

Abstract
Objective
To understand the safety, putaminal coverage, and enzyme expression of adeno‐associated viral vector serotype‐2 encoding the complementary DNA for the enzyme, aromatic L‐amino acid decarboxylase (VY‐AADC01), delivered using novel intraoperative monitoring to optimize delivery.
Methods
Fifteen subjects (three cohorts of 5) with moderately advanced Parkinson's disease and medically refractory motor fluctuations received VY‐AADC01 bilaterally coadministered with gadoteridol to the putamen using intraoperative magnetic resonance imaging (MRI) guidance to visualize the anatomic spread of the infusate and calculate coverage. Cohort 1 received 8.3 × 1011vg/ml and ≤450 μl per putamen (total dose, ≤7.5 × 1011vg); cohort 2 received the same concentration (8.3 × 1011vg/ml) and ≤900 μl per putamen (total dose, ≤1.5 × 1012vg); and cohort 3 received 2.6 × 1012vg/ml and ≤900 μl per putamen (total dose, ≤4.7 × 1012vg). (18)F‐fluoro‐L‐dihydroxyphenylalanine positron emission tomography (PET) at baseline and 6 months postprocedure assessed enzyme activity; standard assessments measured clinical outcomes.
Results
MRI‐guided administration of ascending VY‐AADC01 doses resulted in putaminal coverage of 21% (cohort 1), 34% (cohort 2), and 42% (cohort 3). Cohorts 1, 2, and 3 showed corresponding increases in enzyme activity assessed by PET of 13%, 56%, and 79%, and reductions in antiparkinsonian medication of –15%, –33%, and –42%, respectively, at 6 months. At 12 months, there were dose‐related improvements in clinical outcomes, including increases in patient‐reported ON‐time without troublesome dyskinesia (1.6, 3.3, and 1.5 hours, respectively) and quality of life.
Interpretation
Novel intraoperative monitoring of administration facilitated targeted delivery of VY‐AADC01 in this phase 1 study, which was well tolerated. Increases in enzyme expression and clinical improvements were dose dependent.
ClinicalTrials.gov Identifier: NCT01973543 Ann Neurol 2019;85:704–714
Levodopa is the most effective pharmacological treatment of Parkinson's disease (PD) motor symptoms.1 Clinical benefits of levodopa treatment decline, in part because the enzyme, l‐amino acid decarboxylase (AADC), needed to convert levodopa to dopamine is lost with disease progressesion.2, 3 Dopamine is normally synthesized by nigrostriatal neurons projecting to the putamen and other regions where dopamine is released and binds to its receptors. Dopamine synthesis requires tyrosine hydroxylase to make levodopa, which is converted to dopamine by AADC. Nigrostriatal cells degenerate as PD progresses, and projections to the putamen are largely gone within 5 years of diagnosis,2 leaving reduced levels of AADC and dopamine in the putamen.
As PD progresses, patients require higher doses of levodopa and adjunctive therapies to maintain motor function.4, 5 Motor fluctuations and levodopa‐associated dyskinesias occur in up to 80% of treated PD patients6 and may start within months of initiation.1 Escalating doses of levodopa and other dopaminergic drugs are also associated with nonmotor side effects, including impulse control disorders and psychosis.
Studies in nonhuman primate (NHP) and rodent models of PD have shown that adeno‐associated viral vector serotype‐2 carrying the complementary DNA (cDNA) encoding the enzyme, AADC (AAV2‐AADC), directly administered to the putamen can lead to durable AADC expression primarily in medium spiny neurons of the putamen that do not degenerate in PD,7 bypassing the need for dopamine production in dying nigrostriatal neurons. In the rat 6‐hydroxydopamine model, AAV2‐AADC increased dopamine in microdialysate in response to levodopa, showing that striatal neurons can take up levodopa and then produce and release dopamine.8
Coadministration of a gadolinium contrast agent with an AAV2 vector provides a magnetic resonance imaging (MRI) marker for assessing the location and volume of treated tissue and thus a surrogate marker of the extent of viral transduction.9, 10 Studies in 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐lesioned primates showed that AAV2‐AADC coadministered with gadolinium using real‐time MRI guidance resulted in approximately 35% striatal volumetric coverage and was associated with an enhanced levodopa response and reduced levodopa requirements for a functional motor response lasting at least 8 years.11
Two earlier clinical trials using conventional stereotactic administration of AAV2‐AADC in the putamen showed that gene transfer was well tolerated and could durably increase enzyme levels measured by positron emission tomography (PET) over 5 years.12, 13 These trials used blind stereotactic techniques and low infusion volumes (100 μl/putamen) with limited coverage of the putamen.
This phase 1 dose escalation trial sought to increase putaminal coverage in PD patients with medically refractory motor fluctuations using a novel intraoperative MRI‐guided administration technique to monitor and optimize the delivery of VY‐AADC01 (AAV2‐hAADC) and determine the percentage coverage of the putamen.9 Unlike previous gene therapy trials, which used a fixed volume of 100 μl/putamen,12, 13 we infused larger volumes of up to 900 μl/putamen to increase the percentage of VY‐AADC01‐treated putamen based on subjects’ putaminal anatomy. The primary objective was the safety of the MRI‐guided administration technique. Secondary objectives were assessed AADC activity changes in response to levodopa, clinical outcomes over 12 months, and the durability of those changes after 1 year.
Materials and Methods
Protocol Approval and Patient Consent
The protocol (NCT01973543; PD‐1101) and subject informed consent forms were approved by the Institutional Review Board (IRB) at the University of California, San Francisco (UCSF), the University of Pittsburgh, and the US Food and Drug Administration (FDA); the protocol was also reviewed and approved by the Recombinant DNA Advisory Committee of the National Institutes of Health. The substudy of the effects of intravenous levodopa was approved by the Oregon Health & Science University (OHSU) IRB. A data safety monitoring board reviewed ongoing safety data. All subjects provided written informed consent before trial participation.
Study Procedure
Subjects
Key inclusion criteria included age 40 to 70 years; PD with medically refractory motor fluctuations; modified Hoehn and Yahr Stage ≥2.5 off medication; history and screening examination showing responsiveness to dopaminergic therapy; and optimized and stable antiparkinsonian medication for ≥4 weeks before screening. Key exclusion criteria included atypical parkinsonism; previous stereotactic neurosurgery for PD; unstable psychiatric disorder or other medical problem that would interfere with the safe conduct of the study; or neutralizing antibody titer to AAV2 ≥ 1:1,200 (see www.clinicaltrials.gov for full list of criteria).
