

Abstract
In patients with chronic inflammatory demyelinating polyneuropathy (CIDP), intravenous immunoglobulin (IVIG) is recommended to be periodically reduced to assess the need for ongoing therapy. However, little is known about the effectiveness of restabilization with IVIG in patients who worsen after IVIG withdrawal. In the Polyneuropathy And Treatment with Hizentra (PATH) study, the pre‐randomization period included sudden stopping of IVIG followed by 12 weeks of observation. Those deteriorating were then restabilized with IVIG. Of 245 subjects who stopped IVIG, 28 did not show signs of clinical deterioration within 12 weeks. Two hundred and seven received IVIG restabilization with an induction dose of 2 g/kg bodyweight (bw) IgPro10 (Privigen, CSL Behring, King of Prussia, Pennsylvania) and maintenance doses of 1 g/kg bw every 3 weeks for up to 13 weeks. Signs of clinical improvement were seen in almost all (n = 188; 91%) subjects. During IVIG restabilization, 35 subjects either did not show CIDP stability (n = 21, analyzed as n = 22 as an additional subject was randomized in error) or withdrew for other reasons (n = 14). Of the 22 subjects who did not achieve clinical stability, follow‐up information in 16 subjects after an additional 4 weeks was obtained. Nine subjects were reported to have improved, leaving a maximum of 27 subjects (13%) who either showed no signs of clinical improvement during the restabilization phase and 4 weeks post‐study or withdrew for other reasons. In conclusion, sudden IVIG withdrawal was effective in detecting ongoing immunoglobulin G dependency with a small risk for subjects not returning to their baseline 17 weeks after withdrawal.
Keywords: chronic inflammatory demyelinating polyneuropathy (CIDP), inflammatory neuropathy cause and treatment (INCAT), intravenous immunoglobulin (IVIG), polyneuropathy and treatment with Hizentra (PATH), Privigen
1. INTRODUCTION
Chronic inflammatory demyelinating polyneuropathy (CIDP), a rare immune‐mediated disease of the peripheral nervous system, has a gradually worsening or, less commonly, a relapsing–remitting course.1 Intravenous immunoglobulin (IVIG) is a treatment option as outlined in the European Federation of Neurological Societies and Peripheral Nerve Society (EFNS/PNS) 2010 guidelines.2 While complete remission after short‐term therapy occurs in some patients,3 others require extended therapy. Because patients may go into remission after longer periods of IVIG treatment, and IVIG is expensive, it is recommended that all therapies, and especially IVIG, be periodically reduced or withdrawn to assess ongoing need for continued therapy, or whether the disease has become inactive and therapy is no longer needed. In case symptoms return after immunoglobulin (Ig) withdrawal, treatment is reinstated. To our knowledge, little is known about the outcome of such withdrawal trials and the effectiveness of reinitiation of therapy.
In the Polyneuropathy And Treatment with Hizentra (PATH) study4 before the randomized and blinded portion of the study, subjects underwent a period of sudden, unblinded IVIG withdrawal followed by 13 weeks of close observation and, if they worsened, immediate IVIG restabilization. This was done to ensure that subjects who entered the next phase of the study had active disease. This enrichment strategy was implemented as a consequence of previous trials where IVIG could be reduced without change in symptoms5 or where subjects who were administered placebo after a period of IVIG treatment showed a low rate of relapse.6
The sequelae of stopping IVIG treatment in otherwise stable CIDP patients has not been examined in a prospective study. This report focuses on the questions of whether a sudden treatment stop is feasible, which clinical measures can be used in combination to detect subtle yet meaningful deterioration, how quickly subjects recover after reinitiating therapy, and to what extent recovery is achieved within 17 weeks.
2. METHODS AND MATERIALS
The details of the PATH trial have been previously published.7 In brief, adult subjects with definite or probable CIDP,2 all being treated with IVIG before enrollment were eligible for this study. After screening, IVIG was withheld until clinical deterioration occurred. Subjects who did not show any signs of clinical worsening after 12 weeks of IVIG withdrawal were discontinued from the study as they were assumed to have inactive disease. On deterioration, subjects entered the IVIG (IgPro10, Privigen, CSL Behring) restabilization period to receive one induction dose of 2 g/kg bodyweight (bw) given over 2 to 5 days and an additional 3 to 4 maintenance doses of 1 g/kg bw every 3 weeks. Subjects who were not restabilized were discontinued from the study. A 4‐week post‐study follow‐up was conducted for those subjects. Subjects who recovered and were stable at the last two visits of the IVIG restabilization period were randomized to subcutaneous Ig (SCIG [IgPro20, Hizentra; CSL Behring]) or placebo (Figure 1).
Figure 1.
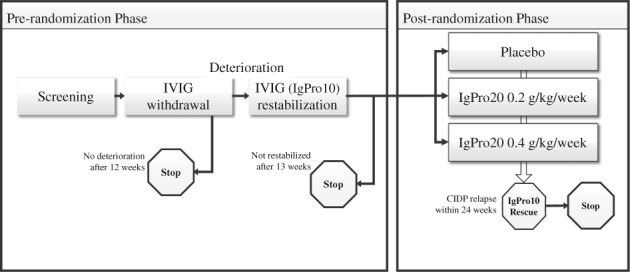
Study design. Abbreviations: CIDP, chronic inflammatory demyelinating polyneuropathy; IgPro10, 10% intravenous immunoglobulin preparation; IgPro20, 20% subcutaneous immunoglobulin preparation; IVIG, intravenous immunoglobulin. Note: “Stop” indicates that subject was discontinued from the study after all study completion assessments were performed
Clinical deterioration was defined as an increase in adjusted inflammatory neuropathy cause and treatment (INCAT) total score by ≥1 point,6, 8 a decrease in inflammatory Rasch‐built overall disability scale (I‐RODS) total score by ≥4 points,9 or a decrease in mean grip strength by ≥8 kilopascals [kPa].10 I‐RODS and grip strength measures were used to detect IVIG dependency only after a protocol change. Clinical improvement was assessed with the same measures including a decrease in the Medical Research Council (MRC) sum score (range 0‐80) by ≥3 points compared with the reference visit (prior to first IVIG restabilization dose).11
Subjects were trained during screening to document I‐RODS score and grip strength measurements daily in a diary and were provided with a Martin Vigorimeter. In addition, subjects' status was assessed every 2 weeks either during a site visit or telephone call. Subjects could also contact the site at any time in case of deterioration outside of the defined contact time points.
This study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines and the Declaration of Helsinki. The study protocol and all other study‐related documents were reviewed and approved by the local Independent Ethics Committees. Written informed consent was obtained from all subjects before they started the study. This study is registered with Clinicaltrials.gov, number NCT01545076.
2.1. Statistical methodology
The pre‐randomization phase (IVIG withdrawal and restabilization) served as an enrichment strategy of the PATH study. The objective of the IgPro10 restabilization period was to investigate the efficacy and safety of IgPro10 (a secondary objective of the PATH trial). No formal sample size determination was performed for the pre‐randomization phase. Efficacy and safety were determined in the pre‐randomization safety data set (PSDS), defined as all subjects enrolled into the study who received at least one dose of IgPro10.
The efficacy endpoints were based on the number and percentage of subjects with improvement for the efficacy scores (adjusted INCAT total score, I‐RODS centile score, mean grip strength, and MRC sum score). Efficacy parameters were summarized by visit, using descriptive statistics, and changes from Reference Visit were calculated. The Reference Visit was the last visit before entering the restabilization period.
The safety analysis was based on the PSDS. Descriptive statistics were calculated for all parameters.
3. RESULTS
3.1. IVIG withdrawal period
When subjects entered the study and passed the screening phase, their regular IVIG treatment was stopped until signs of clinical deterioration were detected. A total of 245 subjects entered this IVIG withdrawal period. Subjects received their last IVIG cycle either at any time before study entry (original protocol) or at the end of the screening period (protocol amendment).
In total, 28 (11.4%) subjects did not show any signs of clinical deterioration and were discontinued from the study (Figure 2). A further 10 subjects withdrew from this phase for other reasons. Clinical deterioration was shown by adjusted INCAT score (≥1 point increase) in 73% of subjects; the remaining subjects showed deterioration by either I‐RODS or grip strength. Deterioration occurred mostly in the first 8 weeks after IVIG withdrawal (median 70 [42–105] days, n = 156 [64%]). In total, 207 subjects entered the IgPro10 restabilization period.
Figure 2.
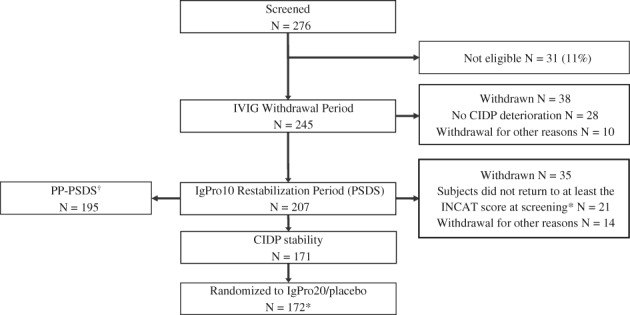
Subject disposition. Per investigator, one additional subject met the criteria to enter the IgPro10 restabilization period (N = 208), but the subject withdrew consent and did not receive any IgPro10 (treated count N = 207). A total of 13 subjects were rescreened. Abbreviations: CIDP, chronic inflammatory demyelinating polyneuropathy; IgPro10, 10% intravenous immunoglobulin preparation; IgPro20, 20% subcutaneous immunoglobulin preparation; IVIG, intravenous immunoglobulin; N, number of subjects; PP‐PSDS, per‐protocol pre‐randomization safety data set; PSDS, pre‐randomization safety data set. *An additional subject did not return to at least the inflammatory neuropathy cause and treatment (INCAT) score at screening, but was randomly allocated in error. †Reasons for exclusion of 12 subjects from the PSDS: five subjects did not demonstrate CIDP deterioration during the IVIG withdrawal period, but were transitioned to the IgPro10 restabilization period; five subjects took prohibited concomitant medication during the pre‐randomization phase; one subject received an increased loading dose during pre‐randomization phase; one subject took prohibited concomitant medication (nonstudy IVIG) during pre‐randomization phase and received an increased loading dose during pre‐randomization phase
3.2. IgPro10 restabilization period
Table 1 shows the subject demographics and disease characteristics of all 207 subjects entering the IgPro10 restabilization period. Approximately 91% of subjects who deteriorated only in adjusted INCAT score during the withdrawal period (n = 151) improved their INCAT score during the restabilization period (Table 2). Of all subjects entering this study period, 91% showed improvement in at least one of the pre‐specified outcome measures (Table 3). Almost all of the subjects who showed improvement had improved after three doses of IgPro10, that is, by Week 10 (99%). Approximately 21% of improving subjects had a better score (adjusted INCAT) compared with screening, prior to IVIG withdrawal. Figure 3 shows the pattern of all four scores over the first 10 weeks of the restabilization period. Subjects showed a mean decline between baseline and IVIG withdrawal and improved mean scores during IVIG restabilization compared with baseline. First improvement, regardless of the outcome measure, occurred within a median time of 23 days, that is, at the first assessment after the first IVIG dose (Table 3).
Table 1.
Demographics and disease characteristics of subjects
| Subjects entering the IVIG withdrawal period, N = 245 | Subjects showing no deterioration within the withdrawal period, N = 28 | Subjects with deterioration entering restabilization, N = 207 (PSDS) | Subjects showing restabilization on IVIG, N = 171 (ITTS, N = 172)a , b | Subjects not showing improvement, N = 22 (PSDS)b | |
|---|---|---|---|---|---|
| Age (years), median (min, max) | 58.2 (24.7, 82.7) | 56.7 (26.8, 82.4) | 58.2 (24.7, 82.7) | 57.8 (24.7, 82.7) | 58.6 (30.8, 77.5) |
| Sex, n (%) | |||||
| Male | 162 (66) | 22 (79) | 131 (63) | 110 (64) | 13 (59) |
| Female | 83 (34) | 6 (21) | 76 (37) | 62 (36) | 9 (41) |
| Weight (kg), median (min, max) | 83 (41.8, 145.8) | 89 (56.5, 118.0) | 82 (41.8, 133.0) | 82 (41.7, 133.0) | 79 (43.7, 111.9) |
| BMIc (kg/m2), median (min, max) | 27 (17.6, 49.4) | 27 (20.8, 35.1) | 27 (17.6, 49.4) | 27 (17.6, 49.4) | |
| Time since initial CIDP diagnosis (years)d | |||||
| Mean (SD) | 4.57 (5.1) | 4.28 (5.0) | 4.66 (5.2) | 4.73 (5.4) | 4.84 (4.6) |
| Median (min, max) | 2.8 (0.1, 33.6) | 2.7 (0.3, 19.2) | 3.0 (0.1, 33.5) | 3.0 (0.1, 33.5) | 3.2 (0.2, 16.4) |
| EFNS/PNS CIDP diagnostic criteria, N (%) | |||||
| Definite | 221 (90) | 26 (93) | 185 (89) | 157 (91) | 19 (86) |
| Probable | 24 (10) | 2 (7) | 22 (11) | 15 (9) | 3 (14) |
| Screening INCAT total score, points | |||||
| Mean (SD) | 2.7 (1.7) | 2.4 (1.6) | 2.7 (1.7) | 2.3 (1.7) | 2.8 (2.3) |
| Median (min, max) | 3.0 (0.0, 8.0) | 2.0 (0.0, 6.0) | 3.0 (0.0, 8.0) | 2.0 (0.0, 7.0) | 3.0 (0.0, 8.0) |
Abbreviations: BMI, body mass index; CIDP, chronic inflammatory demyelinating polyneuropathy; EFNS, European Federation of Neurological Societies; INCAT, inflammatory neuropathy cause and treatment; ITTS, intention to treat set; max, maximum; min, minimum; N, number of subjects; PNS, Peripheral Nerve Society; PSDS, pre‐randomization safety data set; SD, standard deviation.
Data for intention to treat set (ITTS).
Analysis includes one subject who was randomized in error.
BMI at screening = weight at screening (kg) / (height at screening [m])2. For BMI, N = 243 or 205, respectively.
Time since initial diagnosis of CIDP (years) = (date of informed consent – date of initial diagnosis + 1) / 365.25.
Table 2.
CIDP improvement and CIDP stability during IgPro10 restabilization period in relation to results of IVIG withdrawal period (PSDS population)
| IgPro10 restabilization period | Number (%) of subjects, N = 207 | ||
|---|---|---|---|
| IVIG withdrawal period | |||
| CIDP deterioration bya | No CIDP deterioration or withdrawal fromstudy,b | ||
| INCAT total score, N = 151 | I‐RODS total score or mean grip strength, N = 49 | N = 7 | |
| Improvement by adjusted INCAT scorec | 137 (91) | 10 (20) | 4 (57) |
| No improvement by adjusted INCAT scorec | 14 (9) | 39 (80) | 3 (43) |
| Improvement and CIDP stability achieved (based on adjusted INCAT score)c , d | 120 (80) | 9 (18) | 4 (57) |
| No improvement but CIDP stability achieved (based on adjusted INCAT score)c , d | 4 (3) | 32 (65) | 2 (29) |
Abbreviations: CIDP, chronic inflammatory demyelinating polyneuropathy; INCAT, inflammatory neuropathy cause and treatment; I‐RODS, inflammatory Rasch‐built overall disability scale; IVIG, intravenous immunoglobulin; N, number of subjects; PSDS, pre‐randomization safety data set.
CIDP deterioration was defined as an increase in adjusted INCAT total score by ≥1 point, a decrease in I‐RODS total score by ≥4 points, or a decrease in mean grip strength by ≥8 kPa.
Subjects who did not demonstrate CIDP deterioration during the IVIG withdrawal period but entered IgPro10 restabilization period (N = 3) with an additional four subjects who had shown CIDP deterioration but withdrew consent.
Improvement was defined as a decrease in adjusted INCAT score by ≥1 point, an increase in I‐RODS centile score by ≥4 points, an increase in mean grip strength by ≥8 kPa, and an increase in MRC sum score by ≥3 points, as compared to the Reference Visit.
CIDP stability occurred when CIDP status did not show a clinically meaningful difference at the last two consecutive visits during the IgPro10 restabilization period. In addition, to be considered CIDP stable, the CIDP status had to recover back to at least the status at Screening (as assessed by adjusted INCAT score).
Table 3.
Time to first improvement (PSDS population)
| Overall, N = 207 | |||||
|---|---|---|---|---|---|
| INCAT total score | I‐RODS centile score | Mean grip strength (dominant hand) | MRC sum score | First improvement in any criteria | |
| Number of events (improvements), n (%) | 151 (73) | 84 (41) | 123 (59) | 117 (57) | 188 (91) |
| Number of censored observations, n (%)a | 56 (27) | 123 (59) | 84 (41) | 90 (44) | 19 (9) |
| Time to first improvement, daysb | |||||
| Median | 26 | 71 | 65 | 65 | 23 |
| 95% CI | 24, 41 | 66, 86 | 64, 66 | 64, 67 | 22, 23 |
Abbreviations: CI, confidence interval; INCAT, inflammatory neuropathy cause and treatment; I‐RODS, inflammatory Rasch‐built overall disability scale; IVIG, intravenous immunoglobulin; MRC, Medical Research Council; N, number of subjects; PSDS, pre‐randomization safety data set.
Improvement was defined as a decrease in adjusted INCAT score by ≥1 point, an increase in I‐RODS centile score by ≥4 points, an increase in mean grip strength by ≥8 kPa, and an increase in MRC sum score by ≥3 points, as compared to the Reference Visit (prior to first restabilization IVIG dose).
Subjects without improvements were censored at the date of their last visit in the IgPro10 restabilization period.
Using Kaplan‐Meier estimation. Time to first improvement (days) = date of first improvement ‐ date of first IVIG infusion.
Figure 3.
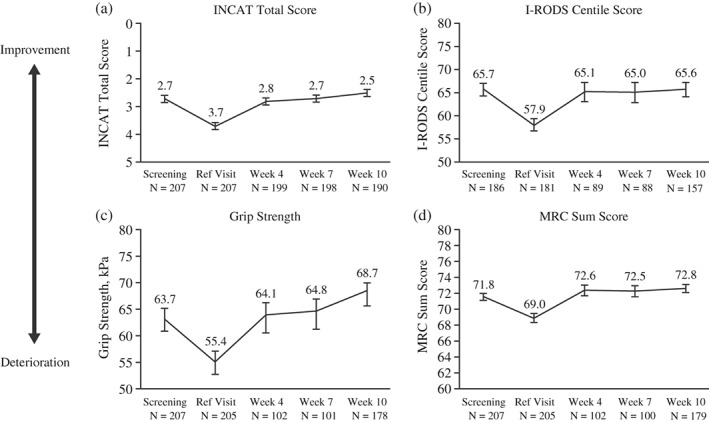
Primary efficacy outputs (PSDS population). Abbreviations: INCAT, inflammatory neuropathy cause and treatment; I‐RODS, inflammatory Rasch‐built overall disability scale; MRC, Medical Research Council; N, number of subjects; PSDS, pre‐randomization safety data set. Mean and SE are plotted
In total, 83% of subjects remained clinically stable at the end of the 10‐ to 13‐week restabilization period by the definition of the protocol, which required the same adjusted INCAT score at the last two visits which could not be worse when compared with their baseline adjusted INCAT score. These stabilized subjects were subsequently randomized to SCIG or placebo.4
Of the subjects treated with IVIG in the restabilization period, 35 (17%) either did not improve (n = 21 [analyzed as n = 22 to include one later identified subject who had not returned to his/her screening INCAT score and was randomized in error]) or could not be stabilized as they withdrew for other reasons (n = 14), while 171 (83%) subjects achieved CIDP stability during this period (Figure 2). Demographic and disease characteristics of the 22 (11%) non‐improving subjects are provided in Table 1. There were no relevant differences observed between the overall restabilized population and those who did not reach CIDP stability. Subjects had a mean adjusted INCAT score of 4.2 (SD 1.6) after IVIG withdrawal, which improved by 0.4 points (SD 1.4) at the last observation. Follow‐up information was obtained 4 weeks post study in 16/22 subjects. In nine subjects, improvement based on clinical judgment was identified. Therefore, 13% (27 subjects) either showed no signs of clinical improvement (n = 13 which includes six subjects who did not respond to the follow‐up questionnaire) during the restabilization phase and 4‐week post‐study follow‐up or withdrew from the study for other reasons (n = 14).
3.3. Safety
Out of 207 subjects in the IVIG restabilization period, 100 (48.3%) experienced a total of 284 adverse events, and causally related adverse events were experienced by 28% of the subjects. The most common adverse events occurring in >5% of subjects were headache, nasopharyngitis, and nausea (Table 4). There were seven related serious adverse events (allergic reaction, pulmonary embolism, increase in diastolic blood pressure, exacerbation of CIDP, worsening of respiratory failure due to chronic obstructive pulmonary disease, rash, and worsening of migraine).
Table 4.
Most common adverse events (PSDS population)
| System organ class preferred term | Number (%) of subjects, N = 207 | Number of events | Rate/infusion, N = 1620a |
|---|---|---|---|
| Any adverse event | 100 (48.3) | 284 | 0.175 |
| Headache | 34 (16.4) | 53 | 0.033 |
| Nasopharyngitis | 12 (5.8) | 12 | 0.007 |
| Nausea | 10 (4.8) | 12 | 0.007 |
| Hemolysisb | 7 (3.4) | 7 | 0.004 |
| Hypertension | 6 (2.9) | 7 | 0.004 |
| Rash | 6 (2.9) | 6 | 0.004 |
| Vomiting | 6 (2.9) | 7 | 0.004 |
| Back pain | 5 (2.4) | 5 | 0.003 |
| Fall | 5 (2.4) | 5 | 0.003 |
| Fatigue | 5 (2.4) | 11 | 0.007 |
| Edema peripheral | 5 (2.4) | 5 | 0.003 |
Abbreviations: N, number of subjects; PSDS, pre‐randomization safety data set.
Number of infusions.
Two out of the seven hemolysis subjects met the laboratory criteria for hemolysis, both from non‐O blood groups.
4. DISCUSSION
The PATH study design mandated an initial determination of IVIG dependence by stopping IVIG treatment for a maximum of 12 weeks and frequently assessing potential clinical deterioration by different outcome measures. Experiences from the IGIV‐C CIDP efficacy (ICE) trial,6 where 58% of placebo subjects did not deteriorate within 24 weeks (equivalent to a blinded IVIG withdrawal) and the randomized, controlled trial of methotrexate for CIDP (RMC) trial,5 where reduction of IVIG dose by ≥20% also did not lead to clinical deterioration, motivated this trial design. With 245 subjects included, the PATH study provided a large cohort in which IVIG withdrawal and restabilization was studied prospectively.
The study was successful in identifying approximately 11% of subjects not requiring immunoglobulin G (IgG) treatment within 12 weeks, that is, showing no signs of clinical deterioration, neither by adjusted INCAT score, I‐RODS, nor daily measured grip strength. An additional 2% of subjects also did not show signs of deterioration but left this study phase due to withdrawal of consent. Reasons for the low number of IVIG‐independent subjects may lie in pre‐selection of subjects and limited observation time.
Most (91%) of the 207 subjects who were treated during the restabilization phase improved in at least one of the clinical measures after treatment with IgPro10, and 83% remained stable according to the clinical measures at the end of this phase. Stabilization was defined as showing the same or better adjusted INCAT score at two consecutive visits after improvement. The restabilization phase was 10 to 13 weeks long as this was considered the minimum time for previously IVIG‐treated subjects to show improvement and be stabilized after previous deterioration.12 However, it may have been too short for subjects who deteriorated later in the IVIG withdrawal period and as such, could not improve in the short timeframe of the IVIG restabilization period. Of the subjects showing improvement, 21% had a better lowest total adjusted INCAT score in the IVIG restabilization period when compared to their INCAT score at screening. This could infer under‐treatment of subjects on stable doses of IVIG. Initially, 22/207 subjects did not return to their baseline status during the 13‐week IgPro10 treatment phase. Of these, at a further follow‐up 4 weeks after completing the study, an additional nine subjects (4%) had shown improvements; and a maximum of 13 subjects (6%) might not have returned to their baseline level since no further information was available.
What are the practical implications of these results? Sudden withdrawal of IVIG in an open fashion is a feasible method to assess ongoing disease activity and the need for IVIG. In our study, we limited the time to assess IgG dependency to 12 weeks, and we therefore might have excluded some IgG‐dependent subjects, who would have only shown dependence beyond 12 weeks. We initially used adjusted INCAT score (≥1 point increase) as a measure of disease impact. A protocol change was introduced to recruit additional subjects whose deterioration was difficult to assess by adjusted INCAT score alone. After the protocol change, adjusted INCAT score along with I‐RODS (≥4 point decrease), or grip strength (≥8 kPa decrease) were used to increase the sensitivity for early detection of worsening and to detect ongoing disease activity. The cutoffs were a compromise between allowing detection of relevant disease worsening and assuring that recurrence of symptoms was temporary, that is, reversible after reinitiation of therapy. The trial was not designed to evaluate the sensitivity or specificity of modified INCAT, I‐RODS, or grip strength with their respective cutoffs for the evaluation of Ig dependence in CIDP after IVIG withdrawal.
The main reason for excluding those with inactive disease was to increase the likelihood for placebo subjects to relapse during the randomized study phase with the SCIG IgPro20. The final study results of PATH,4 with 63% of placebo‐treated subjects showing CIDP relapse (56%) or withdrawal for other reasons (7%) may still seem low but was in the expected pre‐defined range and higher than in an IVIG placebo‐controlled trial (42%, ICE trial6). The IVIG restabilization phase that preceded the SCIG randomized phase may have contributed to this result. All subjects received a 2 g/kg bw induction dose followed by a 1 g/kg bw 3‐weekly maintenance dose, and a carry‐over effect of IVIG into the subcutaneous phase cannot be ruled out, potentially contributing to the placebo response. Similar observations were made in the ICE trial,6 where after a phase of IVIG treatment, only 42% of placebo‐treated subjects had a relapse during the randomized withdrawal phase. The restabilization phase also showed that more than 90% of subjects returned to or improved beyond their baseline level at study entry. The group that did not fully restabilize at least showed a tendency towards improvement, however, we did not treat subjects longer than 13 weeks or formally follow them up for longer than 4 weeks post‐study.
In the Privigen Impact on Mobility and Autonomy (PRIMA) trial12 a similar IgG withdrawal period was performed (up to 10 weeks) for those subjects on a previous regular IVIG regime. Following deterioration, subjects were given an IVIG IgPro10 induction dose (2 g/kg bw) over 2 to 5 days, followed by up to 7 infusions (1 g/kg bw) at 3‐week intervals. In that study, 1/14 subjects (7%) did not show deterioration in adjusted INCAT score and 3/13 (23%) did not return to their baseline adjusted INCAT level. However, all subjects improved in either grip strength or MRC score. In the ICE study, 58% did not show signs of deterioration also measured by adjusted INCAT score at the end of the 24‐week randomized, blinded, withdrawal maintenance phase after responding to IVIG in the first or second treatment phase. It is not known whether these subjects deteriorated in other clinical measures or if their disease was inactive in that period. The fate of relapsed subjects in the ICE study is also not known. In a randomized, placebo‐controlled study comparing IVIG to intravenous methylprednisolone, 38% of subjects on previous IVIG worsened within 5 months after treatment discontinuation (median 4 months, range 1–5 months),13 leading to the conclusion that deterioration after IVIG suspension can occur later than assessment in our study occurred (maximum 12 weeks). After 12 months, 54% of IVIG‐treated subjects did not require further therapy. Long‐term follow‐up of IVIG‐treated subjects revealed that approximately 86% would worsen within a median follow‐up time of 42 months (range 1–57 months) after therapy discontinuation (median time to worsening 4.5 months, range 1–24 months).14
In conclusion, sudden IVIG withdrawal was effective in detecting active disease and ongoing IgG dependency, with approximately five in every six subjects restabilizing during the 13‐week restabilization period following reinitiation of therapy.
ACKNOWLEDGEMENTS
Medical writing support was provided by Dr. Barbara Boggetti of Trilogy Writing and Consulting GmbH, Frankfurt, Germany, funded by CSL Behring. Editorial support was provided by Meridian HealthComms Ltd, funded by CSL Behring. Individual participant data will not be shared.
PATH Study Group: Australia A Sabet, K George (Gold Coast Hospital and Health Service, Southport, QLD). L Roberts, R Carne (St Vincent's Hospital, Melbourne, VIC). S Blum, R Henderson (Royal Brisbane & Women's Hospital, Herston, QLD). Belgium P Van Damme, J Demeestere (UZ Leuven‐Neurologie, Leuven). Canada S Larue, C D'Amour (Hopital Charles LeMoyne, Recherche Sepmus, Greenfield Park, QC). Czech Republic P Kunc, M Valis (Neurologicka klinika, Fakultni nemocnice Hradec Kralove, Hradec Kralove). J Sussova, T Kalous (Neurologicka klinika, Vseobecna fakultni nemocnice v Praze, Prague). R Talab, M Bednar (Privatni ordinace neurologie, Hradec Kralove). Estonia T Toomsoo, I Rubanovits (East Tallinn Central Hospital, Tallinn). K Gross‐Paju, U Sorro (West Tallinn Central Hospital, Tallinn). Finland M Saarela, M Auranen (Helsinki University Central Hospital, Helsinki). France J Pouget, S Attarian (Hôpital de la Timone Neurologi, Marseille). G Le Masson, A Wielanek‐Bachelet (Hôpital Haut‐Lévéque, Service de Neurologie