

Summary
Gender assignment errors are common in some animal species and lead to inaccuracies in downstream analyses. Procedures for detecting gender misassignment are available for array‐based SNP data but are still being developed for genotyping‐by‐sequencing (GBS) data. In this study, we describe a method for using GBS data to predict gender using X and Y chromosomal SNPs. From a set of 1286 X chromosomal and 23 Y chromosomal deer (Cervus sp.) SNPs discovered from GBS sequence reads, a prediction model was built using a training dataset of 422 Red deer and validated using a test dataset of 868 Red deer and Wapiti deer. Prediction was based on the proportion of heterozygous genotypes on the X chromosome and the proportion of non‐missing genotypes on the Y chromosome observed in each individual. The concordance between recorded gender and predicted gender was 98.6% in the training dataset and 99.3% in the test dataset. The model identified five individuals across both datasets with incorrect recorded gender and was unable to predict gender for another five individuals. Overall, our method predicted gender with a high degree of accuracy and could be used for quality control in gender assignment datasets or for assigning gender when unrecorded, provided a suitable reference genome is available.
Keywords: allosomal SNPs, deer, low depth, sex assignment, XX/XY system
Gender misassignment, due to misidentification on examination, data input errors or genotyping errors, occurs in animal species and can hinder genetic gain in breeding programmes (Zhang et al. 2016). Some software packages, such as plink (Purcell et al. 2007) and pygenclean (Lemieux Perreault et al. 2013), contain quality control procedures to detect gender misassignment. These procedures generally use heterozygosity on the X chromosome to distinguish gender, because males, with only one X chromosome, should be hemizygous, whereas females can be heterozygous at some loci as they have two X chromosomes. One exception is the pseudo‐autosomal region (PAR; Johnston et al. 2017), a region of homology between the X and Y chromosomes where males can be heterozygous. Data in the PAR are usually discarded in SNP‐based gender prediction. There are limitations to using the X chromosome as the sole means of predicting gender, as sex anomalies can lead to incorrect assignment. Including Y chromosome information in the prediction may assist in correct assignment (Laurie et al. 2010; Turner et al. 2011; Zhang et al. 2016). Various gender tests have been developed for array‐based SNP calling platforms and typically focus on detecting sample handling errors in human genetic data (Anderson et al. 2010; Laurie et al. 2010; Turner et al. 2011). The high cost of SNP arrays prohibits their widespread use in many animal species, for which a cost‐efficient alternative is genotyping‐by‐sequencing (GBS). In this study, we developed a simple method for building a gender prediction model using X and Y chromosomal SNPs obtained from GBS data, using deer (Cervus sp.) as an example.
A set of allosomal (sex‐linked) SNPs were identified from GBS reads using the following procedure. A set of tag pairs, a collection of sequence reads that differ by a single base pair in the genomic part of a read, were identified using uneak (Lu et al. 2013) from GBS sequence reads in a large and diverse catalogue of deer samples. These tag pairs were aligned to a Red deer reference genome (Bana et al. 2018) using bowtie2 version 2.1.0 (Langmead & Salzberg 2012), with the parameter setting –very‐sensitive and custom Python scripts. SNPs located on tag pairs that aligned to the sex chromosomes were identified as allosomal; 1286 X chromosomal and 23 Y chromosomal SNPs were identified.
A training dataset of 422 deer, consisting predominantly of Red deer (Cervus elaphus) with some Red‐by‐Wapiti (Cervus canadensis) crosses, was used to build a prediction model, and a test dataset, consisting of 619 Red deer and 249 Wapiti, was used to validate the model, in which gender was recorded for all individuals. The animals were managed in accordance with the provisions of the New Zealand Animal Welfare Act 1999 and the Codes of Welfare developed under sections 68–79 of the Act. Tissue samples were collected using ear punches, and DNA was extracted according to Clarke et al. (2014). The GBS libraries were prepared using the restriction enzyme PstI following the method described by Elshire et al. (2011) with variations as outlined by Dodds et al. (2015). The individuals were sequenced at AgResearch, Invermay, Animal Genomics Laboratory on an Illumina HiSeq 2500 v4 chemistry yielding 100‐bp single end reads.