Dosing
Cohort 1 received a concentration of 8.3 × 1011vg (vector genomes)/ml and up to 450 μl per putamen for a total dose of up to 7.5 × 1011vg; cohort 2 received the same concentration of 8.3 × 1011vg/mL and up to 900 μl per putamen for a total dose of up to 1.5 × 1012vg; and cohort 3 received a higher concentration of 2.6 × 1012vg/ml with the same volume as cohort 2 of up to 900 μl per putamen for a total dose of up to 4.7 × 1012vg.
VY‐AADC01 Preparation and Administration
VY‐AADC01 consists of recombinant AAV2 capsids carrying the cDNA of the human AADC gene under the control of the cytomegalovirus immediate early promoter. Both vector and excipient for dilution were manufactured in compliance with FDA current good manufacturing practice at the Children's Hospital of Philadelphia. The AAV vector was produced by transient transfection of an adherent HEK293 cell line using three bacteria‐produced plasmids and then purified by a combination of ion exchange chromatograph and ultracentrifugation.
On the morning of administration, subjects were admitted to the neurosurgical service and taken to the MRI suite. Under general anesthesia, subjects were positioned supine in a carbon fiber head holder. Bilateral frontal burr holes were planned, each capable of accommodating two trajectories to the putamen (one anterior and one posterior) that avoided visible blood vessels, ventricles, and large perivascular spaces; the postcommissural motor region of the putamen was preferentially targeted whenever anatomically feasible. Intraoperative navigation and delivery of VY‐AADC01 used skull mounted SmartFrames with the Clearpoint neuro‐navigational system (MRI Interventions, Inc., Irvine, CA). SmartFlow cannulae with a stepped tip design to resist reflux (MRI Interventions, Inc.) were placed in both putamen (usually in the posterior target first) and simultaneous administration of VY‐AADC01 admixed with gadoteridol (1–2 mM) was performed using convection enhanced delivery. Serial MR images (MP‐RAGE [magnetization‐prepared rapid gradient echo]; 3‐minute acquisition time) were acquired to monitor distribution and assess target coverage (Fig 1). The cannulae were removed after completing the initial infusions and reinserted along a second and, in some cases, a third trajectory for additional infusions. After administration was complete, the cannualae and SmartFrames were removed, burr hole covers placed, and the scalp closed. Participants were admitted for at least 1 night of observation.
Figure 1.
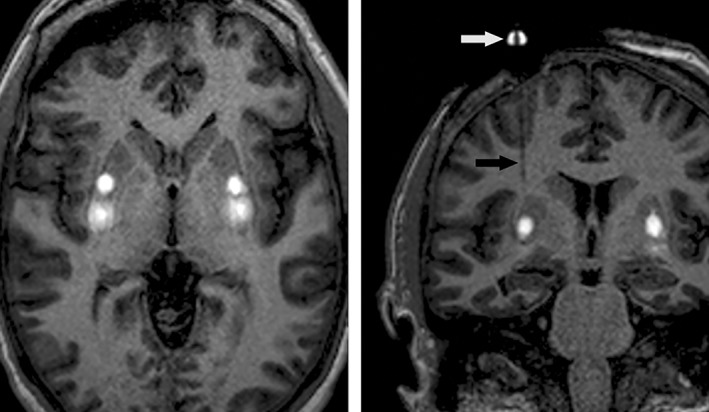
Representative MP‐RAGE images during simultaneous bilateral administration of VY‐AADC01 in the putamen. Axial image (left) shows gadoteridol enhancement as a surrogate marker for vector distribution with two administrations in the posterior portion of each putamen. Coronal oblique image (right) in‐plane with the right infusion cannula during vector delivery; part of the skull mounted SmartFrame aiming device is visible above the burr hole (white arrow), as is the SmartFlow cannula (black arrow). MP‐RAGE = magnetization‐prepared rapid gradient echo.
Putamen Coverage Analysis
Distribution of VY‐AADC01 within the putamen was analyzed using iPlan Flow software (Brainlab AG, Munich, Germany). Putamen segmentation was performed on a preadministration MRI acquisition (VPut) and gadoteridol enhancement volumetrically segmented on end‐of‐administration T1 volume MRI acquisitions (VGad). Volume of VY‐AADC01 distribution within each putamen (Vd) was generated by fusing the MRI images and calculating the intersection of VGad and VPut. VY‐AADC01 distribution across subjects was normalized and presented as percentage putamen coverage.
18F‐Dopa PET Scans
(18)F‐fluoro‐L‐dihydroxyphenylalanine (18F‐dopa) brain PET scans were acquired before and 5 to 6 months after administration of VY‐AADC01. Carbidopa, 2.5 mg/kg (maximum dose, 200 mg), was taken orally 60 to 90 minutes before 18F‐dopa administration. 18F‐dopa (∼3 mCi) was injected as a bolus in an antecubital vein. Acquisition frames captured 65 to 75 minutes after 18F‐dopa administration were analyzed. Data were quantified as previously described,14 yielding the striatum (putamen) to occipital cortex standardized uptake value ratio (SOR‐1) and expressed as percentage change from baseline. The region of interest for the 18F‐dopa analysis was the whole putamen and not limited to the region of the administration.
Clinical Assessments
Subjects were evaluated at baseline and 3, 6, 12, 24, and 36 months post–VY‐AADC01 administration; 18‐ and 30‐month time points were added after study start and are available for some endpoints and subjects. Subjects completed Hauser motor diaries15 for either 2 or 3 consecutive days (after a protocol amendment to lengthen diary duration). The Unified Parkinson's Disease Rating Scale (UPDRS) was measured at baseline and follow‐up visits in the functionally defined off state, 12 or more hours after the last dose of dopaminergic medication(s), and in the fully on state as judged by the patient and investigator 1 hour following the first morning dose; an additional one‐half to one carbidopa/levodopa 25/100 tablet was administered if the subject was not fully “on” as judged by the subject and investigator. Quality of life (QoL) was assessed using the 39‐item PD Questionnaire (PDQ‐39) at baseline and every 6 months.