Centre hospitalier universitaire de Bordeaux, Bordeaux). C Desnuelle, E Delmont (Hôpital Archet 1 Centre de référence maladies neuromusculaires, Nice). P Clavelou, D Aufauvre (Centre hospitalier universitaire Hôpital Gabriel Montpied, Clermont‐Ferrand). Germany J Schmidt, J Zschuentzsch (Universitätsmedizin Göttingen, Göttingen). C Sommer, D Kramer (Universitaetsklinikum Wurzburg, Wurzburg). O Hoffmann, C Goerlitz (St Josefs‐Krankenhaus, Potsdam). J Haas, M Chatzopoulos (Jüdisches Krankenhaus Berlin, Berlin). R Yoon, R Gold (Klinikum der Ruhr‐Universität Bochum, Bochum). P Berlit, A Jaspert‐Grehl (Alfried Krupp Krankenhaus Rüttenscheid, Essen). D Liebetanz, A Kutschenko (Georg‐August‐Universitätsmedizin Göttingen, Göttingen). M Stangel, C Trebst (Medizinische Hochschule Hannover, Hannover). P Baum, F Bergh (Universitaetsklinikum Leipzig, Leipzig). J Klehmet, A Meisel (Klinik und Poliklinik für Neurologie Charité‐Universitätsmedizin Berlin, Berlin). F Klostermann, J Oechtering (Charite Universitaetsmedizin Berlin). H Lehmann, M Schroeter (Universitätsklinikum, Köln). T Hagenacker, D Mueller (Universitätsklinikum Essen, Essen). A Sperfeld, F Bethke (Klinikum Ibbenbüren, Ibbenbüren). Israel V Drory, A Algom (Tel Aviv Sourasky Medical Center, Tel Aviv). D Yarnitsky, B Murinson (Rambam Health Care Campus, Haifa). Italy A Di Muzio, F Ciccocioppo (Policlinico SS Annunziata, Chieti Scalo). S Sorbi, S Mata (Ospedaliero Universitaria Careggi, Firenze). A Schenone, M Grandis (Azienda Ospedaliera Universitaria San Martino di Genova, Genoa). G Lauria, D Cazzato (Fondazione Istituto DiRicovero, Milano). G Antonini, S Morino (Azienda Ospedaliera S Andrea Universita degli Studi di Roma “La Sapienza”, Rome). D Cocito, M Zibetti (Azienda ospedaliero universitaria San Giovanni Battista, Torino). Japan T Yokota, T Ohkubo (Tokyo Medical and Dental University, Tokyo). T Kanda, M Kawai (Yamaguchi University Hospital, Yamaguchi). K Kaida, H Onoue (National Defense Medical Hospital, Saitama). S Kuwabara, M Mori (Chiba University Hospital, Chiba). M Iijima, K Ohyama (Nagoya University Hospital, Nagoya). M Baba, M Tomiyama (Aomori Prefectural Central Hospital, Aomori). K Nishiyama, T Akutsu (Kitasato University Hospital, Kanagawa). K Yokoyama, K Kanai (Juntendo University Hospital, Tokyo). Netherlands I N van Schaik, F Eftimov (Amsterdam University Medical Centers, University of Amsterdam, Amsterdam). N C Notermans, N Visser (University Medical Center Utrecht, Utrecht). C Faber, J Hoeijmakers (Maastricht University Medical Center, Maastricht). Poland K Rejdak, U Chyrchel‐Paszkiewicz (Samodzielny Publiczny Szpital Kliniczny, Lublin). Spain C Casanovas Pons, M Antonia (Universitari de Bellvitge Servicio de Neurología, Barcelona). J Gamez, M Salvado (Hospital Universitario Vall d'Hebron Servicio de Neurología, Barcelona). C Marquez Infante, S Benitez (Hospital Universitario Virgen del Rocío, Seville). United Kingdom M Lunn, J Morrow (National Hospital for Neurology and Neurosurgery, London). D Gosal, T Lavin (Salford Royal Hospital, Salford). United States I Melamed, A Testori (IMMUNOe International Research Centers, Centennial, CO). S Ajroud‐Driss, D Menichella (Northwestern University Feinberg School of Medicine, Chicago, IL). E Simpson, E Chi‐Ho Lai (Methodist Neurological Institute, Houston, TX). M Dimachkie, R J Barohn (University of Kansas Medical Center, Kansas City, KS). S Beydoun, H Johl (University of Southern California Keck School of Medicine, Los Angeles, CA). D Lange, A Shtilbans (Hospital for Special Surgery, New York, NY). S Muley, S Ladha (St Joseph's Hospital and Medical Center, Phoenix, AZ). M Freimer, J Kissel (Wexner Medical Center at the Ohio State University, Columbus, OH). N Latov, R Chin (Weill Medical College of Cornell University, New York, NY). E Ubogu, S Mumfrey (University of Alabama Medical Center Birmingham, Birmingham, AL). T Rao, P MacDonald (The Neurologic Institute, Charlotte, NC). K Sharma, G Gonzalez (University of Miami, Miami, FL). J Allen, D Walk (Department of Neurology, University of Minnesota, Minneapolis, MN). L Hobson‐Webb, K Gable (Duke University Medical Center, Durham, NC).
Mielke O, Bril V, Cornblath DR, et al. Restabilization treatment after intravenous immunoglobulin withdrawal in chronic inflammatory demyelinating polyneuropathy: Results from the pre‐randomization phase of the Polyneuropathy And Treatment with Hizentra study. J Peripher Nerv Syst. 2019;24:72–79. 10.1111/jns.12303
Contributor Information
Orell Mielke, Email: orell.mielke@cslbehring.com.
on behalf of the PATH study group:
A. Sabet, K. George, L. Roberts, R. Carne, S. Blum, R. Henderson, P. Van Damme, J. Demeestere, S. Larue, C. D'Amour, P. Kunc, M. Valis, J. Sussova, T. Kalous, R. Talab, M. Bednar, T. Toomsoo, I. Rubanovits, K. Gross‐Paju, U. Sorro, M. Saarela, M. Auranen, J. Pouget, S. Attarian, G. Le Masson, A. Wielanek‐Bachelet, C. Desnuelle, E. Delmont, P. Clavelou, D. Aufauvre, J. Schmidt, J. Zschuentzsch, C. Sommer, D. Kramer, O. Hoffmann, C. Goerlitz, J. Haas, M. Chatzopoulos, R. Yoon, R. Gold, P. Berlit, A. Jaspert‐Grehl, D. Liebetanz, A. Kutschenko, M. Stangel, C. Trebst, P. Baum, F. Bergh, J. Klehmet, A. Meisel, F. Klostermann, J. Oechtering, H. Lehmann, M. Schroeter, T. Hagenacker, D. Mueller, A. Sperfeld, F. Bethke, V. Drory, A. Algom, D. Yarnitsky, B. Murinson, A. Di Muzio, F. Ciccocioppo, S. Sorbi, S. Mata, A. Schenone, M. Grandis, G. Lauria, D. Cazzato, G. Antonini, S. Morino, D. Cocito, M. Zibetti, T. Yokota, T. Ohkubo, T. Kanda, M. Kawai, K. Kaida, H. Onoue, S. Kuwabara, M. Mori, M. Iijima, K. Ohyama, M. Baba, M. Tomiyama, K. Nishiyama, T. Akutsu, K. Yokoyama, K. Kanai, I.N. van Schaik, F. Eftimov, N.C. Notermans, N. Visser, C. Faber, J. Hoeijmakers, K. Rejdak, U. Chyrchel‐Paszkiewicz, C. Casanovas Pons, M. Antonia, J. Gamez, M. Salvado, C. Marquez Infante, S. Benitez, M. Lunn, J. Morrow, D. Gosal, T. Lavin, I. Melamed, A. Testori, S. Ajroud‐Driss, D. Menichella, E. Simpson, E. Chi‐Ho Lai, M. Dimachkie, R.J. Barohn, S. Beydoun, H. Johl, D. Lange, A. Shtilbans, S. Muley, S. Ladha, M. Freimer, J. Kissel, N. Latov, R. Chin, E. Ubogu, S. Mumfrey, T. Rao, P. MacDonald, K. Sharma, G. Gonzalez, J. Allen, D. Walk, L. Hobson‐Webb, and K. Gable
REFERENCES
- 1. Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry. 2015;86:973‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies . Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society–first revision. J Peripher Nerv Syst. 2010;15:185‐195. [DOI] [PubMed] [Google Scholar]
- 3. Eftimov F, Winer JB, Vermeulen M, de Haan R, van Schaik IN. Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2009;21:CD001797. [DOI] [PubMed] [Google Scholar]
- 4. van Schaik IN, Bril V, van Geloven N, et al. Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy (PATH): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Neurol. 2018;17:35‐46. [DOI] [PubMed] [Google Scholar]
- 5. RMC Trial Group . Randomised controlled trial of methotrexate for chronic inflammatory demyelinating polyradiculoneuropathy (RMC trial): a pilot, multicentre study. Lancet Neurol. 2009;8:158‐164. [DOI] [PubMed] [Google Scholar]
- 6. Hughes RA, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate‐chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo‐controlled trial. Lancet Neurol. 2008;7:136‐144. [DOI] [PubMed] [Google Scholar]
- 7. van Schaik IN, van Geloven N, Bril V, et al. Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy (The PATH Study): study protocol for a randomized controlled trial. Trials. 2016;17:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hughes R, Bensa S, Willison H, et al. Randomized controlled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 2001;50:195‐201. [DOI] [PubMed] [Google Scholar]
- 9. van Nes SI, Vanhoutte EK, van Doorn PA, et al. Rasch‐built Overall Disability Scale (R‐ODS) for immune‐mediated peripheral neuropathies. Neurology. 2011;76:337‐345. [DOI] [PubMed] [Google Scholar]
- 10. Vanhoutte EK, Latov N, Deng C, et al. Vigorimeter grip strength in CIDP: a responsive tool that rapidly measures the effect of IVIG‐‐the ICE study. Eur J Neurol. 2013;20:748‐755. [DOI] [PubMed] [Google Scholar]
- 11. Kleyweg RP, van der Meche FG, Schmitz PI. Interobserver agreement in the assessment of muscle strength and functional abilities in Guillain‐Barre syndrome. Muscle Nerve. 1991;14:1103‐1109. [DOI] [PubMed] [Google Scholar]
- 12. Léger JM, De Bleecker JL, Sommer C, et al. Efficacy and safety of Privigen® in patients with chronic inflammatory demyelinating polyneuropathy: results of a prospective, single‐arm, open‐label Phase III study (the PRIMA study). J Peripher Nerv Syst. 2013;18:130‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nobile‐Orazio E, Cocito D, Jann S, et al. Intravenous immunoglobulin versus intravenous methylprednisolone for chronic inflammatory demyelinating polyradiculoneuropathy: a randomised controlled trial. Lancet Neurol. 2012;11:493‐502. [DOI] [PubMed] [Google Scholar]
- 14. Nobile‐Orazio E, Cocito D, Jann S, et al. Frequency and time to relapse after discontinuing 6‐month therapy with IVIg or pulsed methylprednisolone in CIDP. J Neurol Neurosurg Psychiatry. 2015;86:729‐734. [DOI] [PubMed] [Google Scholar]