Using the training dataset, the identified allosomal SNPs were filtered to remove erroneous SNPs as follows. The X chromosomal SNPs were discarded if they were located in the PAR, if at least 10% of males showed heterozygosity or if at least 10% of females had missing genotypes. Using the data shown in Fig. S1, the PAR was inferred to commence at 170 Mb and extend to the telomere. For the Y chromosome, SNPs were discarded if at least 5% of females had non‐missing genotype calls or if at least 50% of males had heterozygous genotypes. These numbers were liberal to preserve more SNPs on the Y chromosome. Lastly, SNPs with a minor allele frequency less than 0.015 were removed as recommended by Zhang et al. (2016).
The samples for the test and training datasets with the filtered allosomal SNPs were run through kgd (https://github.com/AgResearch/KGD) using default settings, except that samples with a mean depth below 0.3 were discarded. Summary statistics of the filtered data are given in Table 1. For each individual, the proportion of Y chromosomal SNPs with non‐missing genotypes, , and the heterozygosity of the X chromosomal SNPs, H i, were computed. Heterozygosity was adjusted for read depth, using ideas from Dodds et al. (2015), to account for under‐called heterozygotes and was computed as , where n i is the number of heterozygous genotypes for individual i, and d ij is the read depth at SNP j in individual i for j = 1, … , M x. Plots of against H i, which we refer to as gender plots, were produced, where M y is the number of (filtered) Y chromosomal SNPs. The R code and the data used in this analysis are available at https://github.com/AgResearch/GBS_Gender_Predict.
Table 1.
Summary statistics for the training and test datasets after being run through the kgd software
| Dataset | Breed | # deer | # X SNPs | # Y SNPs | Recorded gender | Mean sample depth | |
|---|---|---|---|---|---|---|---|
| Females | Males | ||||||
| Training | Red | 422 | 1026 | 17 | 160 | 261 | 2.89 |
| Test | Red | 619 | 1010 | 16 | 305 | 314 | 1.52 |
| Test | Wapiti | 249 | 131 | 118 | |||
The gender plot for the training dataset is given in Fig. 1a. In general, males were located in the upper left‐hand area of the plot and females along and slightly above the x‐axis. A prediction model was built using the empirical results from the training set such that an individual was classified male if or female if , otherwise gender was unassigned. Under this model, 98.6% of the predicted gender matched the recorded gender. Four recorded females were classified as male and one recorded male classified as female. The gender of these individuals was identified as phenotypic misassignments on re‐examination, with the prediction model correctly classifying each individual. The gender plot for the test dataset (Fig. 1b) gave results similar to those for the training dataset. The predicted gender of the Wapiti matched their recorded gender in all cases, except gender was not assigned for two recorded males. For the Red deer, two recorded males were not assigned gender, one recorded male was predicted female and one recorded female was predicted male. Upon re‐examination the latter two samples were identified as male. In total, 99.3% of the GBS‐predicted gender matched the recorded gender for the test dataset. For all the deer with consistent recorded and predicted gender, no erroneous recorded gender assignments, which become obvious as the animals mature, have been discovered by the breeders.
Figure 1.
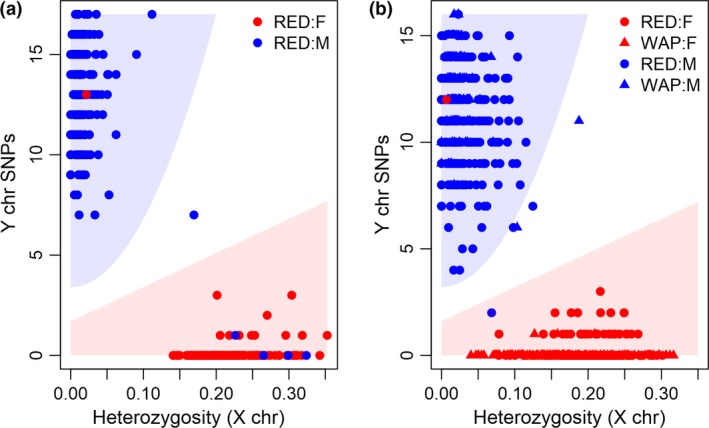
Gender plot for the (a) training dataset and (b) test dataset. Red symbols represent recorded females, blue symbols represent recorded males, circles represent Red deer and triangles represent Wapiti deer. Animals were predicted male if located in the blue region [], were predicted female if located in the red region [] or otherwise were unassigned gender. Interactive plots of (a) and (b) can be downloaded at https://github.com/AgResearch/GBS_Gender_Predict/tree/master/Supplementary/.