Changes in antiparkinsonian medications were allowed following the administration of VY‐AADC01 in response to increased dyskinesia or reduced need for medication based on investigator judgement. Antiparkinsonian medications were standardized to a levodopa equivalent dose (LED).16, 17
Thirteen of the 15 subjects travelled to an independent site (OHSU) both before and approximately 6 months after VY‐AADC01 administration. On sequential days, threshold (0.6 mg/kg/h) or suprathreshold (1.2 mg/kg/h) doses of intravenous levodopa were administered for 2 hours; dosing order was randomized and blinded. Oral carbidopa (25 mg) was administered 1 hour before, during, and 1 hour after the infusion. UPDSR‐III and other clinical and laboratory measures were assessed every 30 minutes for up to 6.5 hours as previously described.18
Statistical Analysis
Observed data and changes from baseline are reported as mean ± standard error of the mean. Analysis of motor diaries was normalized to a 16‐hour waking day. For the intravenous levodopa substudy, area under the curve (AUC) analysis of UPDRS‐III measures were calculated.18
Results
Baseline Characteristics
Three dosing cohorts of 5 subjects each were enrolled and received VY‐AADC01. Of 17 patients screened, 1 was excluded because of elevated anti‐AAV2 antibody titer and 1 because of Hoehn and Yahr score below the inclusion criterion. Demographic and baseline characteristics (Table 1 ) were similar across cohorts except for the UPDRS‐III on medication, which was lower in cohort 1, and dyskinesia (by the Unified Dyskinesia Rating Scale), which was higher in cohort 3. Efficacy and safety data are reported through August 8, 2018, when efficacy data for cohort 1 were available through 36 months, cohort 2 through 24 months, and cohort 3 through 18 months.
Table 1.
Demographic and Baseline Characteristics
| Cohort 1 7.5 × 1011vg (N = 5) | Cohort 2 1.5 × 1012vg (N = 5) | Cohort 3 4.7 × 1012vg (N = 5) | Total (N = 15) | |
|---|---|---|---|---|
| Age (years) | 57.4 (3.2) | 58.4 (3.9) | 57.4 (2.0) | 57.7 (1.7) |
| Sex | 1 female 4 males |
5 males | 1 female 4 males |
2 females 13 males |
| Duration of PD (years) | 9.9 (2.1) | 10.1 (0.7) | 8.5 (1.6) | 9.5 (0.9) |
| UPDRS‐II off medication | 13.6 (0.9) | 16.0 (0.8) | 19.8 (3.5) | 16.5 (1.3) |
| UPDRS‐II on medication | 3.0 (1.3) | 3.6 (0.7) | 5.0 (1.7) | 3.9 (0.7) |
| UPDRS‐III off medication | 37.2 (2.6) | 35.8 (3.4) | 38.2 (4.3) | 37.1 (1.9) |
| UPDRS‐III on medication | 7.6 (2.3) | 17.0 (1.7) | 16.0 (1.4) | 13.5 (1.5) |
| Hauser diary OFF‐time (normalized hours) | 4.9 (0.8) | 4.2 (0.6) | 4.7 (0.5) | 4.6 (0.4) |
| Hauser diary ON‐time (normalized hours) | 10.5 (1.0) | 10.7 (0.8) | 10.3 (0.7) | 10.5 (0.4) |
| Modified H&Y stage | Stage 3: 5 | Stage 3: 5 | Stage 3: 3 Stage 4: 2 | Stage 3: 13 Stage 4: 2 |
| UDysRS Total Score | 19.2 (6.0) | 17.4 (5.6) | 30.2 (3.9) | 22.3 (3.2) |
| LED | 1,467.5 (275.0) | 1,635.5 (307.4) | 1,476.5 (191.9) | 1,526.5 (141.9) |
Data are mean (standard error). Hauser diary data are normalized to a 16‐hour day.
H&Y = Hoehn and Yahr; LED = levodopa equivalent dose; PD = Parkinson's disease; UDyRS = Unified Dyskinesia Rating Scale; UPDRS = Unified Parkinson's Disease Rating Scale; vg = vector genomes.
Safety
The surgical procedure was generally well tolerated; 14 subjects returned home within 2 days of surgery. One participant experienced a deep venous thrombosis (DVT), pulmonary embolus, and subsequent atrial fibrillation, likely attributed to immobility during the surgery. These serious adverse events (SAE) resolved with anticoagulation and a brief course of an antiarrhythmic. DVT prophylaxis with MRI‐compatible sequential compression devices was added to the surgical protocol, and no subsequent events were observed. No vector‐related SAE have been reported. The most common adverse events (Table 2) were expected and temporary. Transient increase in dyskinesias, which resolved with dose reductions of PD medications and/or addition of amantadine, was reported as an adverse event (AE) in 4 subjects.
Table 2.
Subjects Experiencing Treatment Emergent Serious Adverse Events and Adverse Events Occuring in >1 Subject In Any Cohort by MedRA System Organ Class
| Cohort 1 7·5 × 1011vg (N = 5) | Cohort 2 1·5 × 1012vg (N = 5) | Cohort 3 4·7 × 1012vg (N = 5) | Total (N = 15) | |
|---|---|---|---|---|
| Any adverse event | 5 | 5 | 5 | 15 |
| Any serious averse event | 0 | 1a | 0 | 1 |
| Gastrointestinal disorders | ||||
| Constipation | 1 | 2 | 0 | 3 |
| Vomiting | 1 | 1 | 0 | 2 |
| Infections and infestations | ||||
| Tooth abcess | 2 | 0 | 0 | 2 |
| Upper respiratory tract infection | 2 | 0 | 1 | 3 |
| Metabolism and nutrition disorders | ||||
| Vitamin B12 deficiency | 0 | 2 | 0 | 2 |
| Musculoskeletal and connective tissue disorders | ||||
| Back pain | 2 | 2 | 0 | 4 |
| Musculoskeletal pain | 3 | 0 | 1 | 4 |
| Nervous system disorders | ||||
| Dizziness | 2 | 0 | 0 | 2 |
| Dyskinesia | 2 | 1 | 1 | 4 |
| Headache | 5 | 3 | 3 | 11 |
| Hypoaesthesia | 3 | 1 | 3 | 7 |
| Memory impairment | 2 | 0 | 0 | 2 |
| Neuralgia | 2 | 0 | 0 | 2 |
| Psychiatric disorders | ||||
| Depression | 2 | 1 | 0 | 3 |
| Vascular disorders | ||||
| Orthostatic hypotension | 2 | 0 | 0 | 2 |
One participant experienced a deep venous thrombosis, pulmonary embolus, and subsequent atrial fibrillation, likely attributed to immobility during the surgery.
