

Abstract
Aim
To evaluate the long‐term efficacy and safety of ertugliflozin in adults with type 2 diabetes mellitus inadequately controlled on metformin.
Materials and Methods
A 104‐week Phase III, randomized double‐blind study with a 26‐week placebo‐controlled period (Phase A) and a 78‐week period (Phase B) where blinded glimepiride was added to non‐rescued placebo participants with fasting fingerstick glucose ≥6.1 mmol/L. Results through week 104 are reported.
Results
Mean (standard error) change in HbA1c from baseline was −0.7% (0.07) and −1.0% (0.07) at week 52; −0.6% (0.08) and −0.9% (0.08) at week 104 for ertugliflozin 5 and 15 mg. At week 52, 34.8% and 36.6% participants had HbA1c <7.0%, and 24.6% and 33.7% at week 104, for ertugliflozin 5 and 15 mg. Ertugliflozin reduced fasting plasma glucose (FPG), body weight and systolic blood pressure (SBP) from baseline through week 104. The incidence of female genital mycotic infections (GMIs) was higher with ertugliflozin, and symptomatic hypoglycaemia was lower for ertugliflozin versus placebo/glimepiride. Minimal bone mineral density (BMD) changes were observed, similar to placebo/glimepiride, except at total hip where reduction in BMD was greater with ertugliflozin 15 mg versus placebo/glimepiride: difference in least squares means (95% CI) –0.50% (−0.95, −0.04) at week 52 and −0.84% (−1.44, −0.24) at week 104.
Conclusions
Ertugliflozin maintained improvements from baseline in HbA1c, FPG, body weight and SBP through week 104. Ertugliflozin was well tolerated, with non‐clinically relevant changes in BMD. Compared with placebo/glimepiride, ertugliflozin increased female GMIs, but reduced the incidence of symptomatic hypoglycaemia. ClinicalTrials.gov Identifier: NCT02033889.
Keywords: bone mineral density, durability, ertugliflozin, type 2 diabetes mellitus
1. INTRODUCTION
Sodium‐glucose co‐transporter‐2 (SGLT2) inhibitors improve glycaemic control in patients with type 2 diabetes mellitus (T2DM) via inhibition of renal glucose reabsorption and reduction of the renal threshold for glucose excretion.1, 2, 3
Ertugliflozin is a selective SGLT2 inhibitor4, 5 approved by the US Food and Drug Administration6 and the European Medicines Agency7 for the treatment of adults with T2DM. Across the Phase III VERTIS (eValuation of ERTugliflozin effIcacy and Safety) programme, ertugliflozin showed clinically meaningful reductions from baseline in HbA1c, fasting plasma glucose (FPG), body weight and blood pressure.8, 9, 10, 11, 12, 13
The Phase III, randomized, double‐blind VERTIS MET trial (ClinicalTrials.gov: NCT02033889) evaluated ertugliflozin versus placebo in adults with inadequately controlled T2DM receiving metformin monotherapy. The study population was enriched with postmenopausal women to enhance the ability to assess potential effects of ertugliflozin on bone mineral density (BMD). The study consisted of a 26‐week placebo‐controlled period (Phase A) followed by a 78‐week extension (Phase B). At week 26, the primary time point, ertugliflozin was associated with significantly larger reductions from baseline in HbA1c, FPG, blood pressure and body weight than placebo.10 Additionally, a larger proportion of participants with ertugliflozin had HbA1c <7.0% (53 mmol/mol) than placebo. Ertugliflozin was well tolerated, although an increased incidence of genital mycotic infections (GMIs) was observed. There was no adverse impact on BMD with ertugliflozin treatment at week 26.10 Here, we report efficacy results at weeks 52 and 104 (Phase B) and safety results for the overall study (Phases A and B).
2. MATERIALS AND METHODS
This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonisation Good Clinical Practice Guidelines. The final protocol was reviewed and approved by the Institutional Review Board and/or Independent Ethics Committee at each of the investigational centres, and written informed consent was obtained from all study participants. This study was conducted in 14 countries at 103 study centres.
All efficacy hypotheses were tested at week 26 and have been previously published, as have detailed descriptions of the eligibility criteria (Supplementary Methods, see the supporting information), and efficacy and safety assessments.10
2.1. Study design and treatment
The design of the trial is shown in Supporting Information Figure S1; full details have been published previously.10 The study consisted of 2 phases: a double‐blind, placebo‐controlled, 26‐week treatment period (Phase A), and a double‐blind, 78‐week treatment extension period (Phase B). In Phase B, participants with fasting fingerstick glucose ≥6.1 mmol/L (≥110 mg/dL) and not rescued during Phase A received blinded glimepiride (for participants randomized to placebo) or glimepiride placebo (for participants randomized to ertugliflozin). In Phase B, glycaemic rescue therapy with basal insulin was initiated in participants with FPG >11.1 mmol/L (>200 mg/dL) or HbA1c >8.0% (64 mmol/mol) on maximum tolerated doses of glimepiride. Bone rescue therapy with any bone‐active treatment was initiated in participants with a reduction in BMD from baseline of >7% at any anatomical site together with a T‐score below −2.5. Participants receiving any rescue therapy were to continue receiving study medication.
2.2. Study objectives
All primary and secondary efficacy hypotheses were tested at week 26.10 Secondary objectives included assessment of the following at weeks 52 and 104: change from baseline in HbA1c, FPG, body weight, systolic blood pressure (SBP) and diastolic blood pressure (DBP); proportion of participants with HbA1c <7.0% (53 mmol/mol) and <6.5% (48 mmol/mol); proportion of participants who received glycaemic rescue therapy; and change from baseline in BMD at the lumbar spine, femoral neck, total hip and distal forearm. The safety and tolerability of ertugliflozin were assessed through week 104.
2.3. Efficacy assessments
Changes from baseline in HbA1c, FPG, body weight, SBP and DBP were evaluated at week 2610 in Phase A and at weeks 52 and 104 in Phase B. The proportion of participants with HbA1c <7.0% (53 mmol/mol) and <6.5% (48 mmol/mol), and the proportion receiving glycaemic rescue therapy, were also evaluated at week 26,10 week 52 and week 104.
2.4. Safety assessments
Safety assessments included adverse events (AEs), drug‐related AEs, serious AEs, deaths, discontinuations because of AEs, change from baseline over time in estimated glomerular filtration rate (eGFR), physical examination, evaluation of vital signs and laboratory evaluations. See Supplementary Methods for details of events for clinical adjudication.
BMD at the lumbar spine (L1–L4), femoral neck, total hip and distal forearm was measured using dual‐energy X‐ray absorptiometry (DXA). All DXA scans were centrally analysed by an independent Central Evaluation Facility blinded to treatment allocation. Biomarkers of bone turnover (carboxy terminal cross‐linking telopeptides of Type I collagen [CTX] and procollagen type 1N terminal propeptide [P1NP]) and parathyroid hormone (PTH) were also assessed.
GMI by gender, urinary tract infection (UTI), symptomatic hypoglycaemia and hypovolemia were prespecified AEs of special interest. Documented hypoglycaemia (glucose level ≤3.9 mmol/L [≤70 mg/dL], with or without symptoms) and severe hypoglycaemia (episodes that required assistance, medical or non‐medical) were assessed.
2.5. Statistical analyses
The Phase A and B descriptive efficacy and BMD analyses included all randomized and treated participants with baseline data and at least 1 post‐randomization observation for the analysis endpoint. Change from baseline in efficacy endpoints at weeks 52 and 104 was assessed using a longitudinal data analysis (LDA) model for the ertugliflozin groups (with a constraint that baseline is the same, which is valid because of randomization) based on randomized and treated participants with at least 1 assessment at or after baseline. Efficacy endpoints were summarized using the excluding glycaemic rescue approach, i.e. efficacy data obtained after the initiation of glycaemic rescue therapy were censored (treated as missing). Raw mean changes from baseline are presented for the 3 treatment groups, and least squares (LS) mean changes from baseline (consistent with the analysis conducted at week 26 as previously reported10) are presented in Supporting Information Table S1 for the ertugliflozin groups only.
Safety analyses included all randomized and treated participants. With the exception of hypoglycaemia, safety analyses used the including glycaemic rescue approach. GMI by gender, UTI, symptomatic hypoglycaemia and hypovolemia AEs were prespecified for inferential testing without multiplicity control using the Miettinen and Nurminen method.14 Data for BMD and bone turnover biomarkers are presented using an excluding bone rescue approach, i.e. data were censored at the point of a participant taking bone rescue therapy. The percentage changes from baseline in BMD endpoints at weeks 52 and 104 were assessed using an LDA model. An exploratory post hoc analysis also included change from baseline in body weight as a covariate to assess the impact of body weight loss on change in BMD from baseline to week 104.15
Due to the addition of blinded glimepiride in Phase B for previously unrescued participants who were assigned to placebo in Phase A, and who met the fasting fingerstick glucose ≥6.1 mmol/L (≥110 mg/dL) criterion, no formal comparisons were made between the ertugliflozin and placebo/glimepiride groups at weeks 52 and 104 for efficacy or BMD analyses.
3. RESULTS
3.1. Participants
Overall, 621 participants were randomized, and 581 participants entered Phase B and received at least 1 dose of study medication in Phase B. Baseline characteristics were balanced across treatment groups (Supporting Information Table S2).
The proportion of participants who discontinued study medication in the Phase A and B treatment period was higher in the placebo/glimepiride group (25.8%) compared with the ertugliflozin 5 mg (18.8%) and 15 mg (19.0%) groups. Withdrawal by subject was the most common reason for discontinuation from study medication (12.9%, 8.7% and 6.8% for placebo/glimepiride, ertugliflozin 5 mg and ertugliflozin 15 mg, respectively; Supporting Information Table S3). For all other reasons for discontinuation from study medication, the proportions were low and similar across treatment groups (Supporting Information Figure S3).
3.2. Efficacy outcomes
For both ertugliflozin groups, mean reductions in HbA1c were observed at week 26 and maintained at weeks 52 and 104. The observed estimate of the reduction in HbA1c was greater in the ertugliflozin 15 mg group than in the ertugliflozin 5 mg group at each time point. Mean (standard error [SE]) change from baseline in HbA1c at week 104 for the placebo/glimepiride, ertugliflozin 5 mg and ertugliflozin 15 mg groups were −0.6% (0.1), −0.6% (0.1) and −0.9% (0.1), respectively (Figure 1A).
