

Abstract
Objective
Tofacitinib is an oral JAK inhibitor for the treatment of rheumatoid arthritis (RA). The phase III, 24‐month, placebo‐controlled Oral Rheumatoid Arthritis (ORAL) Scan trial was undertaken to evaluate the efficacy, including inhibition of structural progression, and safety of tofacitinib in patients with active RA and an inadequate response to methotrexate (MTX). Month 24 data from the completed study are reported here.
Methods
Patients were randomized 4:4:1:1 to receive tofacitinib 5 mg or 10 mg twice daily, or placebo, switched to tofacitinib 5 mg or 10 mg twice daily, with stable background MTX. Patients receiving placebo switched to tofacitinib at month 3 (nonresponders) or month 6 (remaining patients). Clinical efficacy, structural progression, and treatment‐emergent adverse events were evaluated. Analyses were performed on the full analysis set with observed data or nonresponder imputation with no advancement penalty for clinical efficacy, and imputation by linear extrapolation for structural progression.
Results
Overall, 797 patients were treated; 539 (67.6%) completed 24 months of treatment. Responses according to the American College of Rheumatology criteria for 20% improvement (ACR20), ACR50, and ACR70; the proportion of patients in whom remission or low disease activity was achieved according to the 4‐variable Disease Activity Score in 28 joints using the erythrocyte sedimentation rate, Clinical Disease Activity Index, or Simplified Disease Activity Index; Boolean remission; and Health Assessment Questionnaire disability index scores were maintained from month 12 to 24 and were similar between tofacitinib dosages. Limited structural damage was observed at months 12 and 24. Safety events were similar in type and frequency for both tofacitinib dosages, and were consistent with those previously reported.
Conclusion
Our findings indicate that clinical and radiographic treatment effects are sustained in months 12–24 in patients with RA receiving tofacitinib 5 mg or 10 mg twice daily plus MTX. The safety profile is consistent with that of other tofacitinib studies.
Introduction
Tofacitinib is an oral JAK inhibitor for the treatment of rheumatoid arthritis (RA). The clinical efficacy and safety of tofacitinib 5 mg and 10 mg twice daily administered as monotherapy or in combination with conventional synthetic disease‐modifying antirheumatic drugs, mainly methotrexate (MTX), in patients with RA have been demonstrated in phase III randomized controlled trials (RCTs) of up to 24 months’ duration 1, 2, 3, 4, 5, 6, 7 and in long‐term extension studies with up to 114 months’ observation 8, 9, 10. The phase III Oral Rheumatoid Arthritis (ORAL) Scan RCT was the first trial designed to assess the durability of response, including structural damage progression, and safety in patients with active RA and an inadequate response to MTX who were receiving tofacitinib and stable background MTX for 24 months. Primary end point results from planned interim analyses at month 12 were reported previously 5.
Herein we report efficacy and safety results through month 24 of the completed RCT, focusing on the maintenance of benefit of tofacitinib on clinical efficacy, structural progression, and safety.
Patients and Methods
Study design and patients
Full details of the ORAL Scan study design have been reported previously 5. Briefly, ORAL Scan was a phase III, double‐blind, parallel‐group, placebo‐controlled RCT of tofacitinib 5 mg or 10 mg twice daily with background MTX, compared with continued background MTX plus placebo, in adult patients with RA who had an inadequate response to MTX (ClinicalTrials.gov identifier: NCT00847613; see Appendix A for study investigators). Patients were randomized 4:4:1:1 to receive tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, placebo switched to tofacitinib 5 mg twice daily, or placebo switched to tofacitinib 10 mg twice daily. In the placebo treatment sequences, nonresponders (defined as patients with <20% improvement in swollen and tender joint counts) were advanced in a blinded manner to receive tofacitinib at month 3, according to the prespecified randomization schedule. All remaining patients receiving placebo were advanced in a blinded manner to receive tofacitinib at month 6. Prior to the first dose of study drug, patients must have been receiving MTX continuously for ≥4 months and a stable dose for ≥6 weeks. All patients received stable doses of MTX (≤25 mg weekly) throughout the ORAL Scan study. Stable weekly doses of <15 mg were allowed only in the case of intolerance of higher doses, toxicity from higher doses, or where higher doses would violate the local label.
The trial was approved by the institutional review boards (IRBs) and/or independent ethics committees at each investigational center or a central IRB and conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practice Guidelines. All patients provided written informed consent.
Clinical efficacy assessments
Here we report 24‐month data from the completed RCT (final, locked database). Clinical efficacy parameters evaluated included the American College of Rheumatology criteria for 20% improvement (ACR20), ACR50, and ACR70 responses 11; mean changes from baseline in the 4‐variable Disease Activity Score in 28 joints using the erythrocyte sedimentation rate (DAS28‐ESR) 12; remission defined as DAS28‐ESR <2.6, Clinical Disease Activity Index (CDAI) ≤2.8, or Simplified Disease Activity Index (SDAI) ≤3.3, or by Boolean remission criteria 13; and low disease activity defined as DAS28‐ESR ≤3.2, CDAI ≤10, or SDAI ≤11.
The mean change from baseline in Health Assessment Questionnaire (HAQ) disability index (DI) was a coprimary end point at month 3. Changes from baseline through month 12, for which a step‐down approach was used with coprimary efficacy end points assessed sequentially, have been reported previously 5. Since this article primarily has a safety and efficacy focus, HAQ DI results are briefly presented here for completeness.
Structural progression
Consistent with the 12‐month interim analysis, inhibition of progression of structural damage was assessed by mean changes from baseline in modified Sharp/van der Heijde score (SHS), including erosion and joint space narrowing (JSN) scores, and proportions of patients without radiographic progression (SHS changes from baseline ≤0.5), evaluated for months 12–24. All radiographs were scored independently by 2 readers who were blinded with regard to treatment, visit, and time; these 2 readers’ SHS scores were then averaged for each time point to provide a single composite score for months 12 and 24. Note that all available radiographs were reread for the 24‐month analysis.
Safety
The incidence and severity of all‐cause adverse events (AEs), abnormal clinical laboratory findings, and vital signs were recorded. Treatment‐emergent AEs (TEAEs), serious AEs (SAEs), discontinuations due to AEs, and laboratory evaluations of interest were assessed according to treatment phase: months 0–3, months 3–6, and months 6–24. A Safety End Point Adjudication Committee comprising external independent consultants who were blinded with regard to treatment sequence assignment reviewed all deaths, cardiovascular events, and malignancies. Safety data up to month 12 have been presented previously 5.
Crude exposure‐adjusted incidence rates (with corresponding 95% confidence intervals [95% CIs]) for AEs and serious infections were calculated based on the number of unique patients with events per 100 patient‐years of exposure.
Statistical analysis
All analyses were based on the full analysis set, which included all patients who received ≥1 dose of study drug and had ≥1 postbaseline assessment. Except where noted, data are presented by the randomized treatment sequences. Patients initially treated with tofacitinib who did not experience a decrease of >20% in tender and swollen joints were “advanced” at month 3 to the same dosage of tofacitinib.
Missing ACR responses and data on remission and low disease activity status were addressed by nonresponder imputation with no advancement penalty, i.e., missing values resulting from patients withdrawing from the study for any reason were set to failure, but in patients continuing the study, values measured after advancement were not set to failure. This was done for all patients who met the criterion for advancement, those receiving tofacitinib as well those receiving placebo. This enabled the assessment of responses for patients who were “advanced” from tofacitinib to tofacitinib, and thus had no true change in medication. Analyses with nonresponder imputation with no advancement penalty are presented; specifically, response rates, with corresponding SEs and 95% CIs, based on Z‐scores formed by the normal approximation to the binomial distribution, compared with baseline values within each sequence. P values were not corrected to control for Type I errors.
To maintain blinding at month 3, radiographs were obtained in all patients who advanced at month 3. For radiographic data only, the radiographic scores for month 6 were imputed by linear extrapolation from baseline and month 3. Note that this includes patients receiving tofacitinib as well as patients receiving placebo, with the linear extrapolation value being the best estimate for each randomized treatment. For patients receiving placebo, radiographic scores for month 12 were imputed by linear extrapolation based on baseline and month 3 (for patients who advanced at month 3) or baseline and month 6 (for patients who advanced at month 6). Linear extrapolation was used to impute any missing data at the month 12 and month 24 visits. That is, baseline and non‐missing month 12 data would be used to estimate progression for a missing month 24 visit (radiographic data only).
Results for the clinical efficacy and structural progression end points are provided by their respective imputation methods (nonresponder imputation with no advancement penalty for efficacy and linear extrapolation for radiographic data), as well as observed, without imputation. Changes from baseline in erosion and JSN scores were computed for each patient, and individual values are presented in cumulative probability plots to show the distribution of the changes in individual patients.
The durability of response was assessed for low disease activity and remission as defined by the DAS28‐ESR, after month 6 in the full analysis set and for patients who completed the study, as observed. A flare was recorded at a visit if the DAS28‐ESR worsened by >1.2 from month 6, or, if the DAS28‐ESR was 5.1 at the visit, the score had worsened by >0.6 from month 6. A total of 6 visits, representing 18 months of study time, occurred after month 6.
Each of the structural progression end points were expressed as changes from baseline and analyzed by analysis of covariance applied to the imputed data set, with treatment, baseline values of the end point, and geographic region included as fixed effects.
All nonstructural continuous end points (efficacy variables such as DAS28‐ESR or patient‐reported outcomes such as patient's global assessment of disease activity) were expressed as changes from baseline. Each was analyzed by a linear mixed‐effects repeated‐measures model. Treatment, visit, treatment‐by‐visit interaction, geographic region, and baseline value were included as fixed effects, and patients were included as random effects. In both approaches, estimates of mean changes from baseline were derived from the model as least squares means, with corresponding SEs and 95% CIs, with t‐statistics and P values comparing tofacitinib with placebo. Rates of nonprogression, based on radiographic scores, were calculated based on linear extrapolation as described above.
Results
Patient disposition and demographic characteristics
Of the 800 patients who were randomized, 797 received treatment between March 2009 and February 2012; 539 (67.6%) of these patients completed the 24‐month study. Similar demographic and baseline characteristics were observed for patients in all 4 randomized treatment sequences (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract) 5. The characteristics of the patients continuing the study at month 12 were also generally similar to those of the overall study population (Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract). Patient disposition is summarized in Figure 1, and retention rates for patients receiving tofacitinib 5 mg twice daily and those receiving tofacitinib 10 mg twice daily through month 24 of the study are presented in Supplementary Figure 1 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract).
Figure 1.
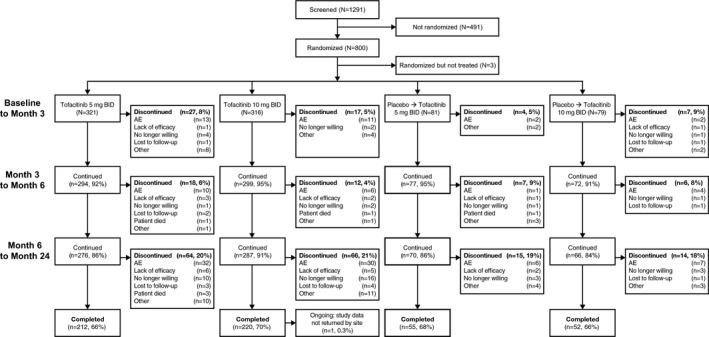
Disposition of the patients from baseline to month 24. Patients were randomized to receive tofacitinib 5 mg twice daily (BID), tofacitinib 10 mg twice daily, or placebo. All patients received background methotrexate, including those in the placebo groups. For patients in the placebo groups, treatment was switched in a blinded manner to tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily at either 3 months (for nonresponders) or 6 months (for all remaining patients in the placebo groups). Patients with <20% improvement in swollen and tender joint counts were considered nonresponders. At month 3, a <20% improvement in swollen and tender joint counts from baseline was found in 84 (26%) of the patients receiving tofacitinib 5 mg twice daily, 56 (18%) of the patients receiving tofacitinib 10 mg twice daily, 42 (52%) of the patients receiving placebo who switched to tofacitinib 5 mg twice daily, and 37 (47%) of the patients receiving placebo who switched to to tofacitinib 10 mg twice daily. Only deaths that occurred during study treatment are included. See Table 3 for details on deaths that occurred after the last dose of study drug. AE = adverse event.
Efficacy
Clinical efficacy
ACR20/50/70 responses, the proportions of patients with disease in remission (DAS28‐ESR <2.6) or low disease activity (DAS28‐ESR ≤3.2), and improvements from baseline in DAS28‐ESR were maintained from month 12 to 24 (Table 1). Similarly, the proportions of patients with disease in remission as defined by CDAI and SDAI scores and those with Boolean‐based remission were maintained from month 12 to 24 (Table 1). Responses were similar between treatment sequences once all patients were receiving tofacitinib. Patients receiving tofacitinib 10 mg twice daily had numerically higher responses than those receiving 5 mg twice daily; however, since the study was not powered for this comparison, no formal statistical comparison between dosages was conducted. Changes in ACR criteria components from baseline to month 24 (observed values) are summarized in Supplementary Table 3 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract).
Table 1.
Clinical efficacy parameters at month 12 and month 24 by treatment cohort (using imputed or observed data in the full analysis set and a longitudinal model based on the full analysis set)a
| Tofacitinib 5 mg twice daily | Tofacitinib 10 mg twice daily | Placebo switching to tofacitinib 5 mg twice daily | Placebo switching to tofacitinib 10 mg twice daily | |||||
|---|---|---|---|---|---|---|---|---|
| 12 months | 24 months | 12 months | 24 months | 12 months | 24 months | 12 months | 24 months | |
| ACR20/50/70 response | ||||||||
| No. of patients | ||||||||
| NRINAP | 309 | 309 | 309 | 309 | 79 | 79 | 75 | 75 |
| Observed | 252 | 211 | 265 | 218 | 67 | 54 | 63 | 52 |
| ACR20 responseb | ||||||||
| NRINAP | 60.8 (2.8) | 50.5 (2.8) | 65.4 (2.7) | 58.3 (2.8) | 62.0 (5.5) | 58.2 (5.5) | 60.0 (5.6) | 57.3 (5.7) |
| Observed | 75.4 (2.7) | 73.5 (3.0) | 78.5 (2.5) | 82.6 (2.6) | 74.6 (5.3) | 83.3 (5.1) | 74.6 (5.5) | 82.7 (5.2) |
| ACR50 responseb | ||||||||
| NRINAP | 37.2 (2.7) | 33.7 (2.7) | 42.1 (2.8) | 46.3 (2.8) | 43.0 (5.6) | 38.0 (5.5) | 33.3 (5.4) | 37.3 (5.6) |
| Observed | 46.0 (3.1) | 48.8 (3.4) | 50.9 (3.1) | 65.1 (3.2) | 52.2 (6.1) | 53.7 (6.8) | 41.3 (6.2) | 53.9 (6.9) |
| ACR70 responseb | ||||||||
| NRINAP | 20.1 (2.3) | 18.8 (2.2) | 26.9 (2.5) | 28.8 (2.6) | 25.3 (4.9) | 21.5 (4.6) | 22.7 (4.8) | 26.7 (5.1) |
| Observed | 24.6 (2.7) | 27.0 (3.1) | 32.5 (2.9) | 40.4 (3.3) | 31.3 (5.7) | 31.5 (6.3) | 27.0 (5.6) | 38.5 (6.7) |
| Remission | ||||||||
| DAS28‐ESR <2.6b | ||||||||
| No. of patients | ||||||||
| NRINAP | 265 | 265 | 257 | 257 | 65 | 65 | 64 | 64 |
| Observed | 218 | 179 | 221 | 179 | 54 | 41 | 54 | 44 |
| NRINAP | 10.6 (1.9) | 11.3 (1.9) | 14.0 (2.2) | 14.8 (2.2) | 10.8 (3.8) | 7.7 (3.3) | 20.3 (5.0) | 15.6 (4.5) |
| Observed | 12.8 (2.3) | 16.8 (2.8) | 18.6 (2.6) | 20.7 (3.0) | 13.0 (4.6) | 12.2 (5.1) | 25.9 (6.0) | 22.7 (6.3) |
| CDAI ≤2.8c | ||||||||
| No. of patients | ||||||||
| NRINAP | 309 | 309 | 308 | 308 | 79 | 79 | 75 | 75 |
| Observed | 252 | 208 | 265 | 218 | 67 | 54 | 63 | 52 |
| NRINAP | 14.2 (2.0) | 12.0 (1.9) | 16.6 (2.1) | 19.8 (2.3) | 12.7 (3.7) | 12.7 (3.7) | 20.0 (4.6) | 22.7 (4.8) |
| Observed | 17.5 (2.4) | 17.8 (2.7) | 20.0 (2.5) | 27.5 (3.0) | 14.9 (4.4) | 18.5 (5.3) | 23.8 (5.4) | 32.7 (6.5) |
| SDAI ≤3.3c | ||||||||
| No. of patients | ||||||||
| NRINAP | 309 | 309 | 308 | 308 | 79 | 79 | 75 | 75 |
| Observed | 252 | 207 | 263 | 216 | 67 | 54 | 63 | 52 |
| NRINAP | 13.9 (2.0) | 14.2 (2.0) | 17.2 (2.2) | 23.4 (2.4) | 12.7 (3.7) | 16.5 (4.2) | 20.0 (4.6) | 28.0 (5.2) |
| Observed | 17.1 (2.4) | 20.8 (2.8) | 20.9 (2.5) | 32.9 (3.2) | 14.9 (4.4) | 22.2 (5.7) | 23.8 (5.4) | 40.4 (6.8) |
| Boolean remission | ||||||||
| No. of patients | ||||||||
| NRINAP | 309 | 309 | 309 | 309 | 79 | 79 | 75 | 75 |
| Observed | 252 | 210 | 265 | 218 | 67 | 54 | 63 | 52 |
| NRINAP | 10.4 (1.7) | 10.0 (1.7) | 13.6 (2.0) | 14.6 (2.0) | 10.1 (3.4) | 13.9 (3.9) | 16.0 (4.2) | 21.3 (4.7) |
| Observed | 12.7 (2.1) | 14.3 (2.4) | 16.6 (2.3) | 20.2 (2.7) | 11.9 (4.0) | 18.5 (5.3) | 19.0 (5.0) | 30.8 (6.4) |
| Low disease activity or remission | ||||||||
| DAS28‐ESR ≤3.2b | ||||||||
| No. of patients | ||||||||
| NRINAP | 265 | 265 | 257 | 257 | 65 | 65 | 64 | 64 |
| Observed | 218 | 179 | 221 | 179 | 54 | 41 | 54 | 44 |
| NRINAP | 23.4 (2.6) | 23.0 (2.6) | 29.2 (2.8) | 30.4 (2.9) | 21.5 (5.1) | 24.6 (5.3) | 31.3 (5.8) | 25.0 (5.4) |
| Observed | 28.4 (3.1) | 33.5 (3.5) | 36.2 (3.2) | 43.0 (3.7) | 25.9 (6.0) | 39.0 (7.6) | 38.9 (6.6) | 36.4 (7.3) |
| CDAI ≤10c | ||||||||
| No. of patients | ||||||||
| NRINAP | 309 | 309 | 308 | 308 | 79 | 79 | 75 | 75 |
| Observed | 252 | 208 | 265 | 218 | 67 | 54 | 63 | 52 |
| NRINAP | 41.1 (2.8) | 40.5 (2.8) | 50.3 (2.9) | 49.4 (2.9) | 48.1 (5.6) | 46.8 (5.6) | 38.7 (5.6) | 45.3 (5.8) |
| Observed | 51.2 (3.2) | 59.1 (3.4) | 60.4 (3.0) | 69.7 (3.1) | 58.2 (6.0) | 66.7 (6.4) | 47.6 (6.3) | 65.4 (6.6) |
| SDAI ≤11c | ||||||||
| No. of patients | ||||||||
| NRINAP | 309 | 309 | 308 | 308 | 79 | 79 | 75 | 75 |
| Observed | 252 | 207 | 263 | 216 | 67 | 54 | 63 | 52 |
| NRINAP | 43.0 (2.8) | 40.5 (2.8) | 52.6 (2.9) | 49.0 (2.9) | 50.6 (5.6) | 48.1 (5.6) | 41.3 (5.7) | 46.7 (5.8) |
| Observed | 53.2 (3.1) | 58.9 (3.4) | 62.7 (3.0) | 69.4 (3.1) | 61.2 (6.0) | 68.5 (6.3) | 50.8 (6.3) | 67.3 (6.5) |
| Change from baselined | ||||||||
| LSM (SE) change in DAS28‐ESRb | −2.2 (0.1) | −2.3 (0.1) | −2.5 (0.1) | −2.6 (0.1) | −2.1 (0.2) | −2.5 (0.2) | −2.4 (0.2) | −2.6 (0.2) |
| No. of patients | 218 | 179 | 220 | 178 | 54 | 41 | 54 | 44 |
| Mean (SE) change in HAQ DI | −0.5 (0.0) | −0.5 (0.0) | −0.6 (0.0) | −0.7 (0.0) | −0.5 (0.1) | −0.6 (0.1) | −0.5 (0.1) | −0.6 (0.1) |
| No. of patients | 251 | 210 | 265 | 218 | 67 | 54 | 63 | 52 |
Except where indicated otherwise, values are the percent of patients (SE). All patients received background methotrexate, including those in the placebo groups. ACR20 = American College of Rheumatology criteria for 20% improvement; NRINAP = nonresponder imputation with no advancement penalty; DAS28‐ESR = Disease Activity Score in 28 joints (4‐variable) using the erythrocyte sedimentation rate; CDAI = Clinical Disease Activity Index; SDAI = Simplified Disease Activity Index; LSM = least squares mean; HAQ DI = Health Assessment Questionnaire disability index.
Values were significantly improved (P ≤ 0.05) at months 12 and 24 versus baseline within each treatment sequence.
P values were not calculated for these outcomes.
Longitudinal model, observed completely.
Improvements in the HAQ DI that were observed from baseline to month 12 5 were maintained through month 24 both for patients receiving tofacitinib 5 mg twice daily and for patients receiving 10 mg twice daily (Table 1). The durability of response for low disease activity and remission according to DAS28‐ESR were analyzed and are shown in Supplementary Table 4 (http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract). Generally, in the full analysis set, patients receiving tofacitinib 10 mg twice daily had low disease activity and disease in remission for a modestly higher number of months, with numerically higher proportions of patients experiencing ≥12 months of uninterrupted low disease activity or remission, compared with the other treatment groups. Patients who switched from placebo to tofacitinib 5 mg twice daily showed a modestly lower total number of months in remission and proportion of patients achieving ≥12 months’ uninterrupted low disease activity or remission versus other groups. These trends remained similar when only patients who completed the study were analyzed. Additionally, no patient in any treatment group had more than 1 flare after month 6.
Structural progression
Baseline radiographs were available for 89.4% of the patients receiving tofacitinib 5 mg twice daily (287 of 321), 94.3% of the patients receiving tofacitinib 10 mg twice daily (298 of 316), and 86.9% of the patients receiving placebo (139 of 160) (values for the placebo groups were imputed, as noted above). At month 24, radiographs were available for most patients who completed the study: 99.1% of those receiving tofacitinib 5 mg twice daily (210 of 212), 98.2% of those receiving tofacitinib 10 mg twice daily (216 of 220), 94.5% of those receiving placebo and then switching to tofacitinib 5 mg twice daily (52 of 55), and 94.2% of those receiving placebo and then switching to tofacitinib 10 mg twice daily (49 of 52). Month 24 structural data were evaluable via linear extrapolation in 287 patients receiving tofacitinib 5 mg twice daily and 298 patients receiving tofacitinib 10 mg twice daily.
In the primary analysis at month 6 5, smaller least squares mean changes in SHS from baseline were reported both for patients receiving tofacitinib 5 mg twice daily (P > 0.05) and for patients receiving tofacitinib 10 mg twice daily (P ≤ 0.05), versus placebo. These effects were consistent throughout the second year of the study.
Actual values, changes from baseline, and changes from month 12 in SHS, erosion, and JSN scores are shown in Figures 2A–C. Patients receiving tofacitinib had minimal progression of structural damage, as determined by change in SHS, erosion score, and JSN score, from baseline to months 12 and 24. The magnitude of progression was similar for patients receiving tofacitinib 5 mg twice daily and those receiving tofacitinib 10 mg twice daily. For patients who initially received placebo and then switched to tofacitinib, there was generally minimal progression of structural damage, as assessed by SHS, erosion score, and JSN score (Supplementary Figure 2, online at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract).
Figure 2.
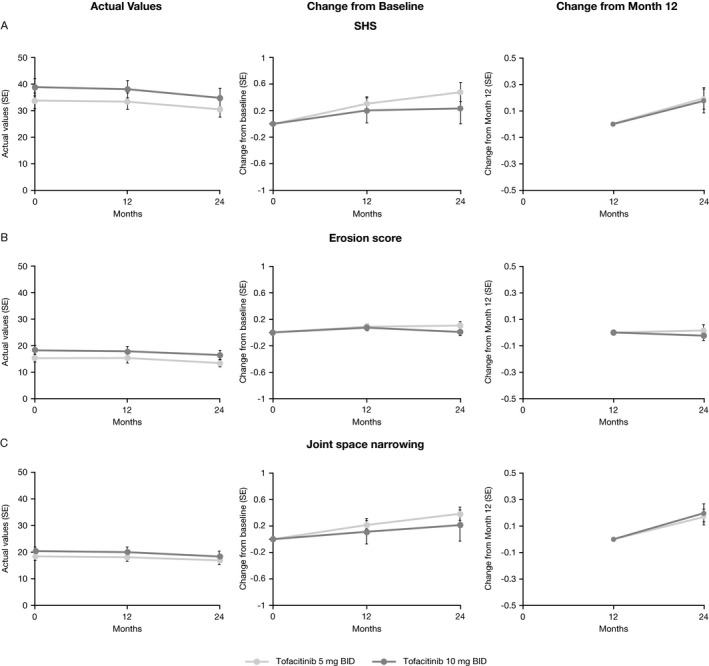
Actual values, change from baseline to months 12 and 24, and change from month 12 to month 24 in A, modified Sharp/van der Heijde score (SHS), B, erosion score, and C, joint space narrowing score in rheumatoid arthritis (RA) patients receiving tofacitinib 5 mg twice daily (BID) and RA patients receiving tofacitinib 10 mg twice daily (as observed in the full analysis set). All patients received background methotrexate. Patients who did not have valid postbaseline radiographs were not included. Data are shown for patients who completed month 12, as observed. Values are the mean ± SEM (n = 287 at baseline, n = 252 at month 12, and n = 210 at month 24 [n = 208 for change from month 12] for tofacitinib 5 mg twice daily; n = 298 at baseline, n = 261 at month 12, and n = 216 at month 24 [n = 214 for change from month 12] for tofacitinib 10 mg twice daily).
Based on linear extrapolation, the proportion of nonprogressors (as determined by SHS) from months 12 to 24 was 91.9% of the patients receiving tofacitinib 5 mg twice daily (295 of 321) and 90.5% of the patients receiving tofacitinib 10 mg twice daily (295 of 326). Furthermore, no new erosions were observed between months 12 and 24 in 97.2% of the patients receiving tofacitinib 5 mg twice daily (312 of 321) and 96.3% of the patients receiving tofacitinib 10 mg twice daily (314 of 326), by linear extrapolation. The mean observed changes in structural parameters (without imputation) for patients receiving tofacitinib 5 mg twice daily and for those receiving tofacitinib 10 mg twice daily are shown in Table 2. The cumulative probability of changes in erosion score and JSN from baseline to month 24 were similar across all treatment sequences (Supplementary Figure 3, online at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract).