MedRA = Medical Dictionary for Regulatory Activities; vg = vector genomes.
Putamen Coverage, AADC Activity, and PD Medications
Intraoperative MRI was used to visualize the distribution of gadoteridol (a surrogate for VY‐AADC01) to monitor delivery (Fig 1). A third cannula placement was used in 4 subjects (two bilateral) to maximize putamen coverage. Postoperatively, end‐of‐administration MR images were evaluated to calculate volumetric coverage of the putamen, which showed 20.7 ± 1.4%, 33.5 ± 2.4%, and 42.3 ± 2.5% coverage in cohorts 1, 2, and 3, respectively.
VY‐AADC01 dose‐related increases were observed in 18F‐dopa PET SOR‐1 assessment of AADC activity with corresponding decreases in LED. At 6 months, 18F‐dopa uptake increased by 13.2 ± 6.6%, 56.1 ± 13.3%, and 79.3 ± 14.7% in cohorts 1, 2, and 3, respectively (Fig 2). Cohort‐related increases in 18F‐dopa signal correlated with VY‐AADC01 (gadoteridol) coverage (r = 0.84; p = 0.0002), suggesting that anatomical coverage of the putamen correlates with the observed increase in AADC activity.
Figure 2.
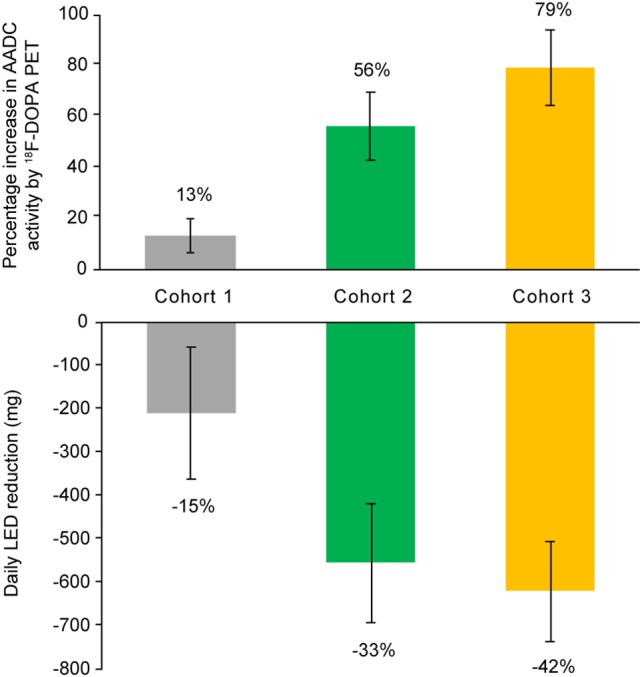
Percentage increase from baseline in 18F‐dopa PET signal of AADC activity and corresponding reductions in levodopa equivalent dose at 6 months. The top of the figure shows the increase in 18F‐dopa PET assessment of AADC activity by cohort with increasing administration volume in cohort 2 and the same volume with an increase in genome concentration in cohort 3. The bottom of the figure shows corresponding reductions in levodopa equivalent dosing. LED = levodopa equivalent dose; PET = positron emission tomography.
Clinical Outcomes
UPDRS scores recorded in the off‐ and on‐medication state at 6, 12, 24, and 36 months showed dose‐dependent improvements (Fig 3). In the on‐medication state at 12 months, UPDRS‐III scores were improved (reduced) by –9.6 ± 1.6 for cohort 2 and –6.8 ± 1.9 for cohort 3. Cohort 1 had a baseline score of 7.6 ± 2.3, which is close to the floor of measurement, and showed a 1.8 ± 2.7 point worsening (increase) post–VY‐AADC01 administration (Fig 3A). In the practically defined off‐medication state, improvement in UPDRS‐III was noted for all cohorts, with mean reductions of –16.4 ± 4.2, –17.0 ± 3.0, and –12.0 ± 2.4 in cohorts 1, 2, and 3, respectively (Fig. 3B). In cohort 1, which had the longest follow‐up, this improvement was sustained through 36 months.
Figure 3.
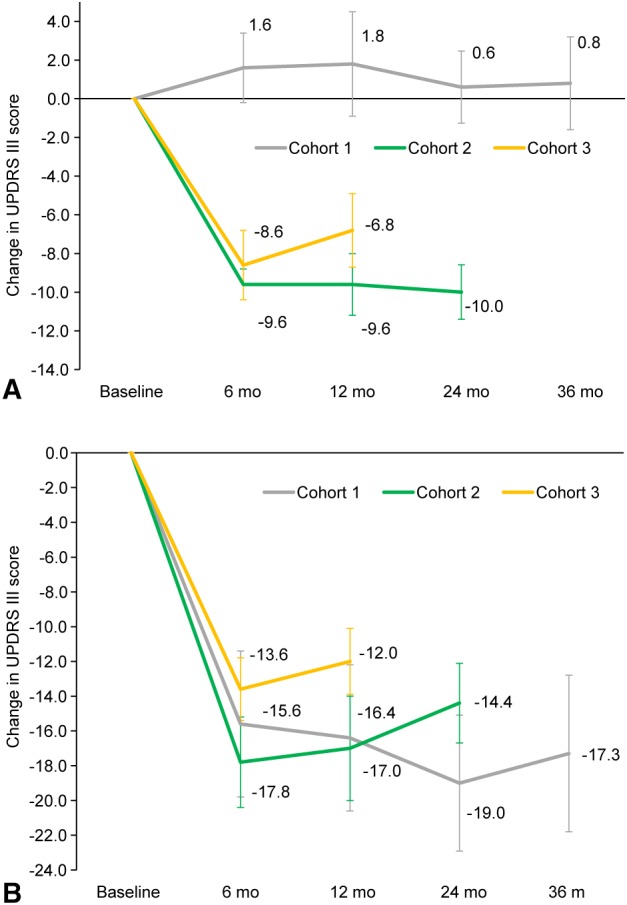
Mean (±SE) change from baseline in UPDRS‐III assessed in the on‐ (A) and off‐medication (B) states. Change in UPDRS‐III scores in the on‐ (A) and off‐medication (B) states. Mean score reductions (improvements) were observed in cohorts 2 and 3 on medication and all three cohorts off medication through last follow‐up. Note that UPDRS data were not collected at month 18 for cohorts 1 and 2. UPDRS = Unified Parkinson's Disease Rating Scale.