Overall, the accuracy of our method was 99.9% with a specificity (true male predicted male) and sensitivity (true female predicted female) of 99.8% and 100% respectively. These measures are similar to those obtained using SNP chip arrays (Zhang et al. 2016) but higher than traditional sexing assays (such as PCR‐based approaches or capillary electrophoresis; Wright et al. 2012; Ristanic et al. 2018) and other sequencing‐based approaches (Stovall et al. 2018). However, unlike SNP chip and traditional sexing assays, the GBS‐based method is applicable across different species, does not require a costly development phase and is a by‐product of having GBS SNP data with their chromosomal positions.
In this work, we developed a method for discovering SNPs located on sex chromosomes in GBS data and using them to predict gender. Results show that the predictions were highly accurate, even across two diverse breeds of deer, and lead to the detection of animals with wrongly recorded gender. Furthermore, the results demonstrate that using both X and Y chromosomes improves accuracy in gender prediction with GBS data compared to using solely the X chromosome information. This is due to the presence of errors (e.g. alignment or sequencing errors) that result in males having non‐zero heterozygosity estimates for X chromosomal SNPs and reads to be called on the Y chromosome for females. Nevertheless, by combining information from the X and Y chromosomes, sufficient differentiation between males and females was obtained in order to predict gender accurately.
One advantage of our method is that it accounts for errors from heterozygous genotypes being called as homozygous in the calculation of heterozygosity, which allows gender prediction on samples with low sample depth. Nevertheless, we discarded individuals with a sample depth below 0.3 because extremely low sample depth can lead to very few or no reads being observed for Y chromosomal SNPs. This would result in males having unassigned gender or even being classified female, as only a few Y chromosomal SNPs were discovered. This problem could be mitigated by using additional sequence reads without SNPs that align to the Y chromosome. However, this raw sequence information is often not available to the end user.
For some individuals, points on the gender plot may fall outside the male and female regions. This may happen if the animal's sample depth is low, as errors can bias heterozygosity estimates for males. In our analysis, all but one of the individuals with unassigned gender were males with a sample depth around 0.6 or lower. Alternatively, these points may represent sex chromosome aneuploidies, such as males with XXY (Laurie et al. 2010; Turner et al. 2011). A female with karyotype XO aneuploidy (Berry et al. 2017) is likely to be classified as female under our model, as , and H i would be expected to equal zero. Another possibility is that sample contamination may lead to unexpected values of and H i, although this would depend on the type and level of contamination present. Consequently, our method could be utilised to detect aneuploidies or contaminated samples. Without any known abnormal animals or samples available in our collection, this could not be tested.
In conclusion, we have developed a method using GBS data that predicts gender in animals with a high degree of accuracy. Although developed in deer, our method can be readily applied to other animal species with an XX/XY sex determination system, provided that a suitable reference genome with X and Y chromosomes is available.
Supporting information
Figure S1 Proportion of heterozygous genotypes for males in the training dataset for X chromosomal SNPs relative to their position on the X chromosome.
Acknowledgements
We thank Focus Genetics (New Zealand) and Landcorp Farming Ltd (New Zealand) for use of their data and acknowledge Mary McEwan and Jamie Ward for their assistance. This work was funded through DEEResearch (New Zealand) via the ‘Hitting Targets’, ‘Deer Progeny Test’ and ‘Deer Select’ research programmes and through the Ministry of Business, Innovation and Employment (New Zealand), Contract C10X1306, ‘Genomics for Production & Security in a Biological Economy’ to AgResearch Ltd.