Table 2.
Changes in radiographic scores from month 12 to month 24 in patients receiving tofacitinib who completed month 12a
| Tofacitinib 5 mgtwice daily(n = 263) | Tofacitinib 10 mgtwice daily (n = 264) | |
|---|---|---|
| SHS | 0.19 ± 1.04 | 0.15 ± 1.23 |
| Erosion score | 0.00 ± 0.49 | −0.04 ± 0.59 |
| JSN score | 0.18 ± 0.83 | 0.19 ± 0.96 |
Values are the mean ± SD change from month 12 to month 24. Treatment groups included patients who received the indicated dosage of tofacitinib from the beginning of the study and patients who received the indicated dosage of tofacitinib after switching from placebo. All patients received background methotrexate. Data are observed (no imputation). SHS = modified Sharp/van der Heijde score; JSN = joint space narrowing.
Safety and tolerability
Exposure to study treatment
The mean duration of study treatment was 572 days (median 709 days) for patients receiving tofacitinib 5 mg twice daily, 600 days (median 711 days) for patients receiving tofacitinib 10 mg twice daily, 580 days (median 714 days) for patients receiving placebo and then switching to tofacitinib 5 mg twice daily, and 569 days (median 714 days) for patients receiving placebo and then switching to tofacitinib 10 mg twice daily. Total exposure was 508 patient‐years and 525 patient‐years for patients receiving tofacitinib 5 mg twice daily and those receiving 10 mg twice daily, respectively, compared with 129 patient‐years and 124 patient‐years for the patients receiving placebo and then switching to tofacitinib 5 mg twice daily and those receiving placebo and then switching to tofacitinib 10 mg twice daily, respectively (Table 3).
Table 3.
Treatment‐emergent AEs (any cause) and AEs of interest throughout the entire study period (months 0–24)a
| Tofacitinib 5 mg twice daily | Tofacitinib 10 mg twice daily | Placebo switching to tofacitinib 5 mg twice daily | Placebo switching to tofacitinib 10 mg twice daily | |
|---|---|---|---|---|
| Treatment‐emergent AEs (any cause) | ||||
| No. of evaluable patients | 321 | 316 | 81 | 79 |
| Duration of treatment, mean (median) days | 572 (709) | 600 (711) | 580 (714) | 569 (714) |
| Total no. of patient‐years of exposure | 508 | 525 | 129 | 124 |
| Number of AEs | 1,510 | 1,562 | 315 | 290 |
| Patients with AEs, no. (%) | 279 (86.9) | 275 (87.0) | 63 (77.8) | 57 (72.2) |
| Crude exposure‐adjusted incidence rate/100 patient‐years (95% CI) | 169 (150.4–190.2) | 167 (148.6–188.2) | 116 (90.8–148.8) | 110 (85.2–143.1) |
| Patients with SAEs, no. (%) | 86 (26.8) | 78 (24.7) | 19 (23.5) | 20 (25.3) |
| Discontinuations due to AEs, no. (%) | 55 (17.1) | 47 (14.9) | 9 (11.1) | 13 (16.5) |
| Deaths, no. (%)b | ||||
| All | 7 (2.2) | 3 (0.9) | 2 (2.5) | 0 (0.0) |
| While receiving study treatmentc | 4 (1.2) | 1 (0.3) | 1 (1.2) | 0 (0.0) |
| Liver enzyme testsd | ||||
| No. of evaluable patients | 318 | 315 | 81 | 78 |
| ALT ≥1× ULN | 143 (45.0) | 151 (47.9) | 22 (27.2) | 36 (46.2) |
| ALT ≥3× ULN | 19 (6.0) | 21 (6.7) | 2 (2.5) | 6 (7.7) |
| AST ≥1× ULN | 134 (42.1) | 148 (47.0) | 27 (33.3) | 32 (41.0) |
| AST ≥3× ULN | 10 (3.1) | 9 (2.9) | 2 (2.5) | 2 (2.6) |
| Total bilirubin ≥1× ULN | 17 (5.3) | 23 (7.3) | 6 (7.4) | 6 (7.7) |
| Total bilirubin ≥3× ULN | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| AEs of interest | ||||
| No. of evaluable patients | 321 | 316 | 81 | 79 |
| Serious infections, no. (%) | 24 (7.5) | 19 (6.0) | 2 (2.5) | 5 (6.3) |
| Crude exposure‐adjusted incidence rate/100 patient‐years (95% CI) | 4.8 (3.2–7.1) | 3.6 (2.3–5.7) | 1.5 (0.4–6.2) | 4.1 (1.7–9.7) |
| Patients with malignancies, no. (%)e | 14 (4.4)f | 10 (3.2)g | 1 (1.2) | 1 (1.3) |
| Excluding nonmelanoma skin cancere | 6 (1.9) | 7 (2.2) | 1 (1.2) | 0 (0.0) |
| Nonmelanoma skin cancer | 8 (2.5) | 3 (0.9) | 0 (0.0) | 1 (1.3) |
| Cardiovascular events, no. (%)e | 9 (2.8) | 9 (2.9) | 2 (2.5)h | 0 (0.0) |
| TB, no. (%)i | 0 (0.0) | 2 (0.6) | 0 (0.0) | 1 (1.3) |
| Herpes zoster, no. (%)j | ||||
| All | 24 (7.5) | 32 (10.1) | 4 (4.9) | 7 (8.9) |
| Disseminated | 0 (0.0) | 1 (0.3) | 0 (0.0) | 0 (0.0) |
| GI perforation, no. (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Lymphopenia, no. (%) | 0 (0.0) | 3 (0.9) | 0 (0.0) | 0 (0.0) |
| Thrombocytopenia, no (%) | 1 (0.3) | 3 (0.9) | 0 (0.0) | 0 (0.0) |
| Neutropenia, no. (%)d, k | ||||
| No. of evaluable patients | 316 | 309 | 79 | 77 |
| Mild (≥1.5 to <2 × 103 cells/μl) | 41 (13.0) | 44 (14.2) | 8 (10.1) | 12 (15.6) |
| Moderate to severe (≥0.5 to <1.5 × 103 cells/μl) | 11 (3.5) | 18 (5.8) | 2 (2.5) | 4 (5.2) |
| Life‐threatening (<0.5 × 103 cells/μl) | 1 (0.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Decrease in hemoglobin, no. (%)d, k | ||||
| No. of evaluable patients | 316 | 309 | 79 | 77 |
| Mild or moderate (decrease of ≥1 gm/dl to ≤2 gm/dl) | 71 (22.5) | 77 (24.9) | 21 (26.6) | 22 (28.6) |
| Severe (decrease of >2 gm/dl to <3 gm/dl or absolute value >7 gm/dl but <8 gm/dl) | 13 (4.1) | 17 (5.5) | 4 (5.1) | 5 (6.5) |
| Life‐threatening (decrease of ≥3 gm/dl or absolute value ≤7 gm/dl) | 7 (2.2) | 8 (2.6) | 1 (1.3) | 2 (2.6) |
Except where indicated otherwise, values are the number (%). All patients received background methotrexate, including those in the placebo groups. Mean changes from baseline over time in hemoglobin level, platelet, neutrophil, and lymphocyte counts, and, low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and serum creatinine levels are shown in Supplementary Figure 4 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract). Changes in ALT and AST are without regard to baseline. 95% CI = 95% confidence interval; SAE = serious adverse event; ULN = upper limit of normal; GI = gastrointestinal.
There was a total of 12 deaths that were adjudicated to be either related or unrelated to study treatment. Three patients died after withdrawing from the study due to AEs considered related to treatment (adult respiratory distress syndrome and viral pneumonia in 1 patient in the tofacitinib 5 mg sequence, metastatic lung cancer in 1 patient in the tofacitinib 5 mg sequence, and acute renal failure in 1 patient in the placebo switching to tofacitinib 5 mg sequence). Two patients died after withdrawing from the study due to non–treatment‐related AEs (multiorgan failure in 1 patient in the tofacitinib 5 mg sequence and chronic obstructive pulmonary disease in 1 patient in the tofacitinib 10 mg sequence). One patient who was randomized to receive tofacitinib 10 mg died before receiving study medication.
In the group receiving tofacitinib 5 mg twice daily, 1 patient each died of pneumonia, probable acute myocardial infarction, cardiac arrest and respiratory arrest, and heart failure. One patient in the tofacitinib 10 mg twice daily group died due to glycerin swab aspiration. One patient who received placebo and then switched to tofacitinib 5 mg twice daily died due to acute renal failure, cardiac arrest, and other AEs.
Based on a value in the specified range being reported for ≥1 sample.
Adjudicated in a blinded manner by a Safety End Point Adjudication Committee.
One patient experienced both an event of nonmelanoma skin cancer (basal cell carcinoma) and a malignancy (prostate cancer).
One patient experienced 2 events of nonmelanoma skin cancer (basal cell carcinoma and squamous cell carcinoma) and a malignancy (melanoma in situ); these events were reported 5 years after the end of the study and were not included in the final study report.
Both events occurred during treatment with tofacitinib.
Includes cases of disseminated and lymph node tuberculosis (TB).
Includes disseminated, ophthalmic herpes zoster.
Decrease from baseline.