In order to measure the time couse of response to standardized levodopa treatments, the UPDRS‐III was performed at fixed 30‐minute intervals before and after an intravenous infusion of “threshold” and “superathreshold” doses of levodopa at an independent site before and 6 months after VY‐AADC01 administration (Fig 4). Across cohorts, the AUC for UPDRS‐III in response to intravenous levodopa showed increases of 168% (threshold levodopa dose) and 67% (suprathreshold levodopa dose) from pre‐ to 6‐months post‐VY‐AADC01 administration.
Figure 4.
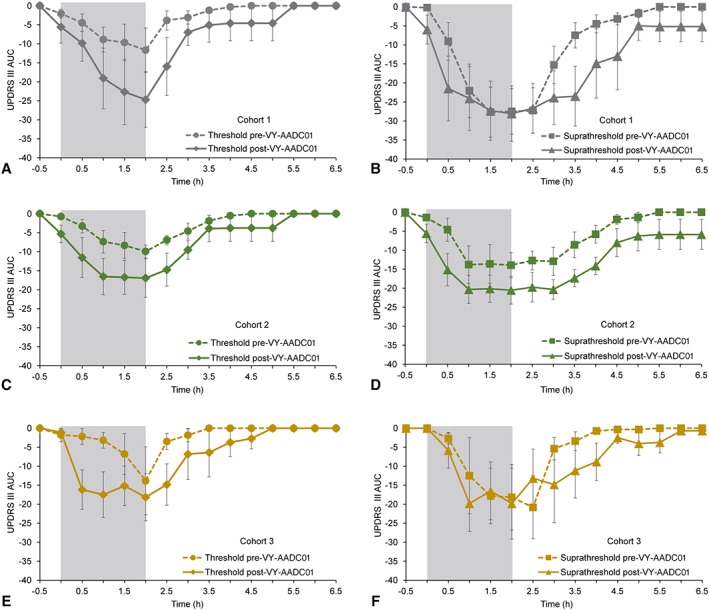
Time‐action curve for UPDRS‐III for threshold and suprathreshold infusions of intravenous levodopa at baseline and approximately 6 months after administration of VY‐AADC01. The figure shows the UPDRS‐III time‐action curves following a threshold (0.6 mg/kg/h; left side) and suprathreshold (1.2 mg/kg/h; right side) intravenous dose of levodopa by cohort (A,B: cohort 1; C,D: cohort 2; E,F: cohort 3). Lower scores indicate improvement. Dashed lines in each figure show data before VY‐AADC01 administration and solid lines postadministration. The gray box shows the actual infusion time. Note in cohort 2 the time‐action curve for threshold (low‐dose) intravenous levodopa post‐VY‐AADC01 administration (C) appears similar to the suprathreshold (high‐dose) intravenous levodopa pre‐VY‐AADC01 administration (D). AUC = area under the curve; UPDRS = Unified Parkinson's Disease Rating Scale.
Patient reported diary data also showed dose‐related outcomes. In each cohort, we observed decreases in OFF time, increases in ON time without troublesome dyskinesia (the sum of ON time without dyskinesia and ON time with nontroublesome dyskinesia), and decreases in ON time with troublesome dyskinesia (Fig 5). At 12 months, cohorts 1, 2, and 3 had increases in ON time without troublesome dyskinesia of 1.6 ± 0.4, 3.3 ± 0.6, and 1.5 ± 0.5 hours, respectively. These results were stable to improved with additional follow‐up. At 36 months, cohort 1 had a 2.1 ± 0.6 hour increase from baseline in ON time without troublesome dyskinesia; at 24 months, cohort 2 had a 2.7 ± 1.4 hour increase; and at 18 months, cohort 3 had a 1.7 ± 1.1 increase.
Figure 5.
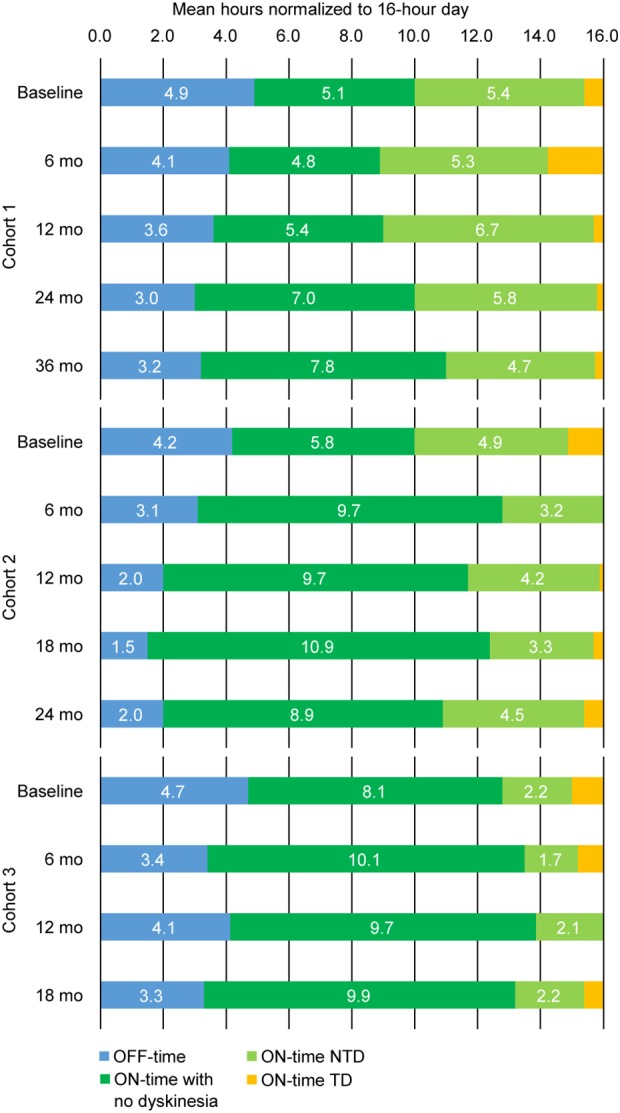
Mean diary ON and OFF time per 16‐hour waking day. Across cohorts, good quality ON time (green bars; ON time with no dyskinesia plus ON time with nontroublesome dyskinesia) increased over time, and both OFF time and ON time with troublesome dyskinesia decreased over time. NTD = nontroublesome dyskinesia; TD = troublesome dyskinesia.