Figure 1.
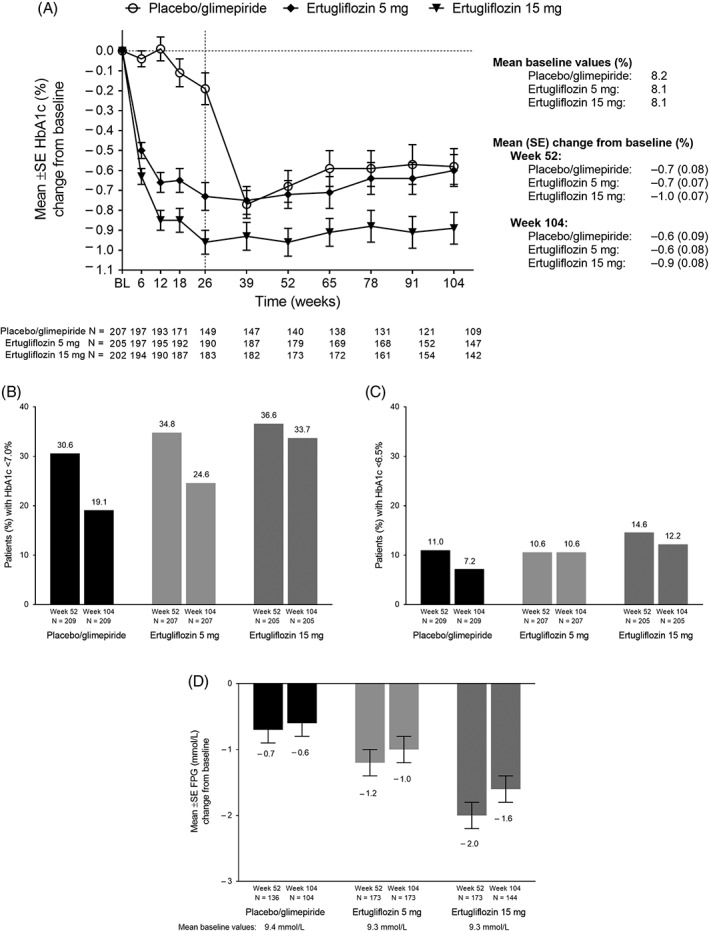
(A) HbA1c change from baseline over time; percentage of participants with HbA1c (B) <7.0% (53 mmol/mol) and (C) <6.5% (48 mmol/mol) at weeks 52 and 104; (D) change in FPG from baseline to weeks 52 and 104. Abbreviations: BL, baseline; FPG, fasting plasma glucose; SE, standard error
The observed estimates of the proportion of participants with HbA1c <7.0% (53 mmol/mol) and <6.5% (48 mmol/mol) were higher in the ertugliflozin groups than in the placebo/glimepiride group at all time points, except at week 52 when the percentage of participants with HbA1c <6.5% (48 mmol/mol) were comparable in the placebo/glimepiride group and in the ertugliflozin 5 mg group (Figure 1B,C). The reductions from baseline to week 26 in FPG observed for both ertugliflozin groups were mostly maintained at weeks 52 and 104 (Supporting Information Figure S3). In the placebo/glimepiride group, 79% of subjects received at least 1 dose of glimepiride; the median dose of glimepiride was 2.0 mg. Metformin doses were stable and comparable between groups throughout the study; the median metformin dose was 2000 mg/day at baseline and at the end of study in all groups.
The observed estimate of the reduction from baseline in body weight was greater for the ertugliflozin treatment groups than for the placebo/glimepiride group at each time point through week 104 (Figure 2A). The mean reductions from baseline to week 26 in body weight achieved with both doses of ertugliflozin were maintained at weeks 52 and 104. Mean (SE) body weight change from baseline was −0.18 kg (0.32), −3.77 kg (0.35) and −3.63 kg (0.32) at week 104, with placebo/glimepiride, ertugliflozin 5 mg and ertugliflozin 15 mg, respectively. At weeks 52 and 104, both doses of ertugliflozin lowered sitting SBP relative to baseline (Figure 2B).
Figure 2.
