De la Rocha P, et al. A phase I/II trial testing immunization of hepatocellular carcinoma patients with dendritic cells pulsed with four alpha‐fetoprotein peptides. Clin Cancer Res 2006;12:2817‐2825. [DOI] [PubMed] [Google Scholar]
- 14. Nakagawa H, Mizukoshi E, Kobayashi E, Tamai T, Hamana H, Ozawa T, et al. Association between high‐avidity T‐cell receptors, induced by alpha fetoprotein‐derived peptides, and anti‐tumor effects in patients with hepatocellular carcinoma. Gastroenterology 2017;152:1395‐1406.e10. [DOI] [PubMed] [Google Scholar]
- 15. Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, Jakobsen BK. Different affinity windows for virus and cancer‐specific T‐cell receptors: implications for therapeutic strategies. Eur J Immunol 2012;42:3174‐3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morgan RA, Chinnasamy N, Abate‐Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti‐MAGE‐A3 TCR gene therapy. J Immunother 2013;36:133‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T‐cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009;114:535‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DN, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011;19:620‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun L, Guo H, Jiang R, Lu L, Liu T, He X. Engineered cytotoxic T lymphocytes with AFP‐specific TCR gene for adoptive immunotherapy in hepatocellular carcinoma. Tumour Biol 2016;37:799‐806. [DOI] [PubMed] [Google Scholar]
- 20. Zhu W, Peng Y, Wang L, Hong Y, Jiang X, Li Q, et al. Identification of alpha‐fetoprotein‐specific T cell receptors for hepatocellular carcinoma immunotherapy. Hepatology 2018;68:574‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, et al. A novel approach to high‐quality postmortem tissue procurement: the GTEx Project. Biopreserv Biobank 2015;13:311‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cole DK, Pumphrey NJ, Boulter JM, Sami M, Bell JI, Gostick E, et al. Human TCR‐binding affinity is governed by MHC class restriction. J Immunol 2007;178:5727‐5734. [DOI] [PubMed] [Google Scholar]
- 23. Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi‐gene deficiency in vivo using a single ‘self‐cleaving’ 2A peptide‐based retroviral vector. Nat Biotechnol 2004;22:589‐594. [DOI] [PubMed] [Google Scholar]
- 24. Yang S, Cohen CJ, Peng PD, Zhao Y, Cassard L, Yu Z, et al. Development of optimal bicistronic lentiviral vectors facilitates high‐level TCR gene expression and robust tumor cell recognition. Gene Ther 2008;15:1411‐1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tagaya H, Fukasawa H, Shoda T, Hoshi K, Hirata S. Novel alpha‐fetoprotein‐V messenger RNA isoforms in humans. Reprod Sci 2009;16:794‐801. [DOI] [PubMed] [Google Scholar]
- 26. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin‐derived HLA‐A1‐presented peptide as a cross‐reactive target for engineered MAGE A3‐directed T cells. Sci Transl Med 2013;5:197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gross G, Eshhar Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off‐tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol 2016;56:59‐83. [DOI] [PubMed] [Google Scholar]
- 28. Border EC, Sanderson JP, Weissensteiner T, Gerry AB, Pumphrey N. Affinity‐enhanced T‐cell receptors for adoptive T‐cell therapy targeting MAGE‐A10: strategy for selection of an optimal candidate. OncoImmunol 2018; doi: 10.1080/2162402X.2018.1532759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci U S A 2010;107:10972‐10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spear TT, Nagato K, Nishimura MI. Strategies to genetically engineer T cells for cancer immunotherapy. Cancer Immunol Immunother 2016;65:631‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spear TT, Foley KC, Garrett‐Mayer E, Nishimura MI. TCR modifications that enhance chain pairing in gene‐modified T cells can augment cross‐reactivity and alleviate CD8 dependence. J Leukoc Biol 2018;103:973‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scholten KB, Kramer D, Kueter EW, Graf M, Schoedl T, Meijer CJ, et al. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin Immunol 2006;119:135‐145. [DOI] [PubMed] [Google Scholar]
- 33. Eck SC, Turka LA. Adoptive transfer enables tumor rejection targeted against a self‐antigen without the induction of autoimmunity. Cancer Res 2001;61:3077‐3083. [PubMed] [Google Scholar]
- 34. Drent E, Themeli M, Poels R, de Jong‐Korlaar R, Yuan H, de Bruijn J, et al. A rational strategy for reducing on‐target off‐tumor effects of CD38‐chimeric antigen receptors by affinity optimization. Mol Ther 2017;25:1946‐1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010;18:843‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sturgeon CM, Duffy MJ, Hofmann BR, Lamerz R, Fritsche HA, Gaarenstroom K, et al. National Academy of Clinical Biochemistry Laboratory Medicine Practice Guidelines for use of tumor markers in liver, bladder, cervical, and gastric cancers. Clin Chem 2010;56:e1‐e48. [DOI] [PubMed] [Google Scholar]
- 37. Schmelzer E, Wauthier E, Reid LM. The phenotypes of pluripotent human hepatic progenitors. Stem Cells 2006;24:1852‐1858. [DOI] [PubMed] [Google Scholar]
- 38. Geissler M, Mohr L, Weth R, Kohler G, Grimm CF, Krohne TU, et al. Immunotherapy directed against alpha‐fetoprotein results in autoimmune liver disease during liver regeneration in mice. Gastroenterology 2001;121:931‐939. [DOI] [PubMed] [Google Scholar]
- 39. Bernuau D, Poliard A, Feldmann G. In situ cellular analysis of alpha‐fetoprotein gene expression in regenerating rat liver after partial hepatectomy. Hepatology 1988;8:997‐1005. [DOI] [PubMed] [Google Scholar]
- 40. Cai X, Zhai J, Kaplan DE, Zhang Y, Zhou L, Chen X, et al. Background progenitor activation is associated with recurrence after hepatectomy of combined hepatocellular‐cholangiocarcinoma. Hepatology 2012;56:1804‐1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Anders K, Kershaw O, Larue L, Gruber AD, Blankenstein T. The immune system prevents recurrence of transplanted but not autochthonous antigenic tumors after oncogene inactivation therapy. Int J Cancer 2017;141:2551‐2561. [DOI] [PubMed] [Google Scholar]
- 42. Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N. Identifying individual T cell receptors of optimal avidity for tumor antigens. Front Immunol 2015;6:582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Purbhoo MA, Sutton DH, Brewer JE, Mullings RE, Hill ME, Mahon TM, et al. Quantifying and imaging NY‐ESO‐1/LAGE‐1‐derived epitopes on tumor cells using high affinity T cell receptors. J Immunol 2006;176:7308‐7316. [DOI] [PubMed] [Google Scholar]
- 44. Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY‐ESO‐1‐reactive T‐cell receptor: long‐term follow‐up and correlates with response. Clin Cancer Res 2015;21:1019‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rapoport AP, Stadtmauer EA, Binder‐Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY‐ESO‐1‐specific TCR‐engineered T cells mediate sustained antigen‐specific antitumor effects in myeloma. Nat Med 2015;21:914‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials