

Summary
Indigenous cattle breeds in northern Eurasia have adapted to harsh climate conditions. The local breeds are important genetic resources with cultural and historical heritages, and therefore, their preservation and genetic characterization are important. In this study, we profiled the whole‐blood transcriptome of two native breeds (Northern Finncattle and Yakutian cattle) and one commercial breed (Holstein) using high‐throughput RNA sequencing. More than 15 000 genes were identified, of which two, 89 and 162 genes were significantly upregulated exclusively in Northern Finncattle, Yakutian cattle and Holstein cattle respectively. The functional classification of these significantly differentially expressed genes identified several biological processes and pathways related to signalling mechanisms, cell differentiation and host–pathogen interactions that, in general, point towards immunity and disease resistance mechanisms. The gene expression pattern observed in Northern Finncattle was more similar to that of Yakutian cattle, despite sharing similar living conditions with the Holstein cattle included in our study. In conclusion, our study identified unique biological processes in these breeds that may have helped them to adapt and survive in northern and sub‐arctic environments.
Keywords: gene expression, Holstein cattle, Northern Finncattle, RNA‐seq, Yakutian cattle
Introduction
As a result of natural and human‐led selection, domestic animals have been able to adapt, survive, be productive and reproduce in challenging environments (Mirkena et al. 2010). Animals in the northern hemisphere (Lapland, northern Russia and Siberia) are used for food production and other socio‐cultural needs and, thus, have been the basis of human life in the North. These animals may have different biological capacities to adapt to extremes in temperature, daylight and feed availability. However, there has been growing interest towards improving local breeds by introducing genetic material from more productive breeds, with a preference towards commercial breeds. Intensive breeding and the replacement of native breeds with commercial breeds may appear to be advantageous at first, but these practices will have long‐lasting consequences. In addition to irreversibly losing the unique genetic resources of native breeds, we will also be losing the cultural and historical heritage associated with those local breeds. The signatures of adaptation, as well as the history of formation, that are encoded in the native breeds may also fade away (Gaouar et al. 2015). Therefore, the local breeds must be genetically characterized and preserved.
Here, we studied two native cattle breeds (Northern Finncattle and Yakutian cattle) and one international breed (Holstein cattle). All these breeds are used for milk and meat production. Previous studies using genetic markers have indicated the genetic distinctiveness among these breeds (Li et al. 2007; Li & Kantanen 2010). Among the three breeds, Holstein cattle have high economic importance and have the shortest adaptation history. Holstein cattle are the most popular dairy breed globally, with an intensive selection programme and high milk production; although they originated in a temperate climate, they have adapted to different parts of the world and can survive in varying climatic conditions. Northern Finncattle and Yakutian cattle have economic, social and cultural values in producing milk and meat in these marginal regions, having roles in local food culture and gastronomy and being cultural symbols (Kantanen et al. 2015). The Northern Finncattle breed is native to Northern Finland and Finnish Lapland. The breed nearly became extinct during the 1970s but is currently maintained (current census is 850 cows) through active in vivo and in vitro conservation activity. Yakutian cattle are characterized by being purebred aboriginal native cattle. Adult Yakutian cows (current population is approximately 1000 animals) typically weigh 350–400 kg, and their height is 111 cm, on average. The animals are well adapted to harsh Siberian conditions, where the temperature falls below −50 °C in long winters (Kantanen et al. 2009).
High‐throughput RNA sequencing (RNA‐seq) has been proven to be an efficient method for studying gene expression (Wang et al. 2009). In recent years, RNA‐seq has been applied to domestic animals, with a major focus on productivity traits (Bai et al. 2016; Li et al. 2016; Silva‐Vignato et al. 2017; Pokharel et al. 2018). A number of transcriptome studies have been conducted in Holstein (Cui et al. 2014; Sandri et al. 2015; Bai et al. 2016; Li et al. 2016; Seo et al. 2016) with a primary focus on milk traits, but to date, there are no reports on gene expression studies in either Yakutian cattle or Northern Finncattle. In this study, we applied RNA‐seq technology and profiled the whole blood transcriptome of the three aforementioned breeds to characterize their genetic differences.
Materials and methods
Sample collection
Animal handling procedures and sample collection were conducted in accordance with legal regulations approved by the Russian authorization board (FS/UVN‐03/163733/07.04.2016) and the Animal Experiment Board in Finland (ESAVI/7034/04.10.07.2015). A 2.5‐ml sample of blood from each of three Yakutian cattle, three Northern Finncattle and three Holstein cattle cows was collected into a PAXgene® Blood RNA IVD tube (Ref. PreAnalytiX®) during the winter period of 2016 and 2017 and stored at −18 °C. Blood samples were collected by jugular venipuncture. All animals included in this study were 4‐ to 8‐year‐old females, except for one 14‐year‐old Holstein cow (HC3).
RNA extraction
The RNA was extracted using the PAXgene® Blood RNA kit (Ref. 762174, Ref. PreAnalytiX®), according to the kit manual, with minor adjustments to the protocol: the samples were thawed at room temperature overnight, the initial centrifugation time at 5000 g was increased to 15 min, the initial pellet was resuspended in double the amount of BR1 buffer and divided into two separate reactions, the proteinase K incubation time was extended to 1 h and the columns were incubated at room temperature in elution buffer for 10 min prior to elution. The concentration and quality of the RNA was measured with a spectrophotometer (NanoDrop ND‐1000), and the integrity of the RNA was measured with an Agilent Bioanalyzer 2100 using the Agilent 6000 RNA Nano kit (Ref. 5067‐1511). All samples selected for sequencing had an RNA integrity number value of at least 7.
Library preparation and sequencing
Library preparation and sequencing tasks were outsourced to the Finnish Functional Genomics Center in Turku, Finland. The library preparation was performed according to Illumina's Truseq® mRNA sample preparation guide protocol. The high quality of the libraries was confirmed with an Advanced Analytical Fragment Analyzer, and the concentrations of the libraries were quantified using Qubit® Fluorometric Quantitation (Life Technologies). The average RNA‐seq library fragments were in the range of 250–350 bp. Only the good libraries with RNA quality number value greater than 7 were sequenced with a paired‐end strategy to generate 75‐bp reads. Illumina's standard bcl2fastq2 software was used for base calling and adapter trimming.
Computational methods
Raw sequence reads were pre‐processed using fastqc v0.11.8 (Andrews n.d.) to determine the quality of the data and to obtain an overview of the sequencing data. The outputs from fastqc were summarized using multiqc v1.5 (Ewels et al. 2016). As the raw data did not have any adapters and the Phred quality score of the reads from all samples was greater than 30, we did not perform further trimming. Processed paired‐end reads were mapped against the latest versions of the cattle reference genome UMD3.1 and transcriptome (Ensembl release 93) using star v2.6 (Dobin et al. 2013). First, the genome indexes were prepared, and mapping was performed with default parameters using star v2.6 (Dobin et al. 2013). Moreover, a gene‐level counts file for each sample was generated as part of the star‐alignment pipeline by specifying the ‘—quantMode GeneCounts’ option. Statistical analysis of read counts were carried out using the deseq2 v1.20.0 (Love et al. 2014) Bioconductor package (Appendix S1). After performing standard differential expression analysis steps using deseq functions, genes that had fewer than five transcript counts were discarded. Differential gene expression between breed groups were tested as pairwise comparisons (Yakutian cattle vs. Northern Finncattle, Yakutian cattle vs. Holstein and Holstein vs. Northern Finncattle). To identify genes significantly differentially expressed between breed groups, we set an adjusted P‐value of 0.05 (Benjamini & Hochberg correction). For the list of significantly differentially expressed genes, we retrieved additional information, such as gene description and chromosomal location, using the biomart Bioconductor package v2.36.1 (Durinck et al. 2009; Smedley et al. 2009). Additional gene information retrieved for differentially expressed genes were based on Ensembl gene annotations for Cow (UMD3.1, Ensembl release 92). Finally, the significantly differentially expressed genes were assessed for their functional roles. Gene Ontology (GO) terms and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway analyses were performed using the cluego v2.5.3 (Bindea et al. 2009) plugin in cytoscape v3.6.1 (Shannon et al. 2003). In addition to the default parameters in cluego, we chose to observe GO terms in the range of level 3 and 5. Similarly, for the GO term or KEGG pathway to be displayed, at least three genes and a minimum of 4% of the total genes needed to be present on our list. We used all 15 960 genes expressed in our data as the background list for comparison. Moreover, the terms or pathways shared by 50% of the genes from our list were grouped together using Kappa statistics (Kappa score threshold of 0.4).
Results and discussion
From nine samples, we obtained 32.2 Gb of RNA‐seq data. More than 87% of the reads from each sample mapped to the cattle reference genome, with Holstein samples having the highest (91.6%) mapping rate followed by Yakutian cattle (88%) and Finncattle (87.9%). The slightly higher mapping rate for Holstein samples could be because that breed is closer to the Hereford breed, which is the source of the reference assembly (Zimin et al. 2009). With sequencing costs becoming cheaper, having breed‐specific reference genomes would be useful in the future.
The 15 960 total number of genes expressed in our data (Table S1) cover 64.8% of known (n = 24 616) cattle genes reported in the annotation file. At the individual level, we observed that both the highest (FC8) and the lowest (FC7) number of genes were expressed in Northern Finncattle samples (Table 1). We believe that a comparatively lower number of reads in the sample FC7 led to a lower number of expressed genes. We did not observe any globin genes among the top most expressed genes. Genes such as MHC class I heavy chain (BOLA), uncoupling protein 2 (UCP2), eukaryotic translation elongation factor 2 (EEF2) and vimentin (VIM) were among the top most expressed genes. These results confirm that RNA extraction using PAXgene® worked well.
Table 1.
Sample summary
| Sample ID | Breed | ENA accession no. | No. of reads (million) | Uniquely mapped reads % | No. of genes expressed |
|---|---|---|---|---|---|
| FC7 | Northern Finncattle | ERS2639473 | 49.4 | 87.2 | 12 106 |
| FC8 | Northern Finncattle | ERS2639474 | 107.6 | 87.1 | 15 038 |
| FC9 | Northern Finncattle | ERS2639475 | 89.4 | 89.7 | 14 811 |
| HC1 | Holstein | ERS2639476 | 104.4 | 91.7 | 14 768 |
| HC2 | Holstein | ERS2639477 | 62.8 | 91.0 | 14 140 |
| HC3 | Holstein | ERS2639478 | 62.4 | 92.4 | 13 831 |
| YC6 | Yakutian | ERS2639479 | 69.2 | 87.3 | 14 047 |
| YC10 | Yakutian | ERS2639480 | 69.8 | 88.2 | 13 736 |
| YC11 | Yakutian | ERS2639481 | 65.8 | 88.5 | 14 371 |
We assessed the co‐expression status of the top 500 most expressed genes (i.e. the genes with highest base mean values) in each of the three breed groups. As seen in the Venn diagram (Fig. 1), 386 of the top expressed genes were present in all three breeds. Holstein samples had the highest number (n = 65) of unique genes, followed by Yakutian cattle (n = 56) and Northern Finncattle (n = 17). Moreover, the results showed that Northern Finncattle had more similar gene expression profiles with Yakutian cattle (53 uniquely shared genes) than with Holstein cattle (44 uniquely shared genes). In contrast, Yakutian cattle and Holstein cattle shared the least number of unique genes.
Figure 1.