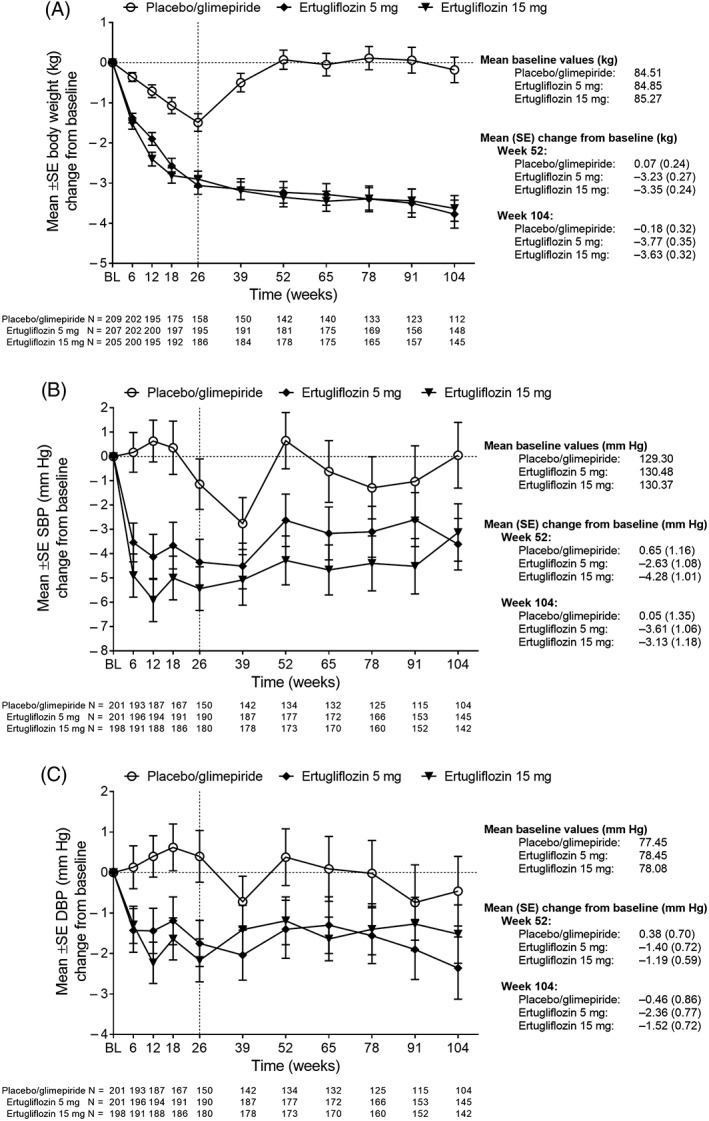
Change in (A) body weight, (B) SBP and (C) DBP over time. Abbreviations: BL, baseline; DBP, diastolic blood pressure; SBP, systolic blood pressure; SE, standard error
The LS mean (95% confidence interval [CI]) for changes from baseline for the efficacy outcomes in the ertugliflozin groups are shown in Supporting Information Table S1.
3.3. Safety outcomes
3.3.1. Overall safety
Across Phase A and B, the incidence of AEs, serious AEs (overall and drug‐related) and discontinuation because of drug‐related AEs were similar across treatment groups. There were 6 deaths during the study: 1 in the ertugliflozin 5 mg group (cause of death: acute cardiac failure), 2 in the ertugliflozin 15 mg group (causes of death: plasma cell myeloma and unknown) and 3 in the placebo/glimepiride group (causes of death: hepatic cancer, cardiac death and myocardial infarction). None of the deaths were considered related to study medication by the investigators (Table 1).
Table 1.
Summary of adverse events
| Participants in population with event, n (%) | Placebo/glimepiride (N = 209) | Ertugliflozin 5 mg (N = 207) | Ertugliflozin 15 mg (N = 205) |
|---|---|---|---|
| ≥1 AE | |||
| Overall | 159 (76.1) | 146 (70.5) | 155 (75.6) |
| Drug‐relateda | 49 (23.4) | 37 (17.9) | 48 (23.4) |
| ≥1 serious AE | |||
| Overall | 20 (9.6) | 19 (9.2) | 20 (9.8) |
| Drug‐relateda | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Discontinued study medication | |||
| Because of AE | 5 (2.4) | 7 (3.4) | 8 (3.9) |
| Because of drug‐relateda AE | 3 (1.4) | 3 (1.4) | 4 (2.0) |
| Because of serious AE | 1 (0.5) | 2 (1.0) | 3 (1.5) |
| Because of serious drug‐relateda AE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Deathsb | 3 (1.4) | 1 (0.5) | 2 (1.0) |
| Prespecified AEs of special interest | |||
| Symptomatic hypoglycaemiac | 28 (13.4) | 12 (5.8) | 12 (5.9) |
| UTI | 15 (7.2) | 9 (4.3) | 22 (10.7) |
| GMI, femaled | 1 (0.9) | 8 (7.3) | 11 (9.8) |
| GMI, male | 2 (2.0) | 5 (5.2) | 5 (5.4) |
| Hypovolemia | 3 (1.4) | 5 (2.4) | 5 (2.4) |
Abbreviations: AE, adverse event; GMI, genital mycotic infection; UTI, urinary tract infection.
Assessed as related to the study drug by the investigator.
Two participants died more than 14 days after the last dose of study medication: 1 in the ertugliflozin 5 mg group (caused by acute heart failure) and another in the placebo/glimepiride group (caused by myocardial infarction). For the 4 deaths that occurred between the first dose of study medication and 14 days after the last dose of study medication, the causes were: plasma cell myeloma and unknown (ertugliflozin 15 mg), and hepatic cancer and cardiac death (placebo/glimepiride). None of the deaths was considered related to study medication by the investigators. For all other AEs, this table contains events that occurred between the first dose of treatment and 14 days after the last dose of treatment.
P = 0.009 for both ertugliflozin groups versus placebo/glimepiride.
P = 0.017 for ertugliflozin 5 mg versus placebo/glimepiride; P = 0.003 for ertugliflozin 15 mg versus placebo/glimepiride.
3.3.2. Prespecified AEs of special interest
The incidence of symptomatic hypoglycaemia was lower in the ertugliflozin 5 mg and 15 mg groups (5.8% and 5.9%, respectively) than in the placebo/glimepiride group (13.4%) (P = 0.009 for both ertugliflozin doses). The incidence of documented hypoglycaemia was 21.1%, 13.5% and 14.1% for placebo/glimepiride, ertugliflozin 5 mg and ertugliflozin 15 mg, respectively. There were no cases of severe hypoglycaemia. The incidence of GMIs was higher in the ertugliflozin groups compared with the placebo/glimepiride group for female participants (P = 0.017 for ertugliflozin 5 mg; P = 0.003 for ertugliflozin 15 mg). One female participant in the ertugliflozin 5 mg group discontinued study medication because of a GMI AE (vulvovaginal mycotic infection). None of the GMI events was serious, all were mild or moderate in intensity. The incidence of UTIs was not notably different among the 3 treatment groups. The incidence of hypovolemia was low and similar across groups; all hypovolemia events were mild or moderate in intensity (Table 1).
3.3.3. Adjudicated AEs
There were fewer confirmed fractures in both ertugliflozin groups than in the placebo/glimepiride group: ertugliflozin 5 mg, 3 fractures in 3 participants (1 high and 2 low trauma); ertugliflozin 15 mg, 2 fractures in 2 participants (both low trauma); placebo/glimepiride, 10 fractures in 7 participants (1 high and 9 low trauma).
One participant in the ertugliflozin 15 mg group experienced a serious AE of diabetic ketoacidosis that met the charter case definition of ‘certain to be ketoacidosis’; the participant had suspected type 1 diabetes (possible Latent Autoimmune Diabetes of Adulthood). Three participants (1 in the ertugliflozin 5 mg group and 2 in the placebo/glimepiride group) had an AE of acute pancreatitis adjudicated as mild acute pancreatitis; none was considered to be caused by the study medication. One participant in the ertugliflozin 15 mg group had 1 renal event that was adjudicated as possibly related to study medication. One participant in the placebo/glimepiride group had an AE of liver injury that was adjudicated as possibly related to study medication.
Results of adjudication for cardiovascular events in this study will be analysed in a programme‐wide manner across ertugliflozin clinical trials, and are not reported here.
3.3.4. Additional safety topics
Three participants, 1 in the ertugliflozin 5 mg and 2 in the ertugliflozin 15 mg groups, underwent a non‐traumatic lower limb amputation; all had either predisposing conditions or risk factors.
Initial mean reductions in eGFR at week 6 were followed by mean increases, returning to baseline levels at week 26. At week 104, the observed estimates of mean eGFR were slightly lower than baseline in the 3 treatment groups (Figure 3).
Figure 3.
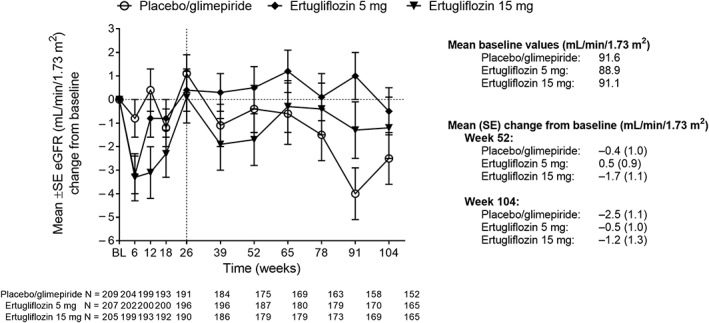
Mean change from baseline in eGFR over time. Abbreviations: BL, baseline; eGFR, estimated glomerular filtration rate; SE, standard error
3.3.5. BMD and bone biomarkers
Ertugliflozin was associated with minimal changes in BMD that were comparable to those observed with the placebo/glimepiride group at all sites, except at the total hip where the reduction in BMD was greater with ertugliflozin 15 mg compared with placebo/glimepiride at weeks 52 and 104 (Figure 4). Comparable findings were observed in the subgroup of women who were postmenopausal for ≥3 years. For this subgroup, the reductions from baseline to weeks 52 and 104 in BMD were similar across the 3 treatment groups except at the total hip where the reduction in BMD was greater with ertugliflozin 15 mg compared with placebo/glimepiride at week 104 (Supporting Information Figure S4). One participant in the ertugliflozin 5 mg group met bone rescue criteria and received bone rescue therapy. In a post hoc exploratory analysis, change in body weight showed a significant association with change in BMD at the total hip (P = 0.0466), and change in body weight appeared to explain ~10% of the observed difference in the percentage reduction of BMD in the total hip between the ertugliflozin 15 mg group and the placebo/glimepiride group at week 10415 (Supporting Information Table S4).
Figure 4.
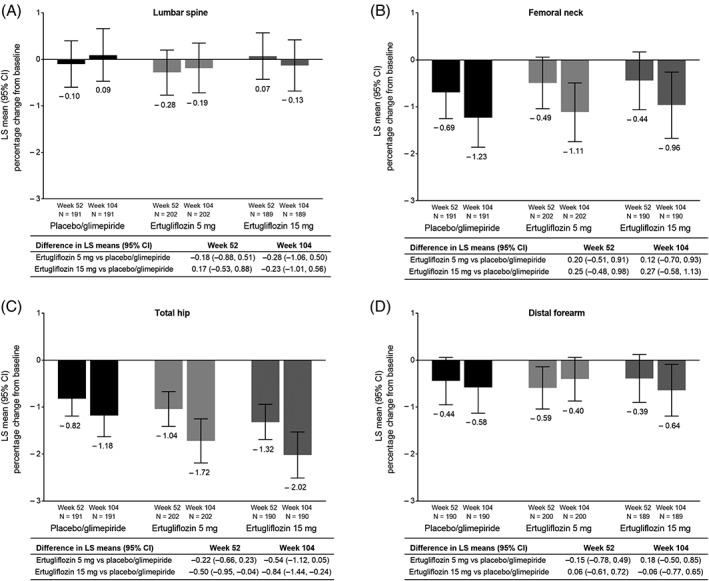
LS mean (95% CI)† percentage change from baseline to weeks 52 and 104 in BMD as measured by DXA at (A) the lumbar spine, (B) femoral neck, (C) total hip and (D) distal forearm. Abbreviations: BMD, bone mineral density; CI, confidence interval; LS, least squares. † Based on longitudinal data analysis model with fixed effects for treatment, time, prior antihyperglycaemic medication (metformin monotherapy or metformin and another antihyperglycaemic agent), baseline estimated glomerular filtration rate (continuous), menopausal status (men, premenopausal women, women who are perimenopausal or <3 years postmenopausal, women who are ≥3 years postmenopausal) and the interaction of time by treatment
The changes in bone biomarkers were modest in the 3 treatment groups. CTX increased from study start in the 3 treatment groups, with the maximum mean percentage change from baseline observed at week 52. The mean percentage increases were higher in the ertugliflozin groups than in the placebo/glimepiride group throughout the study. P1NP also increased from baseline in the 3 treatment groups, and at week 104 the mean percentage increase in P1NP was higher with ertugliflozin 15 mg than with ertugliflozin 5 mg and placebo/glimepiride. PTH increased only slightly in the 3 treatment groups, and the mean percentage increase from baseline to week 104 was higher in the placebo/glimepiride than in the ertugliflozin groups (Supporting Information Table S5).
3.3.6. Laboratory variables
Small mean increases in magnesium and phosphate were observed from baseline at week 26 in the ertugliflozin groups, with no further increases through week 104. Small mean reductions in bicarbonate were observed in all treatment groups at week 26 and the values returned to around baseline at week 104. No meaningful changes were observed in calcium or potassium. Mean decreases from baseline to week 104 in aspartate transaminase and alanine transaminase were observed only in the ertugliflozin groups. Mean increases from baseline in haemoglobin and haematocrit were observed at weeks 12 and 26, respectively, which were maintained through week 104 in the ertugliflozin groups. Mean percentage increases from baseline in low‐ and high‐density lipoprotein‐cholesterol were observed in the ertugliflozin groups at week 26, which were maintained through week 104 (Supporting Information Table S6).
4. DISCUSSION
This study evaluated the long‐term efficacy and safety of ertugliflozin and showed that the clinically meaningful reductions observed at week 26 in HbA1c, FPG, body weight and SBP were maintained through week 104. The number of participants with HbA1c at target at week 104 further highlights the durability of the glycaemic effect of ertugliflozin. No formal comparisons were performed between treatment groups in Phase B of this study because of the addition of glimepiride to the placebo group. Nevertheless, the observed estimates for the reductions in HbA1c at weeks 52 and 104 were comparable or larger in the ertugliflozin groups than in the placebo/glimepiride group, and consistently greater reductions with ertugliflozin were observed for the endpoints of FPG, body weight and SBP.
The long‐term effect of ertugliflozin reflects the fact that it acts independently of β‐cell function and insulin secretion,16 and hence it is not affected by the progressive β‐cell failure observed in patients with T2DM. These data are consistent with glycaemic endpoints in other studies with ertugliflozin8, 9, 10, 11, 12, 13 and other members of the SGLT2 inhibitor class.17 Additionally, as previously established in other studies with ertugliflozin8, 9, 10, 11, 12, 13 and other SGLT2 inhibitors,17 ertugliflozin produces clinically relevant effects on important non‐glycaemic endpoints such as body weight and SBP.
During 2 years of treatment, ertugliflozin was well tolerated. The safety profile, including prespecified AEs of special interest, was consistent with the rest of the ertugliflozin development programme.8, 9, 10, 11, 12, 13
Some AEs are of special interest because of findings in other SGLT2 inhibitors during the conduct of this study: lower limb amputations and ketoacidosis. In this study, 1 participant in the ertugliflozin 5 mg group and 2 in the ertugliflozin 15 mg group experienced a non‐traumatic lower limb amputation and 1 participant in the ertugliflozin 15 mg group had a serious AE adjudicated as certain to be ketoacidosis.
At week 104, participants in the ertugliflozin group experienced minimal, non‐clinically significant changes in BMD that were similar to those observed in the placebo/glimepiride group, except at the hip. Body weight loss has been associated with increases in bone turnover and loss of BMD.18 In this study, ertugliflozin was associated with clinically meaningful body weight loss, significantly greater than the placebo arm at week 26 and maintained throughout the study. In a post hoc analysis, ~10% of the observed difference in the percentage reduction of BMD in the total hip between the ertugliflozin 15 mg group and the placebo group could be explained by changes in body weight.15 However, this finding should be interpreted with caution given its exploratory nature.
In line with the lack of effect of ertugliflozin on BMD observed in this study, the changes in markers of bone metabolism did not consistently support a sustained increase in bone turnover. There were small mean percentage increases in magnesium and phosphate in the ertugliflozin groups but no notable changes in calcium or PTH. Moderate increases in CTX and P1NP were observed in the 3 groups, although there was no consistent pattern across the treatment groups or over time. Importantly, there was no increase in the number of confirmed fractures in the ertugliflozin groups compared with the placebo/glimepiride group. This is consistent with the results of a pooled analysis of 7 Phase III ertugliflozin trials where the incidence of fractures was 0.5% in each of the ertugliflozin groups and 0.6% in the comparator (active and placebo) group.15 It is important to highlight that the current study included a substantial proportion of participants at high risk of BMD loss. More than 40% of the participants were women who had been postmenopausal for at least 3 years. Participants in this study could not be receiving any bone‐active medications, except calcium and vitamin D supplements. This permitted an assessment of BMD without potential confounding. Even in this context, only 1 participant, receiving ertugliflozin 5 mg, met bone rescue criteria. The effects of SGLT2 inhibitors on BMD and fractures have been studied in other members of the class;19, 20, 21, 22, 23 a 182‐patient study found comparable BMD percentage changes between dapagliflozin 10 mg/day and placebo at 52 weeks; at 102 weeks the difference in LS means (95% CI) at the total hip was −0.45 (−1.32, 0.43).20, 23 Canagliflozin treatment resulted in greater percentage reductions in BMD versus placebo at the total hip at all time points in a 104‐week 716‐patient study; at week 104 the difference was −0.9 (−1.5, −0.2) and −1.2 (−1.9, −0.6) for canagliflozin 100 mg and 300 mg versus placebo, respectively.22 The studies cannot be directly compared because of different designs, differences in participants' ages, and in the proportion of women in pre‐, peri‐ and menopausal stages.
In conclusion, in patients with T2DM and inadequate glycaemic control on metformin monotherapy, adding treatment with ertugliflozin 15 mg and 5 mg provided clinically meaningful and durable improvements in glycaemic control, body weight and SBP over 104 weeks. Ertugliflozin was associated with minimal, non‐clinically relevant changes in BMD. Ertugliflozin was generally well tolerated, but with a higher incidence of GMIs in female participants in the ertugliflozin groups relative to the placebo/glimepiride group, while the incidence of symptomatic hypoglycaemia was lower in both ertugliflozin groups than in the placebo/glimepiride group.
CONFLICT OF INTEREST
S. G., A. G., H. S., A. D. and S. G. T. are employees of Pfizer Inc., who may own stock in the Company. S. H. is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, who may own stock in the Company. B. L. was an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, at the time the study was conducted, and who may own stock in the Company. B. C. has received fees for advisory boards (AstraZeneca, Merck‐Sharpe & Dohme, Novo‐Nordisk, Sanofi, Servier) and speaker bureau (AstraZeneca, Lilly, Merck‐Sharpe & Dohme, Novo‐Nordisk, Sanofi, Takeda).
Author contributions
All authors critically reviewed the draft manuscript and approved the final version of the manuscript for publication. A. D., S. G. T. and B. L. were involved in the conception/design of the study. S. G., A. G., H. S. and A. D. were involved in the acquisition of data for the study. All authors were involved in data analysis and interpretation of the data.
Data accessibility
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the United States and/or European Union, or (2) in programmes that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Supporting information
File S1. Supporting information.
ACKNOWLEDGMENTS
Editorial support was provided by Beth Elam, PhD, of Engage Scientific Solutions (Horsham, UK) and was funded by Pfizer Inc., New York, NY, and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ.
Gallo S, Charbonnel B, Goldman A, et al. Long‐term efficacy and safety of ertugliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin monotherapy: 104‐week VERTIS MET trial. Diabetes Obes Metab. 2019;21:1027–1036. 10.1111/dom.13631
Funding information Editorial support was provided by Beth Elam, PhD, of Engage Scientific Solutions (Horsham, UK) and was funded by Pfizer Inc., New York, NY, USA and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
This study was sponsored by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, in collaboration with Pfizer Inc., New York, NY.
Data Availability: Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the United States and/or European Union, or (2) in programmes that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
REFERENCES
- 1. Abdul‐Ghani MA, Norton L, DeFronzo RA. Renal sodium‐glucose cotransporter inhibition in the management of type 2 diabetes mellitus. Am J Physiol Renal Physiol. 2015;309:F889‐F900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733‐794. [DOI] [PubMed] [Google Scholar]
- 3. Scheen AJ. Pharmacokinetics, pharmacodynamics and clinical use of SGLT2 inhibitors in patients with type 2 diabetes mellitus and chronic kidney disease. Clin Pharmacokinet. 2015;54:691‐708. [DOI] [PubMed] [Google Scholar]
- 4. Kalgutkar AS, Tugnait M, Zhu T, et al. Preclinical species and human disposition of PF‐04971729, a selective inhibitor of the sodium‐dependent glucose cotransporter 2 and clinical candidate for the treatment of type 2 diabetes mellitus. Drug Metab Dispos. 2011;39:1609‐1619. [DOI] [PubMed] [Google Scholar]
- 5. Miao Z, Nucci G, Amin N, et al. Pharmacokinetics, metabolism, and excretion of the antidiabetic agent ertugliflozin (PF‐04971729) in healthy male subjects. Drug Metab Dispos. 2013;41:445‐456. [DOI] [PubMed] [Google Scholar]
- 6. FDA . Steglatro (ertugliflozin) Prescribing Information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209803s000lbl.pdf. Accessed June 20, 2018.
- 7. EMA . Ertugliflozin (Steglatro) Summary of Product Characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004315/WC500246918.pdf. Accessed June 20, 2018.
- 8. Dagogo‐Jack S, Liu J, Eldor R, et al. Efficacy and safety of the addition of ertugliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sitagliptin: the VERTIS SITA2 placebo‐controlled randomized study. Diabetes Obes Metab. 2018;20:530‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pratley RE, Eldor R, Raji A, et al. Ertugliflozin plus sitagliptin versus either individual agent over 52 weeks in patients with type 2 diabetes mellitus inadequately controlled with metformin: the VERTIS FACTORIAL randomized trial. Diabetes Obes Metab. 2018;20:1111‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosenstock J, Frias J, Páll D, et al. Effect of ertugliflozin on glucose control, body weight, blood pressure and bone density in type 2 diabetes mellitus inadequately controlled on metformin monotherapy (VERTIS MET). Diabetes Obes Metab. 2018;20:520‐529. [DOI] [PubMed] [Google Scholar]
- 11. Hollander P, Liu J, Hill J, et al. Ertugliflozin compared with glimepiride in patients with type 2 diabetes mellitus inadequately controlled on metformin: the VERTIS SU randomized study. Diabetes Ther. 2018;9:193‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miller S, Krumins T, Zhou H, et al. Ertugliflozin and sitagliptin co‐initiation in patients with type 2 diabetes: the VERTIS SITA randomized study. Diabetes Ther. 2018;9:253‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aronson R, Frias J, Goldman A, Darekar A, Lauring B, Terra SG. Long‐term efficacy and safety of ertugliflozin monotherapy in patients with inadequately controlled T2DM despite diet and exercise: VERTIS MONO extension study. Diabetes Obes Metab. 2018;20:1453‐1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med. 1985;4:213‐226. [DOI] [PubMed] [Google Scholar]
- 15. Hickman A, Frederich R, Patel S, et al. Evaluation of fractures, bone mineral density (BMD), and bone biomarkers in patients with type 2 diabetes mellitus (T2DM) receiving ertugliflozin. American Diabetes Association 78th Scientific Sessions, June 22–26, 2018, Orlando, Florida.
- 16. Brunton SA. The potential role of sodium glucose co‐transporter 2 inhibitors in the early treatment of type 2 diabetes mellitus. Int J Clin Pract. 2015;69:1071‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li J, Gong Y, Li C, Lu Y, Liu Y, Shao Y. Long‐term efficacy and safety of sodium‐glucose cotransporter‐2 inhibitors as add‐on to metformin treatment in the management of type 2 diabetes mellitus: a meta‐analysis. Medicine (Baltimore). 2017;96:e7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shapses SA, Riedt CS. Bone, body weight, and weight reduction: what are the concerns? J Nutr. 2006;136:1453‐1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adil M, Khan RA, Kalam A, et al. Effect of anti‐diabetic drugs on bone metabolism: evidence from preclinical and clinical studies. Pharmacol Rep. 2017;69:1328‐1340. [DOI] [PubMed] [Google Scholar]
- 20. Ljunggren Ö, Bolinder J, Johansson L, et al. Dapagliflozin has no effect on markers of bone formation and resorption or bone mineral density in patients with inadequately controlled type 2 diabetes mellitus on metformin. Diabetes Obes Metab. 2012;14:990‐999. [DOI] [PubMed] [Google Scholar]
- 21. Alba M, Xie J, Fung A, Desai M. The effects of canagliflozin, a sodium glucose co‐transporter 2 inhibitor, on mineral metabolism and bone in patients with type 2 diabetes mellitus. Curr Med Res Opin. 2016;32:1375‐1385. [DOI] [PubMed] [Google Scholar]
- 22. Bilezikian JP, Watts NB, Usiskin K, et al. Evaluation of Bone Mineral Density and Bone Biomarkers in Patients With Type 2 Diabetes Treated With Canagliflozin. J Clin Endocrinol Metab. 2016;101:44‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bolinder J, Ljunggren O, Johansson L, et al. Dapagliflozin maintains glycaemic control while reducing weight and body fat mass over 2 years in patients with type 2 diabetes mellitus inadequately controlled on metformin. Diabetes Obes Metab. 2014;16:159‐169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1. Supporting information.
Data Availability Statement
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the United States and/or European Union, or (2) in programmes that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.