Treatment‐emergent adverse events
TEAEs occurring during months 0−12 have been reported previously 5. TEAEs (any cause) through month 24 are shown in Table 3. Most TEAEs were mild to moderate in severity. The most common TEAEs (affecting ≥5% of patients) for months 0−24 by treatment sequence were: nasopharyngitis, upper respiratory tract infection, and headache for patients receiving tofacitinib 5 mg twice daily; nasopharyngitis, upper respiratory tract infection, urinary tract infection, herpes zoster, and bronchitis for patients receiving tofacitinib 10 mg twice daily; nasopharyngitis, upper respiratory tract infection, and hypertension for patients receiving placebo and then switching to tofacitinib 5 mg twice daily; and upper respiratory tract infection, nasopharyngitis, herpes zoster, increased alanine aminotransferase (ALT) level, increased aspartate aminotransferase (AST) level, stomatitis, and diarrhea for patients receiving placebo and then switching to tofacitinib 10 mg twice daily (Supplementary Table 5, online at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract).
Discontinuations due to TEAEs in months 0−24 were similar across the 4 cohorts (11.1−17.1%) (Table 3). The most frequent TEAEs resulting in discontinuation after month 6 were pneumonia in patients receiving tofacitinib 5 mg twice daily (7 patients [2.2%]), herpes zoster and urinary tract infection in patients receiving tofacitinib 10 mg twice daily (each in 4 patients [1.3%]), and pneumonia and intervertebral disc protrusion in patients receiving placebo and then switching to tofacitinib 10 mg twice daily (each in 2 patients [2.5%]). No preferred term AE resulting in discontinuation occurred on >1 occasion in the group receiving placebo and then switching to tofacitinib 5 mg twice daily.
Eleven patients who received study treatment died during the 24‐month study: 7 patients (2.2%) in the tofacitinib 5 mg twice daily group, 2 patients (0.6%) in the tofacitinib 10 mg twice daily group, and 2 patients (2.5%) in the placebo switched to tofacitinib 5 mg twice daily group (one of whom died during the placebo treatment phase) (Table 3). Five of these deaths occurred during months 12–24. In 6 cases, the cause of death was considered to be related or possibly related to study drug administration; in 3 cases, the cause of death was pneumonia. One patient who was randomized to receive tofacitinib 10 mg twice daily died before receiving treatment. Narratives are provided in Supplementary Table 6 (online at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract).
TEAEs of special interest
Narratives for all opportunistic infections (including tuberculosis [TB]) are provided in Supplementary Table 7 (online at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract). There were 12 opportunistic infections, all of which occurred in female patients in Asia and were considered treatment‐related: in 2 patients receiving tofacitinib 5 mg twice daily (1 was an SAE); 8 patients receiving tofacitinib 10 mg twice daily (3 were SAEs); and 1 patient each in the placebo switched to tofacitinib cohorts (1 was an SAE).
None of the patients with reported opportunistic infections had confirmed lymphocyte counts <500 × 103/ml. One patient receiving tofacitinib 10 mg twice daily had disseminated herpes zoster (not serious), which required temporary discontinuation of the study medication. Herpes zoster (all) was reported in 24 patients receiving tofacitinib 5 mg twice daily, 32 patients receiving tofacitinib 10 mg twice daily, 4 patients receiving placebo and then switching to tofacitinib 5 mg twice daily, and 7 patients receiving placebo and then switching to tofacitinib 10 mg twice daily. Another patient receiving tofacitinib 10 mg twice daily had study medication temporarily discontinued on day 132 due to a treatment‐related SAE of cytomegalovirus viremia, which resolved on day 155. The opportunistic infection reported in the group receiving placebo and then switching to tofacitinib 5 mg twice daily occurred 5 months after treatment was switched, and the opportunistic infection in the group receiving placebo and then switching to tofacitinib 10 mg twice daily occurred 18 months after treatment was switched. Lymph node TB was reported in 2 patients receiving tofacitinib 10 mg twice daily (days 269 and 443), and disseminated TB was reported in 1 patient receiving placebo and then switching to tofacitinib 10 mg twice daily, on day 644, ~18 months after switching to tofacitinib (Supplementary Table 7).
All 3 TB cases were considered treatment related and resulted in discontinuation from the study. All 3 of these patients were negative for TB on screening and had clear chest radiographs at study entry. No patient with known active TB was enrolled in the trial, and no patients with latent or presumed latent TB who received isoniazid developed active TB. Four patients had a medical history of adequately treated TB and were not required to take isoniazid.
The most common malignancies (excluding nonmelanoma skin cancer) were adenocarcinoma and squamous cell carcinoma. There was 1 adenocarcinoma event in the tofacitinib 5 mg twice daily group (stomach), 2 adenocarcinoma events in the tofacitinib 10 mg twice daily group (right breast and stomach/lymph node), and 1 adenocarcinoma event in the placebo switched to tofacitinib 5 mg twice daily group (prostate). There were 2 squamous cell carcinoma events each in the tofacitinib 5 mg twice daily group (metastatic; primary site probably lung and cervical) and tofacitinib 10 mg twice daily group (both cervical) (Supplementary Table 8, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract). Two cases of non‐Hodgkin's lymphoma were reported in patients receiving tofacitinib 10 mg twice daily. Nonmelanoma skin cancer cases included basal cell carcinoma (4 events in the tofacitinib 5 mg group, 3 events in the tofacitinib 10 mg twice daily group, and 1 event in the placebo switched to tofacitinib 10 mg twice daily group), squamous cell carcinoma (3 events in the tofacitinib 5 mg twice daily group and 1 event in the tofacitinib 10 mg twice daily group), and atypical fibroxanthoma (1 event in the tofacitinib 5 mg twice daily group). (One patient in the tofacitinib 10 mg twice daily group had both basal cell carcinoma and squamous cell carcinoma.) One patient had both an event of nonmelanoma skin cancer (basal cell carcinoma) and a malignancy (prostate cancer). Another patient experienced 2 events of nonmelanoma skin cancer (basal cell carcinoma and squamous cell carcinoma) and a malignancy (melanoma in situ); these events were reported 5 years after the end of the study and were not included in the final study report. Further details of adjudicated malignancies are provided in Supplementary Table 8.
Throughout the 24‐month RCT, the number of patients with cardiovascular events meeting adjudication criteria were the same in the tofacitinib 5 mg and tofacitinib 10 mg twice daily groups (9 patients each) (Table 3). Two patients receiving placebo and then switching to tofacitinib 5 mg twice daily experienced cardiovascular AEs, both during treatment with tofacitinib (Table 3). The proportions of patients with stage 1 hypertension (systolic blood pressure [SBP] 140–159/diastolic blood pressure [DBP] 90–99) or stage 2 hypertension (SBP 160–179/DBP 100–109) 14 remained constant throughout 24 months of treatment across all groups.
Clinical laboratory findings
Neutrophil and lymphocyte counts, hemoglobin concentrations, platelet counts, low‐density lipoprotein (LDL) cholesterol, high‐density lipoprotein (HDL) cholesterol, serum creatinine, AST, and ALT concentrations are shown by study period and treatment sequences in Supplementary Figure 4 (online at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract). Following an initial decrease in mean neutrophil and platelet counts, and initial increases in mean lymphocyte count, LDL cholesterol, HDL cholesterol, AST, and ALT levels during the first month of treatment with tofacitinib 5 mg or 10 mg twice daily 5, mean neutrophil and lymphocyte counts, LDL cholesterol and HDL cholesterol levels, hemoglobin concentration, platelets, and AST and ALT concentrations were generally similar in all treatment sequences in months 12–24. Supplementary Table 9 (online at http://onlinelibrary.wiley.com/doi/10.1002/art.40803/abstract) summarizes shifts in maximum LDL cholesterol levels versus baseline levels. Numerically small increases from baseline in mean serum creatinine level were observed (Supplementary Figure 4).
After month 12, there was 1 event of potentially life‐threatening neutropenia (absolute neutrophil count <0.5 × 103/μl) in the tofacitinib 5 mg twice daily group at month 15. There were 3 and 7 potentially life‐threatening events of decreased hemoglobin (defined as a decrease of ≥3 gm/dl versus baseline or an actual value of ≤7 gm/dl) in the tofacitinib 5 mg and tofacitinib 10 mg twice daily groups, respectively, and 1 event each in the group receiving placebo and then switching to tofacitinib 5 mg twice daily and the group receiving placebo and then switching to tofacitinib 10 mg twice daily. Each event was a decrease of ≥3 gm/dl from baseline, rather than an actual hemoglobin value of ≤7 gm/dl.
The proportion of patients across all groups with elevations in ALT or AST level ≥3 times the upper limit of normal (ULN) in ≥1 sample during months 0−24 ranged from 2.5% to 7.7% for ALT level and from 2.5% to 3.1% for AST level. After month 6, one patient each receiving tofacitinib 5 mg and tofacitinib 10 mg twice daily discontinued due to elevations in ALT level and 1 patient in each group discontinued due to elevations in AST level, according to prespecified discontinuation criteria (2 sequential elevations in AST or ALT level >5 times the ULN regardless of total bilirubin or accompanying symptoms). The highest proportion of patients with an elevation in ALT level ≥3 times the ULN was in the group receiving placebo and then switching to tofacitinib 10 mg twice daily, and the highest proportion of patients with an elevation in AST ≥3 times the ULN was in the group receiving tofacitinib 5 mg twice daily (Table 3). There were no confirmed elevations of total bilirubin ≥3 times the ULN in any sample (Table 3).