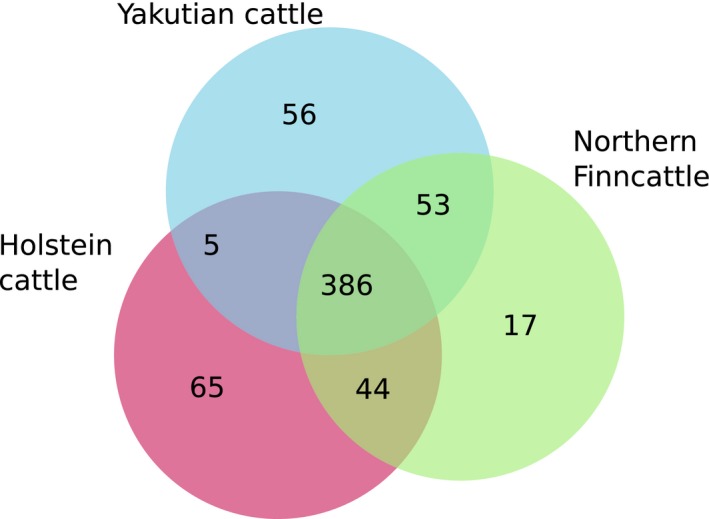
Co‐expression of the top 500 genes among the three breeds.
We then looked more closely into the genes that were differentially expressed between any two of the three breed groups and assessed with what types of biological processes and/or biological pathways those genes were associated.
Differential gene expression between Yakutian cattle and Northern Finncattle
A total of 189 transcripts were significantly differentially expressed between Yakutian cattle and Northern Finncattle (Table S2), of which 108 transcripts were upregulated in Yakutian cattle. The list of differentially expressed genes included a number of cases for which the Ensembl IDs lack gene descriptions. Having a better annotation would certainly help in characterizing the functions of these genes. The GO terms associated with the upregulated genes in Yakutian cattle included ‘natural killer cell chemotaxis’, ‘negative regulation of viral process’, ‘negative regulation of cytokine production’ and ‘granzyme‐mediated apoptotic signalling pathway’ (Fig. 2a & Table S3). The KEGG pathways associated with upregulated genes included ‘antigen processing and presentation’, ‘natural killer cell mediated cytotoxicity’ and ‘graft‐versus‐host disease’ (Table S4). Similarly, the GO terms associated with upregulated genes in Northern Finncattle included ‘regulation of lipid transport’, ‘erythrocyte differentiation’, ‘regulation of cytokine‐mediated signalling pathway’ and ‘regulation of protein maturation’ (Fig. 2b, Table S5). ‘Malaria’ (associated genes HBA, HBB and THBS1) and ‘TCM‐receptor interaction’ (associated genes FN1, GP6 and THBS1) KEGG pathways were related to genes upregulated in Northern Finncattle. The biological processes and pathways related to diseases, immunity and viral processes could be associated with adaptation (Librado et al. 2015; Iso‐Touru et al. 2016).
Figure 2.
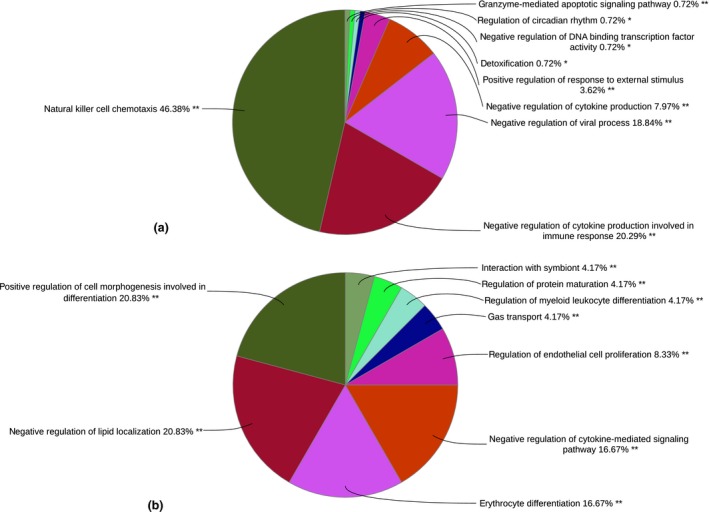
Functional annotation of the significantly differentially expressed genes in Yakutian cattle and Northern Finncattle; GO terms associated with upregulated genes in (a) Yakutian cattle and (b) Northern Finncattle. Similar GO terms were grouped together, and thus, the percentage shown at the end of each representative GO term indicates the total percentages of linked GO terms. The significant GO terms at the P < 0.01 statistical level are indicated by double (**) asterisks and at the P < 0.05 level by a single (*) asterisk.
Differential gene expression between Yakutian cattle and Holstein cattle
The highest number of significantly differentially expressed genes was identified between Yakutian cattle and Holstein cattle (Table S6). Given the phenotypic differences between the breeds, it was not surprising to observe a large number of differentially expressed genes. Yakutian cattle have phenotypic characteristics, such as a small and compact body size and a thick hair coat, that help them survive in a cold environment. In contrast, Holstein cattle have a bigger body size and a thin hair coat and are thus not suitable for a colder climate. In addition, Holstein cattle do not face a shortage of fodder and are maintained inside a barn, whereas Yakutian cattle may suffer from food shortages and are partly outside during the winter. The significant differences between Yakutian cattle and Holstein cattle are in line with earlier reports (Decker et al. 2016; Zinovieva et al. 2016; Yurchenko et al. 2018). Out of 1418 genes that were significantly differentially expressed between the two breeds, 594 genes were upregulated in Yakutian cattle. Because of the high number of differentially expressed genes, we considered only those genes with a log2 fold change greater than or less than 1.5 for GO and KEGG pathway analyses. Genes that were upregulated in Yakutian cattle were associated with 108 GO terms (Fig. 3a & Table S7) and 18 KEGG pathways (Fig. 3b & Table S8). Similarly, the genes that were upregulated in Holstein cattle were associated with 97 GO terms (Fig. 3c & Table S9) and 16 KEGG pathways (Fig. 3d & Table S10).
Figure 3.
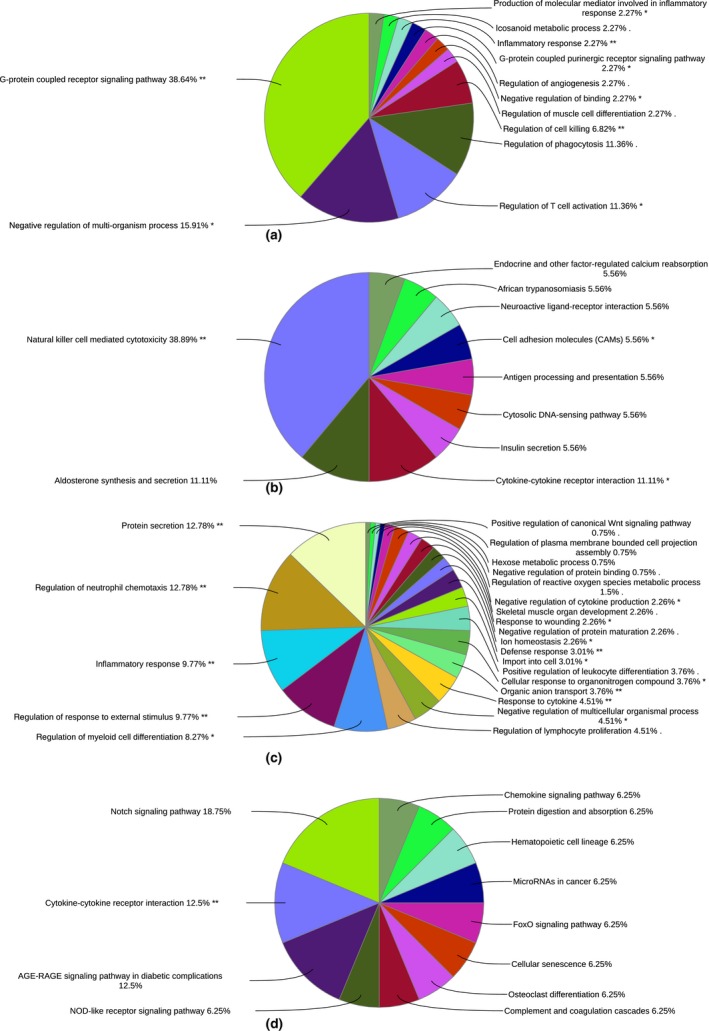
Functional annotation of genes significantly differentially expressed between Yakutian cattle and Holstein cattle. (a) GO terms and (b) KEGG pathways associated with upregulated genes in Yakutian cattle. (c) GO terms and (d) KEGG pathways associated with upregulated genes in Holstein cattle. Similar GO terms and KEGG pathways were grouped together, and thus, the percentage shown at the end of each term indicates the total percentages of linked GO terms and KEGG pathways. The significant GO terms and KEGG pathways at the P < 0.05 and P < 0.01 statistical levels are indicated by single (*) and double (**) asterisks respectively.
Differential expression between Northern Finncattle and Holstein cattle
Between Finncattle and Holstein cattle, we observed 250 significantly differentially expressed transcripts (Table S11), with the majority of the genes (n = 180) being upregulated in Holstein cattle. Only one GO term, ‘eicosanoid metabolic process’ (associated genes ALOX15, ALOX5 and HPGD), and no KEGG pathways were identified for genes that were upregulated in Northern Finncattle. In contrast, 62 GO terms (Fig. 4 & Table S12) and 32 KEGG pathways were associated with upregulated genes in Holstein cattle (Table S13).
Figure 4.
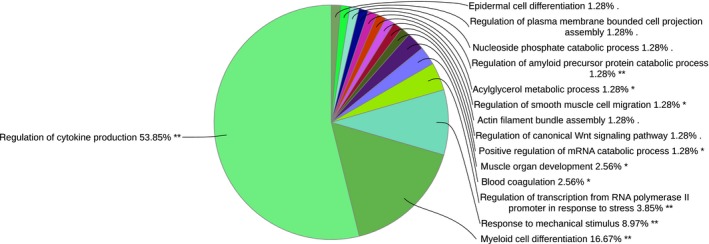
Functional annotation of genes upregulated in Holstein cattle compared to Northern Finncattle. Similar GO terms were grouped together, and thus, the percentage at the end of each term indicates the total percentages of linked GO terms. The significant GO terms at the P < 0.01 statistical level are indicated by double (**) asterisks and at the P < 0.05 level by a single (*) asterisk..
Uniquely differentially expressed genes
We noticed that there was overlap among many genes in more than one comparison (Fig. S1). Therefore, we compiled a list of genes that were uniquely differentially expressed in each breed. The number of uniquely differentially expressed genes (Table 2) in each breed followed a similar pattern to that observed by the top 500 most expressed genes (Fig. 1).
Table 2.
Numbers of uniquely differentially expressed genes in different breeds
| Breed | No. of uniquely differentially expressed genes | Upregulated genes | Downregulated genes |
|---|---|---|---|
| Yakutian cattle | 139 | 89 | 52 |
| Northern Finncattle | 9 | 2 | 7 |
| Holstein cattle | 200 | 162 | 48 |
Yakutian cattle had 139 uniquely differentially expressed genes, of which 89 were reported to be upregulated. We noticed a number of cases in which more than one gene from the same family was upregulated. Such cases could provide higher confidence related to gene functions. The genes or receptors with more than one member are as follows: chemokines (CCL4 and CCL5), carbohydrate sulfotransferases (CHST1 and CHST12), chemokine receptors (CX3CR1 and CXCR6), growth arrests (GAS6 and GAS7), granzymes [GZMB (two paralogs), GZMM and GZMH)], insulin‐like growth factor binding proteins (IGFBP4 and IGFBP7) and natural cytotoxicity triggering receptors (NCR1 and NCR3). Among those on the list were BHLHE40 and PRKCG, which are linked to circadian rhythm. The importance of the circadian clock is particularly applicable to Yakutian cattle, which must be metabolically prepared for food scarcity during long winters (Ebling & Barrett 2008).