The improvements in diary ON time occurred despite reductions in dopaminergic treatments and mirrored the increase in AADC activity (Fig 2). LED dose reductions were in response to improved clinical efficacy and, in some cases, increased dyskinesia. The cohort‐dependent mean LED reductions were –15 ± 10%, –33% ± 3.4%, and –42 ± 7.0%, respectively, at 6 months. LED reductions were maintained through last follow‐up in cohorts 2 and 3, but not in cohort 1 (Table 3). QoL was stable or improved across cohorts. For cohort 1, PDQ‐39 scores changed minimally from baseline (–1.9 ± 2.5 at 12 months, −0.8 ± 2.1 at 24 months, and –0.9 ± 4.6 at 36 months). For cohorts 2 and 3 at 12 months, PDQ‐39 scores improved by –8.4 ± 2.9 and –9.1 ± 3.5, respectively, exceeding the minimum clinically important difference of −4.7.19 QoL was somewhat decreased at 24 months for cohort 2 (3.9 ± 1.9); data for cohort 3 were not collected at 18 months.
Table 3.
Change From Baseline in Levodopa Equivalent Dose (LED) by Cohort
| Cohort 1 7.5 × 1011vg (N = 5) | Cohort 2 1.5 × 1012vg (N = 5) | Cohort 3 4.7 × 1012vg (N = 5) | |
|---|---|---|---|
| Baseline | 1,467.5 (275.0) | 1,635.5 (307.4) | 1,476.5 (191.9) |
| 6 Months | –208.0 (151.4) | –553.2 (137.0) | –617.8 (115.0) |
| 12 Months | –92.5 (191.7) | –627.2 (246.3) | –606.0 (46.7) |
| 18 Months | –176.5 (129.9) | –465.3 (246.0) | –614.1 (87.3) |
| 24 Months | 45.5 (158.8) | –451.9 (249.2) | |
| 36 Months | 347.5 (404.0) |
Data are mean (SE).
LED rose above baseline in cohort 1 at 36 months. This may have been attributed to a single subject taking a supratherapeutic dose (4,500 mg/day) of levodopa or the lower coverage and AADC expression observed in this cohort.
vg = vector genomes.
Discussion
Results from this open‐label trial show that large‐volume administrations of VY‐AADC01 were well tolerated. The novel use of intraoperative MRI‐guided delivery of the vector allowed improved volumetric coverage of the putamen, which, in turn, was closely correlated with increases in AADC activity, as measured by 18F‐dopa PET. Moreover, there were dose‐related improvements in motor fluctuations, UPDRS‐III scores, and QoL measures, despite reductions in dopaminergic medications.
AAV2 vectors have now been used with direct intracranial delivery in over 150 people with PD,20 with no vector‐related serious adverse events reported to date. The most common vector‐related AE in the current trial was transient dyskinesia. Increased dyskinesia generally occurred around 1 month after surgery, when expression of putaminal AADC is expected to reach a steady state, and is consistent with the mechanism of action of this therapy. Dyskinesia responded to reductions in antiparkinsonian medications, and diary data showed a trend toward improvements in troublesome and nontroublesome dyskinesia (Fig 5). There were no AEs attributed to excessive or off‐target dopaminergic stimulation such as impulse control disorders or psychosis. Volumes of administration up to 900 μl per putamen were well tolerated and represent a 9‐fold increase in volume compared to earlier AADC gene therapy trials.12, 13 The only perioperative SAE was a thromboembolic event likely related to immobility during surgery.
In contrast to earlier trials which used smaller volumes and did not use real‐time imaging, we achieved greater coverage of the putamen, as measured by gadoteridol (a gadolinium‐based contrast agent) distribution on intraoperative MRI, which allowed monitoring and optimization of the administration in a manner personalized to each subject's individual putaminal anatomy. This strategy provided a significant advantage over traditional stereotactic techniques used in earlier gene therapy trials,20 and allowed for modification of administration parameters to enhance putamen coverage based on real‐time and post‐hoc evaluations of intraparenchymal distribution resulting in changes in administration volume and technique. This is the first gene‐therapy trial reported to use such a patient‐specific administration strategy.
18F‐dopa PET reflects functional AADC enzyme activity in the medium spiny neurons and the capacity to covert 18F‐dopa to 18F‐dopamine. The anatomical coverage of the putamen achieved during VY‐AADC01 administration was highly correlated with the change in AADC activity assessed by 18F‐dopa PET (r = 0.84; p = 0.0002).
Reductions in dopaminergic medications were generally made from 1 to 6 months after VY‐AADC01 administration in response to participant report of increased dyskinesia, observation of increased dyskinesia by the study neurologist, or participant reported decreased need for medication. The marked reductions in LED in cohorts 2 and 3 did not occur in cohort 1, nor in the two earlier AADC gene therapy trials,20 likely because of lower volumetric coverage of the putamen by the vector. Together with the 18F‐dopa PET data, and the findings from the intravenous levodopa substudy, these LED reductions suggest an increased ability to convert levodopa to dopamine, and a reduced need for dopaminergic medications that is consistent with the expected pharmacological effect of AADC gene therapy.11, 21
We found that the response at 12 months measured by UPDRS‐III in the on‐medication state improved by a clinically meaningful22 –9.6 and –6.8 points in cohorts 2 and 3, respectively. Cohort 1 showed a 1.8‐point worsening at 12 months. This group had a low UPDRS‐III on medication at baseline (Table 1), which may have limited the ability to detect improvement.