Discussion
This phase III, 24‐month ORAL Scan RCT investigated the efficacy, including inhibition of structural damage, and safety of tofacitinib in patients with RA with an inadequate response to MTX. The primary efficacy end points of the study, based on a preplanned interim analysis, were reported previously and demonstrated that tofacitinib 5 mg or 10 mg twice daily plus MTX resulted in statistically significant improvements in ACR20 responses (versus placebo) at month 6. Inhibition of structural progression, disease activity (with improvement defined as DAS28‐ESR <2.6), and physical function were numerically improved in patients receiving tofacitinib 5 mg twice daily, and statistically significant in patients receiving tofacitinib 10 mg twice daily, at month 6 5.
Two hundred twelve (66%) of the patients receiving tofacitinib 5 mg twice daily, 220 (69%) of the patients receiving tofacitinib 10 mg twice daily, 55 (68%) of the patients receiving placebo and then switching to tofacitinib twice daily, and 52 (66%) of the patients receiving placebo and then switching to tofacitinib 10 mg twice daily continued after month 12 through to completion of the study at month 24. These patients generally had demographic characteristics and baseline disease activity similar to the full analysis set. Minimal structural progression, as well as improvements in disease activity, were sustained through 24 months of treatment. For patients receiving tofacitinib, the ACR20/50/70 responses at month 12 were sustained to month 24, as were the proportions of patients with disease in remission or low disease activity.
Similar clinical responses were achieved in patients who received placebo and then switched to tofacitinib and patients who received initial tofacitinib treatment. In general, tofacitinib 10 mg twice daily resulted in numerically higher responses than 5 mg twice daily, and, as expected, responses in the observed population were higher than in the randomized population by nonresponder imputation with no advancement penalty. Nonetheless, even with the strict definition used with nonresponder imputation with no advancement penalty, 41% and 49% of the patients receiving tofacitinib 5 mg twice daily and 10 mg twice daily, respectively, had low disease activity as defined by CDAI ≤10 and SDAI ≤11 at month 24, which is a meaningful and clinically relevant finding. Additionally, low disease activity as defined by DAS28‐ESR ≤3.2 was achieved in 23% and 30% of the patients receiving tofacitinib 5 mg twice daily and 10 mg twice daily, respectively, for nonresponder imputation with no advancement penalty at month 24.
These data show a difference in the proportion of patients with low disease activity, as assessed by CDAI and SDAI, compared with DAS28‐ESR. A previous study analyzing Dutch and Portuguese patient data from the Measurement of Efficacy of Treatment in the Era of Rheumatology (METEOR) database found a discordance between low disease activity defined by DAS28‐ESR and low disease activity defined by SDAI and CDAI 15. These differences may be due to the different weighting of the individual components between the assessment indices, including the heavier weighting of patient’s global assessment of disease activity in the CDAI and SDAI than in the DAS28‐ESR. A durable low disease activity response was generally observed for each treatment group, with no patient experiencing more than 1 flare after month 6.
Between months 12 and 24, ≥90% of the patients receiving tofacitinib showed no progression in structural damage (SHS ≤0.5), and ≥95% did not develop new erosions. Through the 24‐month period, these patients had minimal changes from baseline in SHS and erosion scores.
For context, the efficacy data presented here for tofacitinib, as observed, are generally similar to the efficacy data shown in the intent‐to‐treat patient population of the Abatacept versus Adalimumab Comparison in Biologic‐Naive RA Subjects with Background Methotrexate (AMPLE) study. The AMPLE study was a direct head‐to‐head comparison of 125 mg subcutaneous abatacept weekly versus 40 mg subcutaneous adalimumab every other week in patients receiving background MTX to compare efficacy, safety, and radiographic outcomes over 2 years 16. The AMPLE study showed 2‐year ACR20 responses of 60% each for abatacept and adalimumab, and low disease activity as defined by DAS28 using the C‐reactive protein level, CDAI, and SDAI of 65–66% for abatacept and 68–69% for adalimumab, for the intent‐to‐treat patient populations 16. However, unlike the study presented here, patients in the AMPLE study were naive for biologic disease‐modifying antirheumatic drugs, which precludes any direct comparison of efficacy data between studies.
The safety profile and tolerability of tofacitinib through 24 months was consistent with published findings from the 12‐month analysis 5 and other phase III clinical trials 1, 2, 3, 4, 5, 6, 7. The incidence rates for serious infections and frequency of opportunistic infections were similar through 24 months versus 12 months 5. Exposure‐adjusted incidence rates for serious infections were similar between tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily and comparable with those of the pooled tofacitinib phase III RA patient population 17. In the present study, the length and extent of exposure to tofacitinib was similar between patients randomized to receive tofacitinib 5 mg twice daily and those randomized to receive tofacitinib 10 mg twice daily from study initiation. In the most recent long‐term extension analysis of up to 9 years, there was differential exposure between tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily, due to the chronology of long‐term extension initiation, with a longer median duration of exposure for tofacitinib 5 mg twice daily but greater overall patient‐years of exposure for tofacitinib 10 mg twice daily 8, 10, 17. That study found that AEs were generally stable over time for patients receiving tofacitinib 17.
No dose dependency in rates of herpes zoster was observed in the present RCT; the numerically higher proportions of herpes zoster in the tofacitinib 10 mg twice daily group compared with 5 mg twice daily were consistent with other phase III trials of tofacitinib 18. In this study, patients who tested positive for the interferon‐γ release assay at screening, but with no evidence of active TB, were presumed to have latent TB and received isoniazid for 9 months (tofacitinib was initiated the same day isoniazid was started), and none of these patients developed active TB during the study. The incidence of observed TB was similar to that previously reported 19; in this study 3 patients developed TB, all of whom were negative for TB at baseline, received tofacitinib 10 mg twice daily, and were residents of countries with a high endemic rate of TB.
Causes of death reported during months 12 through 24 were primarily related to cardiovascular events or pneumonia; this was generally similar to causes of death in the first 12 months 5 and in an integrated safety analysis of mortality data across the tofacitinib clinical development program in RA 17. After month 6, one patient in each of the tofacitinib 5 mg and 10 mg twice daily groups was discontinued due to ALT elevation and 1 patient in each group was discontinued due to AST elevation, according to prespecified discontinuation criteria. In accordance with tofacitinib prescribing information 20, and since tofacitinib has not been studied in patients with severe hepatic impairment, use in patients with severe hepatic impairment is not recommended.
A potential limitation of this efficacy analysis is that comparison of longer‐term versus shorter‐term data can be challenging when exposure to placebo is limited to early time points, or following termination of the control arm of a study; this is widely acknowledged as a limitation of this type of analysis 21. For example, while in the primary article the radiographic placebo response was linearly extrapolated to month 12, extrapolating placebo response to month 24 would be even more problematic. However, here we present imputed and observed data, and use longitudinal regression techniques, which allow investigation of longitudinal relationships between variables and account for missing values and variations in time intervals. Additional bias exists, since only those patients who continue to respond and remain in the trial for the long term are assessed; these patients probably represent those achieving the best clinical efficacy and who are least susceptible to AEs that would result in discontinuation 22.
In conclusion, this 24‐month RCT in patients with RA and an inadequate response to MTX receiving tofacitinib 5 mg or 10 mg twice daily plus MTX demonstrated maintenance of efficacy with tofacitinib in those patients with initial responses, including limited structural damage, through 24 months. No new safety signals emerged compared with the initial 12‐month report or with previous clinical RCTs with tofacitinib.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Connell had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Van der Heijde, Strand, Tanaka, Keystone, Kremer, Cardiel, Tegzová, Gruben, Wallenstein, Connell, Fleischmann.
Acquisition of data
Tanaka, Keystone, Kremer, Zerbini, Cardiel, Cohen, Nash, Song, Tegzová, Gruben, Connell, Fleischmann.
Analysis and interpretation of data
Van der Heijde, Strand, Tanaka, Keystone, Kremer, Cardiel, Cohen, Nash, Song, Tegzová, Gruben, Wallenstein, Connell, Fleischmann.
Role of the Study Sponsor
Pfizer, Inc. personnel developed the study protocol, supervised the conduct of the study, and were involved in data analysis and interpretation. Medical writing support under the guidance of the authors was provided by Rebecca J. Douglas, PhD (CMC Connect, a division of McCann Health Medical Communications Ltd, Macclesfield, UK) and was funded by Pfizer, Inc. in accordance with Good Publication Practice (GPP3) guidelines (Battisti WP, Wager E, Baltzer L, Bridges D, Cairns A, Carswell CI, et al. Good publication practice for communicating company‐sponsored medical research: GPP3 Ann Intern Med 2015;163:461–4). The authors had the final decision to submit the manuscript for publication.
Supporting information
Acknowledgments
The authors would like to thank the patients who were involved in this study, the ORAL Scan investigators, and the study team, which included Vivianne Dillon (Clinical Project Manager) and Allison Brailey (Lead Programmer).