References
- Anderson C.A., Pettersson F.H., Clarke G.M., Cardon L.R., Morris A.P. & Zondervan K.T. (2010) Data quality control in genetic case‐control association studies. Nature Protocols 5, 1564–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bana N.Á., Nyiri A., Nagy J. et al (2018) The red deer Cervus elaphus genome CerEla1.0: sequencing, annotating, genes, and chromosomes. Molecular Genetics and Genomics 293, 665–84. [DOI] [PubMed] [Google Scholar]
- Berry D.P., Wolfe A., O'Donovan J., Byrne N., Sayers R.G., Dodds K.G., McEwan J.C., O'Connor R.E., McClure M. & Purfield D.C. (2017) Characterization of an X‐chromosomal non‐mosaic monosomy (59, X0) dairy heifer detected using routinely available single nucleotide polymorphism genotype data. Journal of Animal Science 95, 1042–9. [DOI] [PubMed] [Google Scholar]
- Clarke S.M., Henry H.M., Dodds K.G., Jowett T.W., Manley T.R., Anderson R.M. & McEwan J.C. (2014) A high throughput single nucleotide polymorphism multiplex assay for parentage assignment in New Zealand sheep. PLoS ONE 9, e93392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds K.G., McEwan J.C., Brauning R., Anderson R.M., Stijn T.C., Kristjánsson T. & Clarke S.M. (2015) Construction of relatedness matrices using genotyping‐by‐sequencing data. BMC Genomics 16, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshire R.J., Glaubitz J.C., Sun Q., Poland J.A., Kawamoto K., Buckler E.S. & Mitchell S.E. (2011) A robust, simple genotyping‐by‐sequencing (GBS) approach for high diversity species. PLoS ONE 6, e19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S.E., Huisman J., Ellis P.A. & Pemberton J.M. (2017) A high‐density linkage map reveals sexual dimorphism in recombination landscapes in red deer (Cervus elaphus). G3: Genes, Genomes, Genetics 7, 2859–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. & Salzberg S.L. (2012) Fast gapped‐read alignment with bowtie 2. Nature Methods 9, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie C.C., Doheny K.F., Mirel D.B. et al (2010) Quality control and quality assurance in genotypic data for genome‐wide association studies. Genetic Epidemiology 34, 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux Perreault L.P., Provost S., Legault M.A., Barhdadi A. & Dubé M.P. (2013) pygenclean: efficient tool for genetic data clean up before association testing. Bioinformatics 29, 1704–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F., Lipka A.E., Glaubitz J., Elshire R., Cherney J.H., Casler M.D., Buckler E.S. & Costich D.E. (2013) Switchgrass genomic diversity, ploidy, and evolution: novel insights from a network‐based SNP discovery protocol. PLoS Genetics 9, e1003215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd‐Brown K. et al (2007) plink: a tool set for whole‐genome association and population‐based linkage analyses. American Journal of Human Genetics 81, 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristanic M., Stanisic L., Maletic M., Glavinic U., Draskovic V., Aleksic N. & Stanimirovic Z. (2018) Bovine foetal sex determination—different DNA extraction and amplification approaches for efficient livestock production. Reproduction in Domestic Animals 53, 947–54. [DOI] [PubMed] [Google Scholar]
- Stovall W.R., Taylor H.R., Black M., Grosser S., Rutherford K. & Gemmell N.J. (2018) Genetic sex assignment in wild populations using genotyping‐by‐sequencing data: a statistical threshold approach. Molecular Ecology Resources 18, 179–90. [DOI] [PubMed] [Google Scholar]
- Turner S., Armstrong L.L., Bradford Y. et al (2011) Quality control procedures for genome‐wide association studies. Current Protocols in Human Genetics 68, 1.19.1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright C.F., Wei Y., Higgins J.P. & Sagoo G.S. (2012) Non‐invasive prenatal diagnostic test accuracy for fetal sex using cell‐free DNA a review and meta‐analysis. BMC Research Notes 5, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang I.L., Couldrey C. & Sherlock R.G. (2016) Using genomic information to predict sex in dairy cattle. Proceedings of the New Zealand Society of Animal Production 76, 26–30. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Proportion of heterozygous genotypes for males in the training dataset for X chromosomal SNPs relative to their position on the X chromosome.