Four granzyme transcripts and perforin were upregulated in Yakutian cattle. Granzymes are serine proteases that are used by cytotoxic lymphocytes to destroy malignant and virus‐infected cells. Granzymes are transported into the cytoplasm of the target cell by perforin 1 (PRF1), after which they cleave specific proteins and trigger apoptosis (MacDonald et al. 1999; Russell & Ley 2002; Johnson et al. 2003). It has been suggested that the evolution of granzymes is related to species‐specific immune challenges (humans have five granzyme genes and mice have 10), and they are maintained by gene duplications and alterations in substrate specificity (Kaiserman et al. 2006). Three out of the six granzymes known in cattle (Yang et al. 2018), with two nearby paralogs of granzyme B (Fig. S2) and PRF1, are all upregulated in Yakutian cattle. These findings suggest that Yakutian cattle have a very solid granzyme‐mediated immune system.
The number of upregulated genes was sufficient to identify significant GO terms. The GO terms associated with the upregulated genes included ‘negative regulation of cytokine production’, ‘regulation of lymphocyte‐mediated immunity’, ‘positive regulation of leukocyte chemotaxis’ and ‘cellular response to interferon‐gamma’ (Fig. 5a & Table S14). Similarly, pathways such as ‘natural killer cell‐mediated cytotoxicity’, ‘inflammatory bowel disease (IBD)’, ‘African trypanosomiasis’ and ‘cytosolic DNA‐sensing pathway’ were related to uniquely upregulated genes in Yakutian cattle (Fig. 5b & Table S15). Two GO terms, ‘regulation of lipid transport’ (associated genes ABCA1, ABCG1, IRS2 and THBS1) and ‘regulation of endothelial cell proliferation’ (associated genes CCL24, ECM1 and THBS1), and no KEGG pathways were found for the downregulated genes.
Figure 5.
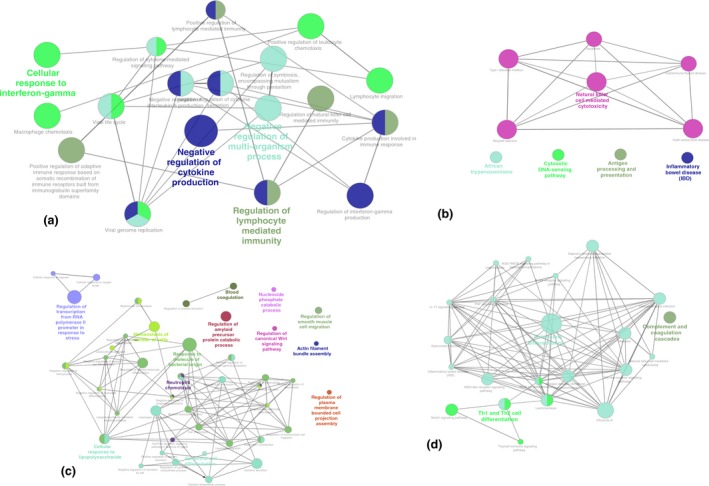
Network representation of GO and KEGG pathways associated with uniquely differentially expressed genes. Nodes represent GO terms or KEGG pathways, with node size corresponding to the significance of term enrichment, and functionally related groups partially overlap. Network of (a) GO terms and (b) KEGG pathways associated with the genes upregulated in Yakutian cattle. Network of (c) GO terms and (d) KEGG pathways associated with upregulated genes in Holstein cattle. The representative GO terms and KEGG pathways are highlighted in bold.
The majority (162/200) of the uniquely differentially expressed genes in Holstein cattle were found to be upregulated. The list also includes a number of cases in which more than one member of the gene family was present. Genes encoding the solute carrier protein family (SLC16A12, SLC16A6, SLC25A37, SLC28A3, SLC2A9, SLC45A4, SLC6A6 and SLC23A1), ring finger proteins (RNF149 and RNF24), notch proteins (NOTCH1, NOTCH2 and NOTCH3), lysine demethylases (KDM4B and KDM6B), interferon receptors (IFNAR1, IFNAR2 and IFNGR1), CD proteins (CD101, CD164, CD300a and CD55) and others were upregulated in Holstein cattle samples. The GO terms associated with the upregulated genes included ‘cellular response to lipopolysaccharide’, ‘response to molecule of bacterial origin’, ‘negative regulation of leukocyte differentiation’, ‘regulation of transcription from RNA polymerase II promoter in response to stress and phosphatidylinositol binding’ (Fig. 5c & Table S16). Similarly, KEGG pathways such as ‘osteoclast differentiation’, ‘Th1 and Th2 cell differentiation’ and ‘complement and coagulation cascades’ were related to the upregulated genes (Fig. 5d & Table S17). We did not find any GO terms or KEGG pathways associated with the downregulated genes.
Among other functions, membrane‐bound solute carriers are key to maintaining physiological processes, including nutrient uptake, waste removal and ion transport (Hediger et al. 2004). The upregulated expression of eight solute carrier proteins in Holstein cattle samples suggests the highly important function of these genes. A recent study showed that, during sloughing, amphibians increase the rate of ion uptake to maintain internal homeostasis (Wu et al. 2017). We speculate that, with a comparatively bigger body size and a thin hair coat, Holstein cattle may have developed a similar system for maintaining internal homeostasis in response to lower temperatures. The upregulation of three notch proteins (NOTCH1, NOTCH2 and NOTCH3) and the biological processes associated with hypoxia (‘cellular response to oxygen levels’ and ‘cellular response to hypoxia’) is in line with earlier findings showing that the ‘notch signalling pathway’ was activated in response to hypoxia (Hiyama et al. 2011). This finding also suggests that Holstein cattle may not have adapted well to winter conditions, as hypoxia is linked to low temperatures (Rocha & Branco 1998). ‘Notch signalling pathway’ is also known to have important roles in milk lactose metabolism, mammary gland development and lactation, which are all associated with the high milk production traits of Holstein cattle (Politi et al. 2004; Yalcin‐Ozuysal et al. 2010). Moreover, NOTCH1 and NOTCH2 are part of the ‘Ras protein signal transduction’ and ‘regulation of ERK1 and ERK2 cascade’ processes.