Appendix A. ORAL Scan Study Investigators
The ORAL Scan Study investigators are as follows: in Australia, Dr. Stephen Hall, Dr. David Nicholls, and Dr. Maureen Rischmueller; in Canada, Dr. Milton F. Baker, Dr. Louis Bessette, Dr. Alfred A Cividino, Dr. Boulos Haraoui, Dr. Henry Niall Jones, Dr. Edward Keystone, Dr. Majed Khraishi, and Dr. J. Thorne; in the US, Dr. Charles Allen Birbara, Dr. Herbert Stuart Block Baraf, Dr. Joan Marie Bathon, Dr. Alan Lawrence Brodsky, Dr. John Joseph Cush, Dr. Ara Hagop Dikranian, Dr. Erdal Diri, Dr. Paul Andrew Dura, Dr. Kristine Marie Lohr, Dr. Roy Fleischmann, Dr. Robert Michael Griffin, Jr., Dr. Dale George Halter, Dr. Jody Kay Hargrove, Dr. David William Bouda, Dr. Suresh Kumar Reddy Pasya, Dr. Geneva Louise Hill, Dr. Raymond Edward Jackson, Dr. Shelly Pearl Kafka, Dr. Jeffrey Louis Kaine, Dr. Paul L. Katzenstein, Dr. Kevin James Kempf, Dr. Karen Sue Kolba, Dr. Joel Kremer, Dr. Selden Longley, III, Dr. Steven D. Mathews, Dr. Ami Charise Milton, Dr. Richard James Misischia, Dr. Haydon Anthony Moorman, Dr. Larry Wayne Moreland, Dr. Mark William Niemer, Dr. William Rodney Palmer, Dr. Michael Eugene Sayers, Dr. Patrick Thomas Schuette, Dr. Talha Shamim, Dr. William Julius Shergy, Dr. David Hilton Sikes, Dr. Joel Charles Silverfield, Dr. Chokkalingam Siva, Dr. James D. Taborn, Dr. Bridget Tyrell Walsh, Dr. Alvin Francis Wells, and Dr. Sanford Mayer Wolfe; in Asia, Dr. Prabha Adhikari (India), Dr. Kouichi Amano (Japan), Dr. Sang‐Cheol Bae (Korea), Dr. Srikantiah Chandrashekara (India), Dr. Arvind K. Chopra (India), Dr. Ping‐Ning Hsu (Taiwan), Dr. Mitsuhiro Iwahashi (Japan), Dr. Jugal Kishore Kadel (India), Dr. Yojiro Kawabe (Japan), Dr. Eun‐Mi Koh (Korea), Dr. Joung‐liang Lan (Taiwan), Dr. Soo‐Kon Lee (Korea), Dr. Hsiao‐Yi Lin (Taiwan), Dr. Lieh‐bang Liou (Taiwan), Dr. Ming‐Fei Liu (Taiwan), Dr. Kiyoshi Migita (Japan), Dr. Toshiaki Miyamoto (Japan), Dr. Nobuyuki Miyasaka (Japan), Dr. Shunsuke Mori (Japan), Dr. Yasuhiko Munakata (Japan), Dr. Shuji Ohta (Japan), Dr. Won Park (Korea), Dr. Sung‐Hwan Park (Korea), Dr. Uppuluri Ramakrishna Rao (India), Dr. Seung Cheol Shim (Korea), Dr. Vineeta Shobha (India), Dr. Yeong‐Wook Song (Korea), Dr. Yoshinari Takasaki (Japan), Dr. Tsutomu Takeuchi (Japan), Dr. Yoshiya Tanaka (Japan), Dr. Shigeto Tohma (Japan), Dr. Wen‐Chan Tsai (Taiwan), Dr. Yukitaka Ueki (Japan), Dr. Sarath Chandra Mouli Veeravalli (India), Dr. Shrikant Wagh (India), Dr. Hisashi Yamanaka (Japan), and Dr. Bin Yoo (Korea); in Europe, Dr. Anastas Batalov (Bulgaria), Dr. Daniela Bichoversuska (Bulgaria), Dr. Zdenek Dvorak (Czech Republic), Dr. Ivan Goranov (Bulgaria), Dr. Halyna M. Hrytsenko (Ukraine), Dr. Jana Kopackova (Czech Republic), Dr. Zdenka Mosterova (Czech Republic), Dr. Boycho Oparanov (Bulgaria), Dr. Andriy Petrov (Ukraine), Dr. Ines Pokrzywnicka‐Gajek (Poland), Professor Vladyslav V. Povoroznyuk (Ukraine), Dr. Jan Rosa (Czech Republic), Dr. Zofia Ruzga (Poland), Professor Loukas Settas (Greece), Professor Mykola A. Stanislavchuk (Ukraine), Dr. Dana Tegzová (Czech Republic), Dr. Vira Iosypivna Tseluyko (Ukraine), and Dr. Petr Vitek (Czech Republic); in Latin America, Dr. Cristiano A. F. Zerbini (Brazil), Dr. Joao Carlos Tavares Brenol (Brazil), Dr. Mario H. Cardiel (Mexico), Dr. William Jose Otero Escalante (Colombia), Dr. Maria Concepcion Maldonado‐Lopez (Colombia), Dr. Juan Jose Jaller Raad (Colombia), Dr. Javier Dario Marquez Hernandez (Colombia), Dr. Edwin Antonio Jauregui (Colombia), Dr. Mauro W. Keiserman (Brazil), Dr. Ana Claudia Cauceglia Melazzi (Brazil), Dr. Virginia Pascual‐Ramos (Mexico), Dr. Luciana Teixeira Pinto (Brazil), Dr. Sebastiao C. Radominski (Brazil), Dr. Antonio Carlos Ximenes (Brazil), and Dr. Sol Villegas de Morales (Venezuela).
ClinicalTrials.gov identifier: NCT00847613.
Presented in part at the 76th Annual Scientific Meeting of the American College of Rheumatology, Washington, DC, November 2012.
Supported by Pfizer, Inc.
1Désirée van der Heijde, MD, PhD: Leiden University Medical Center, Leiden, The Netherlands; 2Vibeke Strand, MD: Biopharmaceutical Consultant, Portola Valley, California; 3Yoshiya Tanaka, MD, PhD: University of Occupational and Environmental Health, Kitakyushu, Japan; 4Edward Keystone, MD: Mount Sinai Hospital, Toronto, Ontario, Canada; 5Joel Kremer, MD: Albany Medical College, Albany, New York; 6Cristiano A. F. Zerbini, MD: Centro Paulista de Investigação Clinica, Sao Paulo, Brazil; 7Mario H. Cardiel, MD: Centro de Investigacion Clinica de Morelia, Morelia, Mexico; 8Stanley Cohen, MD, Roy Fleischmann, MD: Metroplex Clinical Research Center, Dallas, Texas; 9Peter Nash, PhD: Nambour General Hospital, Nambour, Queensland, Australia, and University of Queensland, Brisbane, Queensland, Australia; 10Yeong‐Wook Song, MD, PhD: Seoul National University Hospital, Seoul, Republic of Korea; 11Dana Tegzová, MD: Institute of Rheumatology, Prague, Czech Republic; 12David Gruben, PhD, Gene Wallenstein, PhD, Carol A. Connell, PhD: Pfizer, Inc., Groton, Connecticut.
Dr. van der Heijde has received consulting fees, speaking fees, and/or honoraria from AbbVie, Amgen, Astellas, AstraZeneca, Bristol‐Myers Squibb, Boehringer Ingelheim, Celgene, Daiichi, Galapagos, Gilead, GlaxoSmithKline, Janssen, Eli Lilly, Merck, Novartis, Pfizer, Inc., Regeneron, Roche, Sanofi, Takeda, and UCB (less than $10,000 each). Dr. Strand has received consulting fees from AbbVie, Alder, Amgen, Bristol‐Myers Squibb, Boehringer Ingelheim, Celgene, Celltrion, Corrona, Crescendo, GlaxoSmithKline, Janssen, Eli Lilly, Merck, Novartis, Pfizer, Inc., Protagen, Regeneron, Samsung, Sandoz, Sanofi, and UCB (less than $10,000 each). Dr. Tanaka has received consulting fees, speaking fees, and/or honoraria from Chugai, Janssen, Eli Lilly, Sanofi, UCB, and YL Biologics (less than $10,000 each) and from Astellas, Bristol‐Myers Squibb, Daiichi Sankyo, Mitsubishi Tanabe, and Pfizer, Inc. (more than $10,000 each). Dr. Keystone has received consulting fees, speaking fees, and/or honoraria from Abbott, AstraZeneca, Biotest, Bristol‐Myers Squibb, Celltrion, Crescendo, Genentech, Gilead, Janssen, Merck, Pfizer, Inc., Roche, Sanofi, Sandoz, and UCB (less than $10,000 each) and from AbbVie, Amgen, and Lilly (more than $10,000 each), and research support from Abbott, Amgen, Bristol‐Myers Squibb, Gilead, Janssen, Eli Lilly, Pfizer, Inc., Roche, and Sanofi. Dr. Kremer has received consulting fees, speaking fees, and/or honoraria from AbbVie, Bristol‐Myers Squibb, Genentech, GlaxoSmithKline, Eli Lilly, Novartis, and Pfizer, Inc. (less than $10,000 each) and owns stock or stock options in Corrona. Dr. Zerbini has received consulting fees, speaking fees, and/or honoraria from Pfizer, Inc. (less than $10,000). Dr. Cardiel has received consulting fees, speaking fees, and/or honoraria from Gilead, Pfizer, Inc., Eli Lilly, and Roche (less than $10,000 each) and has served as an expert witness on behalf of Eli Lilly and Pfizer, Inc. Dr. Cohen has received consulting fees, speaking fees, and/or honoraria from Gilead and Eli Lilly (less than $10,000 each) and from Amgen and Pfizer, Inc. (more than $10,000 each). Dr. Nash has received consulting fees and/or speaking fees from AbbVie, Bristol‐Myers Squibb, Celgene, Janssen, JCB, Eli Lilly, Novartis, Pfizer, Inc., Roche, and Sanofi (less than $10,000 each). Dr. Tegzová has received consulting fees, speaking fees, and/or honoraria from AbbVie, Novartis, and UCB (less than $10,000 each). Dr. Fleischmann has received consulting fees, speaking fees, and/or honoraria from Amgen, AbbVie, Bristol‐Myers Squibb, GlaxoSmithKline, and Eli Lilly (less than $10,000 each) and from Pfizer, Inc. (more than $10,000). Drs. Gruben, Wallenstein, and Connell own stock or stock options in Pfizer, Inc. No other disclosures relevant to this article were reported.