We assume that the administration of the AADC gene to the putamen would increase striatal AADC enzyme activity and therefore augment the effects of administered levodopa, with a similar result to administering a larger dose of levodopa. The effects of larger intravenous doses of levodopa are to shorten the latency to onset and prolong the duration of response. Larger intravenous levodopa doses do not increase the magnitude of the clinical response; the response is essentially all or none.23 In the intravenous levodopa substudy, 6 months after AADC gene therapy, the threshold (Fig 4, left panels) and suprathreshold (Fig 4, right panels) infusions produced a more rapid improvement in UPDRS‐III scores. Before AADC administration, threshold infusions produced no, incomplete, or brief responses to the infusions, which, when plotted as mean responses, showed a delayed and intermediate improvement. Thus, there appears to be a substantial difference in the magnitude of effect of the levodopa infusions after AADC administration when all subjects responded fully to the threshold infusions. This is what accounts for the apparently more robust effect of AADC gene therapy on threshold infusions.
We also observed complementary large improvements in off medication UPDRS‐III scores that were not dose related, as was observed in earlier AAV‐AADC studies.12, 13 Although it is mechanistically less clear why UPDRS‐III off medication scores would also respond to AADC gene therapy, as do on medication scores, it is possible that AADC treatment allows for improved conversion of endogenous levodopa in remaining dopaminergic nigrostriatal axons, as has been observed in NHPs.24 It is also possible that AADC treatment increases the long‐duration response of levodopa, perhaps by allowing more efficient and uniform conversion to dopamine in the putamen.1
Consistent with the improved motor function measured by UPDRS‐III, patient‐reported motor diary ON and OFF time also improved over time (Fig 5). The gains in ON time without troublesome dyskinesia stem from reductions in both OFF time and ON time with troublesome dyskinesia. The ON time with nontroublesome dyskinesia was also reduced, which further demonstrates an overall increase of good‐quality ON time with the renewed availability of AADC to convert levodopa to dopamine. The pattern of response observed in the current trial may result from increased dopamine production alone or in combination with reduced doses of dopaminergic drugs to offset dyskinesia. At 12 months, cohort 2 had clinically meaningful22 increases in ON time of nearly 5 hours and reduced troublesome dyskinesia compared to baseline. More modest gains were noted in cohort 3, possibly attributed to the greater dyskinesia at baseline (Table 1) and more marked reductions in dopaminergic medications.
In general, participant‐reported improvement in motor diaries showed improvements (longer ON time and shorter OFF time) at 6 months, which increased at the later timepoints (Fig 5). Similar findings have been noted in MPTP‐lesioned NHPs that received AADC gene transfer, where there was progressive improvement in the response to levodopa that plateaued after 12 months.25
In preclinical studies, optimal motor control after AADC gene transfer occurred at approximately one‐tenth the dose of intramuscular levodopa.24 In clinical practice, however, low doses of levodopa may lead to inconsistent absorption and variable motor control. Cohort 3 of this trial received the highest dose of VY‐AADC01 and showed greater increases in 18F‐dopa PET and larger reductions in dopaminergic medications than cohorts 1 and 2. The average LED reduction in cohort 3 was more than 600 mg (Table 3). The marked reductions in dopaminergic medications in cohort 3 reduced dyskinesia, but may have led to lesser improvements in OFF time than cohort 2.
The stable to improved motor outcomes observed over time are encouraging for the potential durability of the treatment effect. Cohort 1 diary data showed continued improvement out to 36 months, which may reflect underlying motor circuit plasticity in response to increased dopamine production,6 or continued increases in AADC expression. Earlier clinical trials with more limited vector distribution showed stable increases in AADC activity assessed by FMT‐PET (6‐[18F]Fluoro‐l‐m‐tyrosine PET) for up to 4 years after administration.26 NHP data have shown stable expression for up to 15 years.27
Open‐label data in PD trials must be interpreted with caution given that randomized, blinded, placebo‐controlled trials have often failed to replicate findings from open‐label trials. Even so, there are key differences between this and other open‐label gene therapy trials in PD. First, intraoperative MRI monitoring was used to visualize coverage of the putamen to ensure delivery of a dose believed to be sufficient for therapeutic efficacy based on preclinical models. Second, there is objective imaging evidence of dose‐related increases in AADC activity. Third, across different endpoints and independent ratings (blinded intravenous levodopa response), there is evidence for both dose‐ and time‐related treatment effects that are consistent with preclinical findings and are distinct from placebo effects, which often peak shortly after treatment.28
Based on these open‐label results, a randomized trial using a placebo‐surgery control is underway to further understand the safety and efficacy of VY‐AADC. The recently initiated phase 2 study (NCT03562494) randomizes subjects to either continued optimized medical management, including levodopa, or optimized medical management plus VY‐AADC. The results of this new study will allow us to both better understand the safety and efficacy of VY‐AADC and to understand its efficacy relative to optimal medical management alone.
Author Contributions
C.W.C., K.S.B., J.N., B.R., A.P.K., and P.S.L. contributed to the concept and design of the study. C.W.C., K.S.B., A.D.V.L., R.M.R., B.R., A.P.K., B.B., A.J.M., J.N., M.E.T., and P.S.L. contributed to the acquisition and analysis of data. C.W.C., K.S.B., A.D.V.L., R.M.R., B.R., A.P.K., B.B., J.N., and P.S.L. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
The authors report the following potential conflicts of interest during the conduct of this study: C.W.C. and J.N. received grants from Voyager Therapeutics, Inc.; K.S.B. received grants and personal fees from Voyager Therapeutics and holds a patent on the use of AAV2‐AADC for the treatment of Parkinson's disease licensed to Genzyme and Voyager Therapeutics, and a patent on the step cannula for gene delivery with royalties paid by MRI Interventions to UCSF; A.D.V.L., R.M.R., and A.J.M. received grants from Voyager Therapeutics; B.R. was an employee of Voyager Therapeutics and has a patent on AADC gene transfer in Parkinson's disease pending; A.P.K. was an employee of Voyager Therapeutics; B.B. was an employee of Voyager Therapeutics; M.E.T. received grants and nonfinancial support from Voyager Therapeutics; and P.S.L. received grants from Voyager Therapeutics and nonfinancial support from MRI Interventions, the company that makes the surgical delivery platform and step cannula used for gene delivery.
Acknowledgment
The Michael J. Fox Foundation (for Parkinson's research) and Sponsor provided funding for the study, data collection, and analysis.
We thank Alexander Sedkof, MS, of Voyager Therapeutics, Inc. for statistical support and Marc de Somer, MD, and Elisabeth Fine, PhD, both of Voyager Therapeutics, for their careful review of the manuscript. We are grateful to the people with PD and their families who participated in this study, and the study support teams at UCSF, University of Pittsburgh Medical Center, and OHSU.
References
- 1. Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson's disease. N Engl J Med 2004;351:2498–2508. [DOI] [PubMed] [Google Scholar]
- 2. Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 2013;136(pt 8):2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ciesielska A, Samaranch L, San Sebastian W, et al. Depletion of AADC activity in caudate nucleus and putamen of Parkinson's disease patients: implications for ongoing AAV2‐AADC gene therapy trial. PLoS One 2017;12:e0169965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chan PL, Nutt JG, Holford NH. Pharmacokinetic and pharmacodynamic changes during the first four years of levodopa treatment in Parkinson's disease. J Pharmacokinet Pharmacodyn 2005;32:459–484. [DOI] [PubMed] [Google Scholar]
- 5. Gray R, Ives N, Rick C, et al. Long‐term effectiveness of dopamine agonists and monoamine oxidase B inhibitors compared with levodopa as initial treatment for Parkinson's disease (PD MED): a large, open‐label, pragmatic randomised trial. Lancet 2014;384:1196–1205. [DOI] [PubMed] [Google Scholar]
- 6. Voon V, Napier TC, Frank MJ, et al. Impulse control disorders and levodopa‐induced dyskinesias in Parkinson's disease: an update. Lancet Neurol 2017;16:238–250. [DOI] [PubMed] [Google Scholar]
- 7. Bankiewicz KS, Eberling JL, Kohutnicka M, et al. Convection‐enhanced delivery of AAV vector in parkinsonian monkeys: in vivo detection of gene expression and restoration of dopaminergic function using pro‐drug approach. Exp Neurol 2000;164:2–14. [DOI] [PubMed] [Google Scholar]
- 8. Sánchez‐Pernaute R, Harvey‐White J, Cunningham J, Bankiewicz KS. Functional effect of adeno‐associated virus mediated gene transfer of aromatic L‐amino acid decarboxylase into the striatum of 6‐OHDA‐lesioned rats. Mol Ther 2001;4:324–330. [DOI] [PubMed] [Google Scholar]
- 9. Su X, Kells AP, Salegio EA, et al. Real‐time MR imaging with Gadoteridol predicts distribution of transgenes after convection‐enhanced delivery of AAV2 vectors. Mol Ther 2010;18:1490–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.10.Richardson RM, Kells AP, Martin AJ, et al. Novel platform for MRI‐guided convection‐enhanced delivery of therapeutics: preclinical validation in nonhuman primate brain. Stereotact Funct Neurosurg 2011;89:141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hadaczek P, Eberling JL, Pivirotto P, et al. Eight years of clinical improvement in MPTP‐lesioned primates after gene therapy with AAV2‐hAADC. Mol Ther 2010;18:1458–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christine CW, Starr PA, Larson PS, et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 2009;73:1662–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Muramatsu S, Fujimoto K, Kato S, et al. A phase I study of aromatic L‐amino acid decarboxylase gene therapy for Parkinson's disease. Mol Ther 2010;18:1731–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dhawan V, Ma Y, Pillai V, et al. Comparative analysis of striatal FDOPA uptake in Parkinson's disease: ratio method versus graphical approach. J Nucl Med 2002;43:1324–1330. [PubMed] [Google Scholar]
- 15. Hauser RA, Friedlander J, Zesiewicz TA, et al. A home diary to assess functional status in patients with Parkinson's disease with motor fluctuations and dyskinesia. Clin Neuropharmacol 2000;23:75–81. [DOI] [PubMed] [Google Scholar]
- 16. Tomlinson CL, Stowe R, Patel S, et al. Systematic review of levodopa dose equivalency reporting in Parkinson's disease. Mov Disord 2010;25:2649–2653. [DOI] [PubMed] [Google Scholar]
- 17. Espay AJ, Pagan FL, Walter BL, et al. Optimizing extended‐release carbidopa/levodopa in Parkinson disease: consensus on conversion from standard therapy. Neurol Clin Pract 2017;7:86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brodsky MA, Park BS, Nutt JG. Effects of a dopamine agonist on the pharmacodynamics of levodopa in Parkinson disease. Arch Neurol 2010;67:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horváth K, Aschermann Z, Kovács M, et al. Changes in quality of life in Parkinson's disease: how large must they be to be relevant? Neuroepidemiology 2017;48:1–8. [DOI] [PubMed] [Google Scholar]
- 20. Bartus RT, Weinberg MS, Samulski RJ. Parkinson's disease gene therapy: success by design meets failure by efficacy. Mol Ther 2014;22:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Forsayeth J, Bankiewicz KS. Transduction of antigen‐presenting cells in the brain by AAV9 warrants caution in preclinical studies. Mol Ther 2015;23:612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shulman LM, Gruber‐Baldini AL, Anderson KE, et al. The clinically important difference on the Unified Parkinson's Disease Rating Scale. Arch Neurol 2010;67:64–70. [DOI] [PubMed] [Google Scholar]
- 23. Nutt JG. On‐off phenomenon: relation to levodopa pharmacokinetics and pharmacodynamics. Ann Neurol 1987;22:535–540. [DOI] [PubMed] [Google Scholar]
- 24. Forsayeth JR, Eberling JL, Sanftner LM, et al. A dose‐ranging study of AAV‐hAADC therapy in Parkinsonian monkeys. Mol Ther 2006;14:571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bankiewicz KS, Forsayeth J, Eberling JL, et al. Long‐term clinical improvement in MPTP‐lesioned primates after gene therapy with AAV‐hAADC. Mol Ther 2006;14:564–570. [DOI] [PubMed] [Google Scholar]
- 26. Mittermeyer G, Christine CW, Rosenbluth KH, et al. Long‐term evaluation of a phase 1 study of AADC gene therapy for Parkinson's disease. Hum Gene Ther 2012;23:377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sehara Y, Fujimoto KI, Ikeguchi K, et al. Persistent expression of dopamine‐synthesizing enzymes 15 years after gene transfer in a primate model of Parkinson's disease. Hum Gene Ther Clin Dev 2017;28:74–79. [DOI] [PubMed] [Google Scholar]
- 28. Lidstone SC. Great expectations: the placebo effect in Parkinson's disease. Handb Exp Pharmacol 2014;225:139–147. [DOI] [PubMed] [Google Scholar]