Keratin 72 (KRT72, ENSBTAG00000007904) and transmembrane protein 8 A (TMEM8A, ENSBTAG00000016588) were the only genes that were upregulated exclusively in Northern Finncattle. KRT72 is involved in hair formation, and the function of TMEM8A is unknown. Out of seven downregulated genes, four IDs (ENSBTAG00000038233, ENSBTAG00000039691, ENSBTAG00000046611 and ENSBTAG00000000930) did not have gene descriptions available at the time of this writing. The remaining three genes were acetylcholinesterase (ACHE), chemokine (C‐C motif) ligand 3 (CCL3) and Bos taurus regulator of G‐protein signalling 2, 24 kDa (RGS2). The RGS family of proteins is involved in fine‐tuning the signalling activities of G‐protein‐coupled receptors (Berman et al. 1996; Blumer 2004). An increased level of RGS2 expression is linked to the regulation of numerous biological activities, such as immune responses, bone formation, cardiovascular function and anxiety (Zhang & Mende 2014). CCL3, CCL4 and CCL5 encode members of the CC chemokine family of proteins, which exhibit proinflammatory activities. Moreover, CCL3 inhibits the proliferation of haematopoietic stem/progenitor cells (Cook 1996). ACHE has numerous functions, and its role in muscle development, neuritogenesis, cell adhesion, the activation of dopamine neurons and amyloid fibre assembly has been reviewed earlier (Soreq & Seidman 2001).
Limitations of the study
Finally, we would like to highlight some of the limitations of this study that we are aware of. First, having more samples could have given higher confidence to the results. Comparisons involving samples from temperate climates would be equally interesting. Due to the quality of the gene annotations, many genes could not be functionally interpreted. Moreover, due to the large number of differentially expressed genes, we did not look in detail at all the genes, and therefore, many interesting genes may not have been highlighted in this study. Moreover, the differences in gene expression level we observed between the breeds could be affected by other factors such as the physiological status of the animal, management and environmental differences. Although we tried to interpret our results based on model species, primarily human studies, some of the interpretation may not be accurate. Particularly, most of the studies were based on model species with more controlled settings, whereas the animals in our study, especially Yakutian animals, live in harsh environmental conditions.
Conclusions
In conclusion, using blood as a starting material, we were able to capture more than 60% of cattle genes to assess the differential gene expression in three breeds. Gene expression profiles of two important indigenous cattle breeds that are adapted to the northern and even sub‐arctic climate have been reported for the first time. The gene expression profiles of Northern Finncattle appeared to be more similar to Yakutian cattle than to Holstein cattle, despite sharing similar living conditions with the Holstein cows that were analysed in this study. Moreover, we identified breed‐specific differences in maintaining immunity and disease resistance mechanisms among the breeds included in our study. Some of our results, based on Holstein cattle, explain how alternative biological processes can help imported animals cope with challenging environmental conditions. Studies such as this one could identify genetic markers that may assist in animal breeding and the sustainable utilization and conservation practices of animal genetic resources in changing northern Eurasian environments. One of the main limitations of our study is that we were able to see differentially expressed genes based entirely on only one tissue. This study might have missed many important genes, and the genes that were found to be differentially expressed may have been associated with some external factors (such as living conditions, seasonal difference and feeding). Therefore, future studies based on additional tissues with a more controlled experimental design will certainly provide a better picture of genetic basis of adaptation in northern Eurasian conditions.
Author contributions
J.K. conceived the analysis. J.K., M.H., T.R., J.P., H.L., R.P. and S.Z. participated in sample collection. H.H. extracted mRNA from blood samples. K.P. and M.W. performed RNA‐seq data analyses. K.P. wrote the manuscript. All authors read, revised and approved the final manuscript.
Conflict of interests
The authors declare that they have no competing interests.
Supporting information
Figure S1 Venn diagram based on the list of significantly differentially expressed genes following all three possible comparisons.
Figure S2 Illustration of the closely located Granzyme B in the genome.
Table S1 Expression levels for each sample.
Table S2 Significantly differentially expressed genes between Yakutian cattle and Northern Finncattle.
Table S3 GO terms associated with upregulated genes in Yakutian cattle when compared with Northern Finncattle.
Table S4 KEGG pathways associated with upregulated genes in Yakutian cattle when compared with Northern Finncattle.
Table S5 GO terms associated with upregulated genes in Northern Finncattle when compared with Yakutian cattle.
Table S6 Summary of genes significantly differentially expressed between Yakutian cattle and Holstein cattle.
Table S7 GO terms associated with upregulated genes in Yakutian cattle when compared with Holstein cattle.
Table S8 KEGG pathways associated with upregulated genes in Yakutian cattle when compared with Holstein cattle.
Table S9 GO terms associated with upregulated genes in Holstein cattle when compared with Yakutian cattle.
Table S10 KEGG pathways associated with upregulated genes in Holstein cattle when compared with Yakutian cattle.
Table S11 Summary of significantly genes differentially expressed between Northern Finncattle and Holstein cattle.
Table S12 GO terms associated with upregulated genes in Holstein cattle when compared with Northern Finncattle.
Table S13 KEGG pathways associated with upregulated genes in Holstein cattle when compared with Northern Finncattle.
Table S14 GO terms associated with the genes that were exclusively upregulated in Yakutian cattle.
Table S15 KEGG pathways associated with the genes that were exclusively upregulated in Yakutian cattle.
Table S16 GO terms associated with the genes that were exclusively upregulated in Holstein cattle.
Table S17 KEGG pathways associated with genes that were exclusively upregulated in Holstein cattle.
Appendix S1 R script used to identify and annotate differentially expressed genes in this study.
Acknowledgements
We thank our colleague Tuula Marjatta Hamama from the Natural Resources Institute Finland (Luke) for laboratory assistance and Innokenty Ammosov for valuable assistance in sampling in Yakutia. This study is part of the Arctic‐Ark project, funded by the Academy of Finland (decision no. 286040). The authors wish to acknowledge the CSC – IT Center for Science, Finland, for computational resources. This study was supported by the Finnish Functional Genomics Centre, University of Turku and Åbo Akademi and Biocenter Finland. Special thanks to the owners of the experimental animals for letting us collect the blood samples.
Data availability
The raw sequence reads (FASTQ files) have been deposited in the European Nucleotide Archive (ENA) under accession no. PRJEB28074 (please refer to Table 2 for sample‐specific accessions). The script used during the differential expression analysis is available in Appendix S1.
References
- Andrews S. (n.d.) fastqc . Babrahaham Bioinformatics, Cambridge. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Bai X., Zheng Z., Liu B., Ji X., Bai Y. & Zhang W. (2016) Whole blood transcriptional profiling comparison between different milk yield of Chinese Holstein cows using RNA‐seq data. BMC Genomics 17(Suppl 7), 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman D.M., Wilkie T.M. & Gilman A.G. (1996) GAIP and RGS4 are GTPase‐activating proteins for the Gi subfamily of G protein alpha subunits. Cell 86, 445–52. [DOI] [PubMed] [Google Scholar]
- Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W.‐H., Pagès F., Trajanoski Z. & Galon J. (2009) cluego: a Cytoscape plug‐in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumer K.J. (2004) The need for speed. Nature 427, 20–1. [DOI] [PubMed] [Google Scholar]
- Cook D.N. (1996) The role of MIP‐1 alpha in inflammation and hematopoiesis. Journal of Leukocyte Biology 59, 61–6. [DOI] [PubMed] [Google Scholar]
- Cui X., Hou Y., Yang S., Xie Y., Zhang S., Zhang Y., Zhang Q., Lu X., Liu G.E. & Sun D. (2014) Transcriptional profiling of mammary gland in Holstein cows with extremely different milk protein and fat percentage using RNA sequencing. BMC Genomics 15, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker J.E., Taylor J.F., Kantanen J., Millbrooke A., Schnabel R.D., Alexander L.J. & MacNeil M.D. (2016) Origins of cattle on Chirikof Island, Alaska, elucidated from genome‐wide SNP genotypes. Heredity 116, 502–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. & Gingeras T.R. (2013) star: ultrafast universal RNA‐seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S., Spellman P.T., Birney E. & Huber W. (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomart . Nature Protocols 4, 1184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebling F.J.P. & Barrett P. (2008) The regulation of seasonal changes in food intake and body weight. Journal of Neuroendocrinology 20, 827–33. [DOI] [PubMed] [Google Scholar]
- Ewels P., Magnusson M., Lundin S. & Käller M. (2016) multiqc: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaouar S.B.S., Da Silva A., Ciani E., Kdidi S., Aouissat M., Dhimi L., Lafri M., Maftah A. & Mehtar N. (2015) Admixture and local breed marginalization threaten Algerian sheep diversity. PLoS ONE 10, e0122667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger M.A., Romero M.F., Peng J.‐B., Rolfs A., Takanaga H. & Bruford E.A. (2004) The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers Archiv: European Journal of Physiology 447, 465–8. [DOI] [PubMed] [Google Scholar]
- Hiyama A., Skubutyte R., Markova D., Anderson D.G., Yadla S., Sakai D., Mochida J., Albert T.J., Shapiro I.M. & Risbud M.V. (2011) Hypoxia activates the notch signaling pathway in cells of the intervertebral disc: implications in degenerative disc disease. Arthritis & Rheumatism 63, 1355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso‐Touru T., Tapio M., Vilkki J., Kiseleva T., Ammosov I., Ivanova Z., Popov R., Ozerov M. & Kantanen J. (2016) Genetic diversity and genomic signatures of selection among cattle breeds from Siberia, eastern and northern Europe. Animal Genetics 47, 647–57. [DOI] [PubMed] [Google Scholar]
- Johnson H., Scorrano L., Korsmeyer S.J. & Ley T.J. (2003) Cell death induced by granzyme C. Blood 101, 3093–101. [DOI] [PubMed] [Google Scholar]
- Kaiserman D., Bird C.H., Sun J., Matthews A., Ung K., Whisstock J.C., Thompson P.E., Trapani J.A. & Bird P.I. (2006) The major human and mouse granzymes are structurally and functionally divergent. Journal of Cell Biology 175, 619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantanen J., Ammosov I., Li M.‐H., Osva A. & Popov R. (2009) A cow of the permafrost In: Sakha Ynaga: Cattle of the Yakuts (Ed. by Granberg L., Kantanen J. & Soini K.), pp. 19–44. Finnish Academy of Science and Letters, Helsinki. [Google Scholar]
- Kantanen J., Løvendahl P., Strandberg E., Eythorsdottir E., Li M.‐H., Kettunen‐Praebel A., Berg P. & Meuwissen T. (2015) Utilization of farm animal genetic resources in a changing agro‐ecological environment in the Nordic countries. Frontiers in Genetics 6, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.‐H. & Kantanen J. (2010) Genetic structure of Eurasian cattle (Bos taurus) based on microsatellites: clarification for their breed classification. Animal Genetics 41, 150–8. [DOI] [PubMed] [Google Scholar]
- Li M.‐H., Tapio I., Vilkki J. et al (2007) The genetic structure of cattle populations (Bos taurus) in northern Eurasia and the neighbouring Near Eastern regions: implications for breeding strategies and conservation. Molecular Ecology 16, 3839–53. [DOI] [PubMed] [Google Scholar]
- Li C., Cai W., Zhou C., Yin H., Zhang Z., Loor J.J., Sun D., Zhang Q., Liu J. & Zhang S. (2016) RNA‐seq reveals 10 novel promising candidate genes affecting milk protein concentration in the Chinese Holstein population. Scientific Reports 6, 26813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P., Der Sarkissian C., Ermini L. et al (2015) Tracking the origins of Yakutian horses and the genetic basis for their fast adaptation to subarctic environments. Proceedings of the National Academy of Sciences of the United States of America 112, E6889–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W. & Anders S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with deseq2. Genome Biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald G., Shi L., Vande Velde C., Lieberman J. & Greenberg A.H. (1999) Mitochondria‐dependent and ‐independent regulation of granzyme B‐induced apoptosis. Journal of Experimental Medicine 189, 131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkena T., Duguma G., Haile A., Tibbo M., Okeyo A.M., Wurzinger M. & Sölkner J. (2010) Genetics of adaptation in domestic farm animals: a review. Livestock Science 132, 1–12. [Google Scholar]
- Pokharel K., Peippo J., Honkatukia M. et al (2018) Integrated ovarian mRNA and miRNA transcriptome profiling characterizes the genetic basis of prolificacy traits in sheep (Ovis aries). BMC Genomics 19, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi K., Feirt N. & Kitajewski J. (2004) Notch in mammary gland development and breast cancer. Seminars in Cancer Biology 14, 341–7. [DOI] [PubMed] [Google Scholar]
- Rocha P.L. & Branco L.G. (1998) Seasonal changes in the cardiovascular, respiratory and metabolic responses to temperature and hypoxia in the bullfrog Rana catesbeiana . Journal of Experimental Biology 201, 761–8. [PubMed] [Google Scholar]
- Russell J.H. & Ley T.J. (2002) Lympphocyte‐mediated cytotoxicity. Annual Review of Immunology 20, 323–70. [DOI] [PubMed] [Google Scholar]
- Sandri M., Stefanon B. & Loor J.J. (2015) Transcriptome profiles of whole blood in Italian Holstein and Italian Simmental lactating cows diverging for genetic merit for milk protein. Journal of Dairy Science 98, 6119–27. [DOI] [PubMed] [Google Scholar]
- Seo M., Lee H.‐J., Kim K., Caetano‐Anolles K., Jeong J.Y., Park S., Oh Y.K., Cho S. & Kim H. (2016) Characterizing milk production related genes in Holstein using RNA‐seq. Asian‐Australasian Journal of Animal Science 29, 343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B. & Ideker T. (2003) cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research 13, 2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva‐Vignato B., Coutinho L.L., Cesar A.S.M., Poleti M.D., Regitano L.C.A. & Balieiro J.C.C. (2017) Comparative muscle transcriptome associated with carcass traits of Nellore cattle. BMC Genomics 18, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D., Haider S., Ballester B., Holland R., London D., Thorisson G. & Kasprzyk A. (2009) biomart – biological queries made easy. BMC Genomics 10, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreq H. & Seidman S. (2001) Acetylcholinesterase – new roles for an old actor. Nature Reviews Neuroscience 2, 294–302. [DOI] [PubMed] [Google Scholar]
- Wang Z., Gerstein M. & Snyder M. (2009) RNA‐seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N.C., Cramp R.L. & Franklin C.E. (2017) Living with a leaky skin: upregulation of ion transport proteins during sloughing. Journal of Experimental Biology 220, 2026–35. [DOI] [PubMed] [Google Scholar]
- Yalcin‐Ozuysal Ö., Fiche M., Guitierrez M., Wagner K.‐U., Raffoul W. & Brisken C. (2010) Antagonistic roles of Notch and p63 in controlling mammary epithelial cell fates. Cell Death and Differentiation 17, 1600–12. [DOI] [PubMed] [Google Scholar]
- Yang J., Vrettou C., Connelley T. & Morrison W.I. (2018) Identification and annotation of bovine granzyme genes reveals a novel granzyme encoded within the trypsin‐like locus. Immunogenetics 70, 585–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenko A., Yudin N., Aitnazarov R. et al (2018) Genome‐wide genotyping uncovers genetic profiles and history of the Russian cattle breeds. Heredity 120, 125–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P. & Mende U. (2014) Functional role, mechanisms of regulation, and therapeutic potential of regulator of G protein signaling 2 in the heart. Trends in Cardiovascular Medicine 24, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin A.V., Delcher A.L., Florea L. et al (2009) A whole‐genome assembly of the domestic cow, Bos taurus . Genome Biology 10, R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinovieva N.A., Dotsev A.V., Sermyagin A.A., Wimmers K., Reyer H., Sölkner J., Deniskova T.E. & Brem G. (2016) Study of genetic diversity and population structure of five Russian cattle breeds using whole‐genome SNP analysis. Sel'skokhozyaistvennaya Biologiya 51, 788–800. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Venn diagram based on the list of significantly differentially expressed genes following all three possible comparisons.
Figure S2 Illustration of the closely located Granzyme B in the genome.
Table S1 Expression levels for each sample.
Table S2 Significantly differentially expressed genes between Yakutian cattle and Northern Finncattle.
Table S3 GO terms associated with upregulated genes in Yakutian cattle when compared with Northern Finncattle.
Table S4 KEGG pathways associated with upregulated genes in Yakutian cattle when compared with Northern Finncattle.
Table S5 GO terms associated with upregulated genes in Northern Finncattle when compared with Yakutian cattle.
Table S6 Summary of genes significantly differentially expressed between Yakutian cattle and Holstein cattle.
Table S7 GO terms associated with upregulated genes in Yakutian cattle when compared with Holstein cattle.
Table S8 KEGG pathways associated with upregulated genes in Yakutian cattle when compared with Holstein cattle.
Table S9 GO terms associated with upregulated genes in Holstein cattle when compared with Yakutian cattle.
Table S10 KEGG pathways associated with upregulated genes in Holstein cattle when compared with Yakutian cattle.
Table S11 Summary of significantly genes differentially expressed between Northern Finncattle and Holstein cattle.
Table S12 GO terms associated with upregulated genes in Holstein cattle when compared with Northern Finncattle.
Table S13 KEGG pathways associated with upregulated genes in Holstein cattle when compared with Northern Finncattle.
Table S14 GO terms associated with the genes that were exclusively upregulated in Yakutian cattle.
Table S15 KEGG pathways associated with the genes that were exclusively upregulated in Yakutian cattle.
Table S16 GO terms associated with the genes that were exclusively upregulated in Holstein cattle.
Table S17 KEGG pathways associated with genes that were exclusively upregulated in Holstein cattle.
Appendix S1 R script used to identify and annotate differentially expressed genes in this study.
Data Availability Statement
The raw sequence reads (FASTQ files) have been deposited in the European Nucleotide Archive (ENA) under accession no. PRJEB28074 (please refer to Table 2 for sample‐specific accessions). The script used during the differential expression analysis is available in Appendix S1.