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices for indications that have been approved in the US and/or European Union or in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Contributor Information
Carol A. Connell, Email: carol.a.connell@pfizer.com.
the ORAL Scan Investigators:
Stephen Hall, David Nicholls, Maureen Rischmueller, Milton F. Baker, Louis Bessette, Alfred A Cividino, Boulos Haraoui, Henry Niall Jones, Edward C. Keystone, Majed Khraishi, J. Thorne, Charles Allen Birbara, Herbert Stuart Block Baraf, Joan Marie Bathon, Alan Lawrence Brodsky, John Joseph Cush, Ara Hagop Dikranian, Erdal Diri, Paul Andrew Dura, Kristine Marie Lohr, Roy Mitchell Fleischmann, Robert Michael Griffin, Dale George Halter, Jody Kay Hargrove, David William Bouda, Suresh Kumar Reddy Pasya, Geneva Louise Hill, Raymond Edward Jackson, Shelly Pearl Kafka, Jeffrey Louis Kaine, Paul L. Katzenstein, Kevin James Kempf, Karen Sue Kolba, Joel Marc Kremer, Selden Longley, Steven D. Mathews, Ami Charise Milton, Richard James Misischia, Haydon Anthony Moorman, Larry Wayne Moreland, Mark William Niemer, William Rodney Palmer, Michael Eugene Sayers, Patrick Thomas Schuette, Talha Shamim, William Julius Shergy, David Hilton Sikes, Joel Charles Silverfield, Chokkalingam Siva, James D. Taborn, Bridget Tyrell Walsh, Alvin Francis Wells, Sanford Mayer Wolfe, Prabha Adhikari, Kouichi Amano, Sang‐Cheol Bae, Srikantiah Chandrashekara, Arvind K. Chopra, Ping‐Ning Hsu, Mitsuhiro Iwahashi, Jugal Kishore Kadel, Yojiro Kawabe, Eun‐Mi Koh, Joung‐liang Lan, Soo‐Kon Lee, Hsiao‐Yi Lin, Lieh‐bang Liou, Ming‐Fei Liu, Kiyoshi Migita, Toshiaki Miyamoto, Nobuyuki Miyasaka, Shunsuke Mori, Yasuhiko Munakata, Shuji Ohta, Won Park, Sung‐Hwan Park, Uppuluri Ramakrishna Rao, Seung Cheol Shim, Vineeta Shobha, Yoshinari Takasaki, Tsutomu Takeuchi, Yoshiya Tanaka, Shigeto Tohma, Wen‐Chan Tsai, Yukitaka Ueki, Sarath Chandra Mouli Veeravalli, Shrikant Wagh, Hisashi Yamanaka, Bin Yoo, Anastas Batalov, Daniela Bichoversuska, Zdenek Dvorak, Ivan Goranov, Halyna M. Hrytsenko, Jana Kopackova, Zdenka Mosterova, Boycho Oparanov, Andriy Petrov, Ines Pokrzywnicka‐Gajek, Vladyslav V. Povoroznyuk, Jan Rosa, Zofia Ruzga, Loukas Settas, Mykola A. Stanislavchuk, Vira Iosypivna Tseluyko, Petr Vitek, Cristiano Augusto de Freitas Zerbini, Joao Carlos Tavares Brenol, Mario H. Cardiel‐Rios, William Jose Otero Escalante, Maria Concepcion Maldonado‐Lopez, Juan Jose Jaller Raad, Javier Dario Marquez Hernandez, Edwin Antonio Jauregui, Mauro W. Keiserman, Ana Claudia Cauceglia Melazzi, Virginia Pascual‐Ramos, Luciana Teixeira Pinto, Sebastiao C. Radominski, Antonio Carlos Ximenes, and Sol Villegas de Morales
References
- 1. Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley J, Gruben D, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 2014;370:2377–86. [DOI] [PubMed] [Google Scholar]
- 2. Fleischmann R, Strand V, Wilkinson B, Kwok K, Bananis E. Relationship between clinical and patient‐reported outcomes in a phase 3 trial of tofacitinib or MTX in MTX‐naïve patients with rheumatoid arthritis. RMD Open 2016;2:e000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burmester GR, Blanco R, Charles‐Schoeman C, Wollenhaupt J, Zerbini C, Benda B, et al. Tofacitinib (CP‐690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised Phase 3 trial. Lancet 2013;381:451–60. [DOI] [PubMed] [Google Scholar]
- 4. Kremer J, Li ZG, Hall S, Fleischmann R, Genovese M, Martin‐Mola E, et al. Tofacitinib in combination with nonbiologic disease‐modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med 2013;159:253–61. [DOI] [PubMed] [Google Scholar]
- 5. Van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, et al. Tofacitinib (CP‐690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve‐month data from a twenty‐four–month phase III randomized radiographic study. Arthritis Rheum 2013;65:559–70. [DOI] [PubMed] [Google Scholar]
- 6. Fleischmann R, Kremer J, Cush J, Schulze‐Koops H, Connell CA, Bradley JD, et al. Placebo‐controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 2012;367:495–507. [DOI] [PubMed] [Google Scholar]
- 7. Van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 2012;367:508–19. [DOI] [PubMed] [Google Scholar]
- 8. Wollenhaupt J, Silverfield J, Lee EB, Curtis JR, Wood SP, Soma K, et al. Safety and efficacy of tofacitinib, an oral Janus kinase inhibitor, for the treatment of rheumatoid arthritis in open‐label, longterm extension studies. J Rheumatol 2014;41:837–52. [DOI] [PubMed] [Google Scholar]
- 9. Yamanaka H, Tanaka Y, Takeuchi T, Sugiyama N, Yuasa H, Toyoizumi S, et al. Tofacitinib, an oral Janus kinase inhibitor, as monotherapy or with background methotrexate, in Japanese patients with rheumatoid arthritis: an open‐label, long‐term extension study. Arthritis Res Ther 2016;18:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wollenhaupt J, Silverfield J, Lee EB, Terry K, Kwok K, Strengholt S, et al. Tofacitinib, an oral Janus kinase inhibitor, in the treatment of rheumatoid arthritis: safety and efficacy in open‐label, long‐term extension studies over 9 years [abstract]. Arthritis Rheumatol 2017;69 Suppl 10 URL: https://acrabstracts.org/abstract/tofacitinib-an-oral-janus-kinase-inhibitor-in-the-treatment-of-rheumatoid-arthritis-safety-and-efficacy-in-open-label-long-term-extension-studies-over-9-years/. [Google Scholar]
- 11. Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38:727–35. [DOI] [PubMed] [Google Scholar]
- 12. Prevoo ML, van ‘t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty‐eight–joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 13. Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, et al. American College of Rheumatology/European League Against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Arthritis Rheum 2011;63:573–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. National Heart, Lung, and Blood Institute: National Institutes of Health . JNC 7 complete report: the science behind the new guidelines. 2014. URL: https://www.nhlbi.nih.gov/health-pro/guidelines/current/hypertension-jnc-7/complete-report.
- 15. Canhao H, Rodrigues AM, Gregorio MJ, Dias SS, Melo Gomes JA, Santos MJ, et al. Common evaluations of disease activity in rheumatoid arthritis reach discordant classifications across different populations. Front Med (Lausanne) 2018;5:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schiff M, Weinblatt ME, Valente R, van der Heijde D, Citera G, Elegbe A, et al. Head‐to‐head comparison of subcutaneous abatacept versus adalimumab for rheumatoid arthritis: two‐year efficacy and safety findings from AMPLE trial. Ann Rheum Dis 2014;73:86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cohen SB, Tanaka Y, Mariette X, Curtis JR, Lee EB, Nash P, et al. Long‐term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheum Dis 2017;76:1253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Winthrop KL, Yamanaka H, Valdez H, Mortensen E, Chew R, Krishnaswami S, et al. Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Arthritis Rheumatol 2014;66:2675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Winthrop KL, Park SH, Gul A, Cardiel MH, Gomez‐Reino JJ, Tanaka Y, et al. Tuberculosis and other opportunistic infections in tofacitinib‐treated patients with rheumatoid arthritis. Ann Rheum Dis 2016;75:1133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xeljanz (tofacitinib) prescribing information. New York (NY): Pfizer; 2017. URL: http://labeling.pfizer.com/ShowLabeling.aspx?id=959. [Google Scholar]
- 21. Buch MH, Aletaha D, Emery P, Smolen JS. Reporting of long‐term extension studies: lack of consistency calls for consensus. Ann Rheum Dis 2011;70:886–90. [DOI] [PubMed] [Google Scholar]
- 22. Landewé R, van der Heijde D. Follow up studies in rheumatoid arthritis. Ann Rheum Dis 2002;61:479–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials