

Abstract
Mesityl gold(I) carbenes lacking heteroatom stabilization or shielding ancillary ligands have been generated and spectroscopically characterized from chloro(mesityl)methylgold(I) carbenoids bearing JohnPhos‐type ligands by chloride abstraction with GaCl3. The aryl carbenes react with PPh3 and alkenes to give stable phosphonium ylides and cyclopropanes, respectively. Oxidation with pyridine N‐oxide and intermolecular C−H insertion to cyclohexane have also been observed. In the absence of nucleophiles, a bimolecular reaction, similar to that observed for other metal carbenes, leads to a symmetrical alkene.
Keywords: carbenes, carbenoids, density-functional calculations, gold, structure elucidation
Gold(I) carbenes have attracted great interest from the outset of the homogeneous gold‐catalysis era1 and have been the center of debate regarding their structures and, in particular, the degree of stabilization provided by the π‐backdonation from gold(I) to the carbon center.2, 3 Indeed, recent studies on well‐characterized gold(I) carbene complexes have shown that these species can be considered anything from metal‐stabilized carbocations to metal carbenes depending on the nature of the ancillary donating ligand(s) on gold(I).3, 4, 5, 6, 7 However, these stabilized complexes require the presence of either heteroatoms4 or highly conjugated systems5 at the carbene moiety and shielding ancillary ligands6 or bidentate phosphines7 at the metal center and, with very few exceptions,8 do not display the reactivity proposed for the intermediate species involved in catalysis.
Metal carbenoids are species in which the metal is bound to an sp3‐carbon center bearing a good leaving group that can generate a metal carbene upon ionization (Scheme 1 a).3a, 9 Based on this relationship, we have recently developed a method for the preparation of chloromethylgold(I) carbenoids, which, after chloride abstraction, display typical reactivities of gold(I) carbenes in solution.10 During the course of our investigation, preliminary results suggested that [JohnPhosAuCHPhCl] (1), after chloride abstraction, generates stilbene (2 a), which undergoes cyclopropanation by an undetected phenyl gold(I) carbene intermediate (Scheme 1 b).9 The observed cyclopropanation closely resembles the reactivity displayed by aryl gold(I) carbenes (5) generated by a gold(I)‐catalyzed retro‐Buchner reaction of cycloheptatrienes (4; Scheme 1 c).11 Although these arylidene complexes (5) have never been observed in solution, such species have been detected in the gas phase.12, 13
Scheme 1.
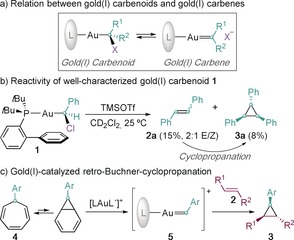
General concepts and reactivity of [JohnPhosAuCHPhCl] (1) carbenoid. Tf=trifluoromethanesulfonyl, TMS=trimethylsilyl.
Herein, we report the generation and spectroscopic characterization of mesityl gold(I) carbenes from stable carbenoids (1 a–e) bearing JohnPhos‐type ligands. These gold(I) carbenes display the same reactivity towards alkenes as the intermediates of the retro‐Buchner reaction and many other gold(I)‐catalyzed transformations.10 Importantly, the gold(I) carbenes undergo bimolecular homocoupling to form the corresponding symmetrical alkenes by a process reminiscent to that shown by electrophilic and nucleophilic metal carbenes.
Gold(I) carbenoids (1 a–e) were obtained by reaction of the complexes 6 a–e with mesityl diazomethane, and their structures were confirmed by X‐ray diffraction (Scheme 2).14 Remarkably, upon addition of GaCl3 to 1 a in CD2Cl2 at −90 °C, the solution turned deep red, suggesting the formation of the mesityl gold(I) carbene 5 a, which was characterized by NMR techniques (Table 1). Although gold(I) carbenoids bearing JohnPhos and different substitution patterns in the aryl group were tested under the same reaction conditions, the generation of gold(I) carbenes could not be detected by NMR spectroscopy in those cases.15 Although 5 a partially decomposed at −90 °C, the carbenes 5 b–e were stable until −70 °C. These are the first monosubstituted gold(I) carbenes that have been characterized spectroscopically. The complexes 5 a–e present a characteristic signal in 1H NMR spectroscopy at δ=11.74–12.67 ppm corresponding to H1 (Scheme 3, Table 1). The carbenic carbon centers (C1) resonate at δ=284.6–290.0 ppm as a doublet [2 J (13C‐31P)=96.8–99.8 Hz], within the range of previously characterized gold(I) carbenes (δ=225–321 ppm).4, 5, 6, 7 In addition, clear correlations between the carbenic carbon (C1) and proton (H1) were observed in the 1H‐13C HSQC spectra.16 DFT calculations for 1 b′–c′ and 5 b′–c′ complexes at the B3LYP‐D3/6‐31G(d,p) + SDD(Au) level of theory led to computed 1H and 13C NMR chemical shifts for H1, C1, and 1 J(1H1‐13C1) constants consistent with the experimental results (Table 1). Furthermore, 5 a–e were detected in the gas phase by ESI‐MS from 1 a–e.
Scheme 2.
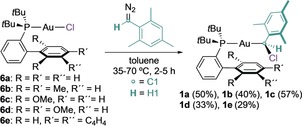
Synthesis of the chloro(mesityl)methylgold(I) carbenoids 1 a–e.
Table 1.
Selected measured and calculated NMR data.[a]
| Complex | Method | H1 (ppm) | C1 (ppm) | 1 J (1H1‐13C1) [Hz] |
|---|---|---|---|---|
| 5 a | Exp[b] | 12.35 (4.92) | 290.0 (69.7)[d] | – |
| DFT[c] | 12.12 (4.22) | 280.8 (72.2) | 125.2 (144.2) | |
| 5 b | Exp[b] | 12.67 (4.28) | 287.9 (71.8) | 129.8 (143.9) |
| DFT[c] | 12.17 (4.24) | 280.2 (71.2) | 124.4 (143.4) | |
| 5 c | Exp[b] | 12.32 (4.17) | 290.0 (73.7) | 127.8 (144.3) |
| DFT[c] | 12.09 (4.30 | 278.9 (75.4) | 126.1 (144.6) | |
| 5 d | Exp[b] | 12.57 (4.24) | 289.0 (71.4)[d] | – |
| DFT[c] | 12.03 (4.69) | 274.6 (74.0) | 125.6 (144.4) | |
| 5 e | Exp[b] | 11.74 (3.26) | 284.6 (70.7)[d] | – |
| DFT[c] | 11.06 (−) | 272.4 (−) | 125.4 (144.4) |
[a] Data for the corresponding carbenoid precursor given within parentheses. [b] −90 °C in CD2Cl2. [c] Computed at 25 °C in CH2Cl2. [d] 25 °C in [D8]toluene.
Scheme 3.
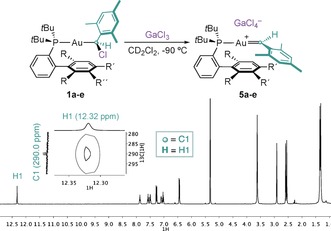
Generation of the mesityl gold(I) carbenes 5 a–e. 1H NMR spectrum and selected region of 1H‐13C HSQC spectra of 5 c in CD2Cl2 at −90 °C.
The calculated structure 5 c′ shows the mesitylidene fragment parallel to the lower ring of the biaryl ligand plane (Figure 1, top).17 The computed Au‐C1 (2.026 Å) and C1‐C2 (1.401 Å) bond distances for 5 c′ are slightly shorter than those for the precursor 1 c, Au‐C1 (X‐ray: 2.085 Å, computed: 2.095 Å) and C1‐C2 (X‐ray: 1.498 Å, computed: 1.502 Å). The reactivity of electrophilic gold(I) carbenes is dominated by the LUMO orbital, which in the case of 5 c′ is located over the occupied dxz(Au) orbital at gold and the 2pπ‐system of the carbene ligand (Figure 1, bottom left). An NBO analysis for 5 c′ revealed two main electronic contributions involved in the π‐orbital stabilization of C1. First, a small π‐backdonation from dxz(Au) to 2pπ(C1) in a donor–acceptor interaction (24.4 kcal mol−1) and 3.7 % contribution of 2pπ(C1) in the corresponding NLMO (Figure 1, bottom right), along with a lone pair at C2 [2pπ(C2)] highly polarized towards C1 [23.5 % on 2pπ(C1)].18 The calculated energy barrier to Au‐C1 (ΔG ≠=1.3 kcal mol−1) and C1‐C2 (ΔG ≠=18.6 kcal mol−1) bond rotation supported the low Au‐C1 bond π‐character and a significant C1‐C2 π‐bond. Although for these species the benzyl carbocation structure predominates, back‐donation from gold is expected to be higher for other alkyl‐substituted gold(I) carbenes generated under catalytic conditions.
Figure 1.
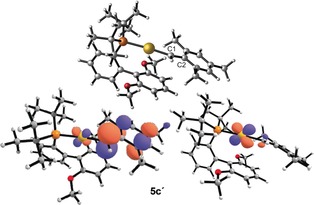
Top: Optimized geometry for 5 c′. Bottom left: plot for the LUMO orbital for 5 c′. Bottom right: plot for the NLMO associated to Au‐to‐C1 π‐backdonation for complex 5 c′. Cutoff: 0.05.
Gold(I) carbenes (5 b–c) were trapped by PPh3 at −90 °C, affording the phosphonium‐ylide complexes 7 a,b, whose structures were confirmed by X‐ray diffraction (Scheme 4 a). 7 a,b resemble the phosphonium‐ylide gold adducts previously used to detect aryl gold(I) carbenes in the gas phase.11a–11c, 12 In our reactivity studies, small amounts of 2,4,6‐trimethylbenzaldehyde (8) were detected, corresponding to the oxidation of gold(I) carbenes with oxygen.19 Indeed, we observed instantaneous formation of 8 as the only organic product when oxygen was bubbled through solutions of 5 a–d at −90 °C.20 Clean oxygen‐atom transfer also took place by treatment of 5 a–d with pyridine N‐oxide at −90 °C (Scheme 4 b). Similar reactivity has been observed for other gold(I) carbenes.5
Scheme 4.
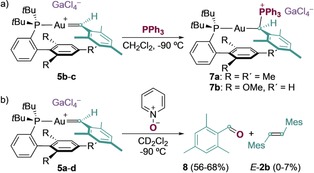
a) Synthesis of the phosphonium‐ylide complexes 7 a,b. b) Oxygen transfer from pyrine N‐oxide. Yields determined by 1H NMR spectroscopy using Ph2CH2 as an internal standard. Mes=2,4,6‐trimethylphenyl.
To date, none of the isolated gold(I) carbenes have been shown to undergo C−H insertion into alkanes. In contrast, 5 a–e, generated at −30 °C from the corresponding 1 a–e in the presence of cyclohexane, undergo clean C−H insertion to give 9 in 39–69 % yield (Scheme 5).
Scheme 5.
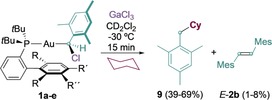
Gold(I)‐promoted C−H insertion. Yields determined by 1H NMR spectroscopy using Ph2CH2 as an internal standard. Mes=2,4,6‐trimethylphenyl.
In the reactions of 5 a–d with pyridine N‐oxide or cyclohexane, traces of (E)‐1,2‐dimesitylethene [(E)‐2 b] were also detected (Schemes 4 b and 5). (E)‐2 b was cleanly formed, together with the corresponding chloride‐bridged digold(I) complexes (10 a–e),21 when solutions of 5 a–e in CD2Cl2 were warmed up from −90 °C to either −30 or 0 °C (Table 2). Significant amounts of the cyclopropane 3 b were also obtained in some cases (Table 1, entries 1,3, and 5). Analysis of the reactivity of 5 c 22 shows that (E)‐2 b was initially formed between −70 and −40 °C. Then, around −40 °C, 3 b and small amounts of (Z)‐2 b were also detected. The formation of (E)‐2 b follows a second‐order dependence on the concentration of 5 b.20 Related bimolecular couplings to form symmetrical alkenes have been observed as common decomposition pathways for rhenium methylidenes23, 24 and ruthenium carbenes,25, 26 as well as typical Fischer27 and Schrock carbenes.28
Table 2.
Alkene and cyclopropane formation.[a]
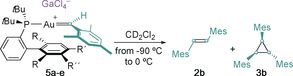
| Entry | Complex | T [°C] |
2 b
Yield [%] |
E/Z |
3 b
Yield [%] |
|---|---|---|---|---|---|
| 1 | 5 a | −10 | 25 | 12:1 | 36 |
| 2 | 5 b | −20 | 68 | >60:1 | <1 |
| 3 | 5 c | −10 | 27 | 9:1 | 39 |
| 4 | 5 d | −30 | 62 | >60:1 | <1 |
| 5 | 5 e | 0 | 7 | >60:1 | 83 |
[a] Yields were determined by 1H NMR spectroscopy using 1,3,5‐tris(trifluoromethyl)benzene as the internal standard. Mes=2,4,6‐trimethylphenyl.
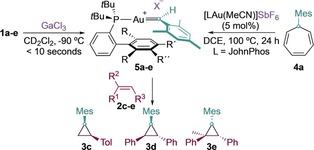
As suggested by the competitive cyclopropanation observed in the formal dimerization of the carbene fragments leading to 2 b (Table 2), the substituted alkenes 2 c–e readily react at −90 °C with 5 a–e to form the corresponding cyclopropanes 3 c–e (Table 3, entries 1–5).29 Importantly, the gold(I)‐catalyzed retro‐Buchner reaction10, 30 of the aryl cycloheptatriene 4 a in the presence of 2 c,d also leads to cyclopropanes (3 c–e; entry 6), and strongly suggests that 5 a is also generated under catalytic conditions. The lower yields of 3 c–e obtained under catalytic conditions can be explained by the steric hindrance introduced by the mesityl group in the rate‐determining cleavage of the cyclopropane bond of the norcaradiene in tautomeric equilibrium with the cycloheptatriene.
Table 3.
Cyclopropanation by 5 a–e from 1 a–e (left) or through decarbenation of 4 a (right).[a]
| Entry | Precursor | Yield [%][b] | ||
|---|---|---|---|---|
| 3 c | 3 d | 3 e | ||
| 1 | 1 a | 99 | 80 | 99 (2.3:1) |
| 2 | 1 b | 87 | 90 | 99 (3:1) |
| 3 | 1 c | 99 | 99 | 99 (1:1.2) |
| 4 | 1 d | 99 (1:1.5)31 | 99 | 72 (1:2.1) |
| 5 | 1 e | 99 | 90 | 99 (3.5:1) |
| 6 | 4 a | 32 | 38 | 17 (1:7.5) |
[a] Yields determined by 1H NMR spectroscopy using Ph2CH2 as the internal standard. [b] Diastereomeric ratio given within parenthesis. DCE=1,2‐dichloroethane.
To get a deeper insight into the mechanism of this rate‐limiting bimolecular coupling, we performed DFT calculations with 5 b′ (Scheme 6). Our calculations suggest that the association complex IntI, which is stabilized by face‐to‐face π–π interactions between the two mesitylidenes fragments and shows a relatively short C1‐C1 distance (3.13 Å), is preferentially formed (ΔG=−3.3 kcal mol−1) and can evolve through TSIntI‐IntIII (ΔG ≠=12.1 kcal mol−1) to form a (η 2‐Z‐5 a)gold(I) complex (IntIII) in a highly exothermic process (ΔG=−48.2 kcal mol−1). Alternatively, the less stable association complex IntII (ΔG=1.9 kcal mol−1), with the optimal orientation of the mesitylidenes for trans C1–C1 bond formation, can evolve via TSIntII‐IV (ΔG ≠=6.2 kcal mol−1) to form (E)‐2 b. At −35 °C, the calculated (E/Z)‐2 b ratio (ΔΔG ≠=0.6 kcal mol−1, 3:1 E/Z) is in close agreement with the experimental results (2.4:1 E/Z). Related mechanisms have been proposed for the formation of alkenes from rhenium21 and ruthenium23 carbenes.
Scheme 6.
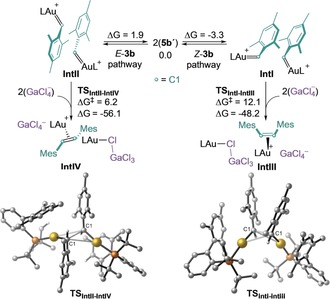
Calculated reaction profile for the bimolecular coupling of 5 b′. L=MeJohnPhos. DFT calculations were carried out at the B3LYP‐D3/6‐31G(d,p) + SDD on Au and Ga. CH2Cl2 was represented with the PCM. Free energies in kcal mol−1.
We also examined theoretically the reaction pathways for the cyclopropanation of (Z)‐2 b and (E)‐2 b by 5 b′.32 As we have found before for intermolecular cyclopropanations of intermediate gold(I) carbenes,33 these reactions proceed by an asynchronous concerted mechanism with an activation barrier for (E)‐2 b (ΔG ≠=12.7 kcal mol−1), 8.5 kcal mol−1 lower than that for (Z)‐2 b (ΔG ≠=21.2 kcal mol−1).29
In summary, we have generated and characterized spectroscopically monosubstituted gold(I) carbenes for the first time in solution, and they undergo representative transformations, such as cyclopropanation, oxidation, and C−H insertion reactions, of intermediate gold(I) carbenes formed under catalytic conditions. These aryl gold(I) carbenes correspond to the intermediates generated in the gold(I)‐catalyzed decarbenation of cycloheptatrienes (retro‐Buchner reaction). In the absence of other reagents, we observed dimerization to form preferentially the E‐configured alkene by a process similar to that followed by other well‐known metal carbenes, and places these highly electrophilic species among the metal carbene family, despite the weak back donation from gold(I) to the carbenic carbon center.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Agencia Estatal de Investigación (AEI)/FEDER, UE (CTQ2016‐75960‐P and FPI predoctoral fellowship to C.G.‐M.), H2020‐Marie Sklodowska‐Curie program (postdoctoral fellowship to X.‐L.P.), Swiss National Science Foundation (Early Postdoc Mobility fellowship to J.M.S.T.), the AGAUR (2017 SGR 1257), and CERCA Program/Generalitat de Catalunya for financial support. We also thank the ICIQ NMR and X‐ray diffraction units for technical support and Ángel L. Mudarra (ICIQ) for helpful discussions.
C. García-Morales, X.-L. Pei, J. M. Sarria Toro, A. M. Echavarren, Angew. Chem. Int. Ed. 2019, 58, 3957.
Dedicated to Professor Pablo Espinet on the occasion of his 70th birthday
References
- 1.
- 1a. Fürstner A., Chem. Soc. Rev. 2009, 38, 3208–3221; [DOI] [PubMed] [Google Scholar]
- 1b. Shapiro N., Toste F. D., Synlett 2010, 675–691; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Hashmi A. S. K., Angew. Chem. Int. Ed. 2010, 49, 5232–5241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5360–5369; [Google Scholar]
- 1d. Liu L.-P., Hammond G. B., Chem. Soc. Rev. 2012, 41, 3129–3139; [DOI] [PubMed] [Google Scholar]
- 1e. Obradors C., Echavarren A. M., Acc. Chem. Res. 2014, 47, 902–912; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1f. Fensterbank L., Malacria M., Acc. Chem. Res. 2014, 47, 953–965; [DOI] [PubMed] [Google Scholar]
- 1g. Dorel R., Echavarren A. M., Chem. Rev. 2015, 115, 9028–9072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1h. Pflästerer D., Hashmi A. S. K., Chem. Soc. Rev. 2016, 45, 1331–1367; [DOI] [PubMed] [Google Scholar]
- 1i. Echavarren A. M., Muratore M., López-Carrillo V., Escribano-Cuesta A., Huguet N., Obradors C., Org. React. 2017, 92, 1. [Google Scholar]
- 2.
- 2a. Fürstner A., Morency L., Angew. Chem. Int. Ed. 2008, 47, 5030–5033; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 5108–5111; [Google Scholar]
- 2b. Hashmi A. S. K., Angew. Chem. Int. Ed. 2008, 47, 6754–6756; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6856–6858; [Google Scholar]
- 2c. Seidel G., Mynnot R., Fürstner A., Angew. Chem. Int. Ed. 2009, 48, 2510–2513; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2548–2551; [Google Scholar]
- 2d. Benitez D., Shapiro N. D., Tkatchouk E., Wang Y., Goddard W. A., Toste F. D., Nat. Chem. 2009, 1, 482–486; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Echavarren A. M., Nat. Chem. 2009, 1, 431–433. [DOI] [PubMed] [Google Scholar]
- 3.Reviews on the topic:
- 3a. Wang Y., Muratore M. E., Echavarren A. M., Chem. Eur. J. 2015, 21, 7332–7339; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Harris R. J., Widenhoefer R. A., Chem. Soc. Rev. 2016, 45, 4533–4551. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Fañanás-Mastral M., Aznar F., Organometallics 2009, 28, 666–668; [Google Scholar]
- 4b. Ung G., Bertrand G., Angew. Chem. Int. Ed. 2013, 52, 11388–11391; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11599–11602; [Google Scholar]
- 4c. Brooner R. E. M., Widenhoefer R. A., Chem. Commun. 2014, 50, 2420–2423; [DOI] [PubMed] [Google Scholar]
- 4d. Seidel G., Gabor B., Goddard R., Heggen B., Thiel W., Fürstner A., Angew. Chem. Int. Ed. 2014, 53, 879–882; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 898–901; [Google Scholar]
- 4e. Seidel G., Fürstner A., Angew. Chem. Int. Ed. 2014, 53, 4807–4811; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4907–4911; [Google Scholar]
- 4f. Ciancaleoni G., Biasiolo L., Bistoni G., Macchioni A., Tarantelli F., Zuccaccia D., Belpassi L., Chem. Eur. J. 2015, 21, 2467–2473; [DOI] [PubMed] [Google Scholar]
- 4g. Wang J., Cao X., Lv S., Zhang C., Xu S., Shi M., Zhang J., Nat. Commun. 2017, 8, 14625–14635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harris R. J., Widenhoefer R. A., Angew. Chem. Int. Ed. 2014, 53, 9369–9371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9523–9525. [Google Scholar]
- 6. Hussong M. W., Rominger F., Krämer P., Straub B. F., Angew. Chem. Int. Ed. 2014, 53, 9372–9375; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9526–9529. [Google Scholar]
- 7.
- 7a. Joost M., Estévez L., Mallet-Ladeira S., Miqueu K., Amgoune A., Bourissou D., Angew. Chem. Int. Ed. 2014, 53, 14512–14516; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14740–14744; [Google Scholar]
- 7b. Zeineddine A., Rekhroukh F., Carrizo E. D. S., Mallet-Ladeira S., Miqueu K., Amgoune A., Bourissou D., Angew. Chem. Int. Ed. 2018, 57, 1306–1310; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1320–1324. [Google Scholar]
- 8.Among all the isolated gold(I) carbenes,[4–7] only three of these species display reactivity related to real intermediates in catalysis. In particular, cyclopropanation of styrenes,[4e, 7b] oxidation with H2O2 [4g] N-pyridine-oxide[5] and B−H and O−H insertion.[7b]
- 9. Caballero A., Pérez P. J., Chem. Eur. J. 2017, 23, 14389–14393. [DOI] [PubMed] [Google Scholar]
- 10. Sarria Toro J. M., García-Morales C., Raducan M., Smirnova E. S., Echavarren A. M., Angew. Chem. Int. Ed. 2017, 56, 1859–1863; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1885–1889. [Google Scholar]
- 11.
- 11a. Solorio-Alvarado C. R., Echavarren A. M., J. Am. Chem. Soc. 2010, 132, 11881–11883; [DOI] [PubMed] [Google Scholar]
- 11b. Herlé B., Holstein P. M., Echavarren A. M., ACS Catal. 2017, 7, 3668–3675; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Mato M., Herlé B., Echavarren A. M., Org. Lett. 2018, 20, 4341–4345. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Fedorov A., Moret M.-E., Chen P., J. Am. Chem. Soc. 2008, 130, 8880–8881; [DOI] [PubMed] [Google Scholar]
- 12b. Fedorov A., Chen P., Organometallics 2009, 28, 1278–1281; [Google Scholar]
- 12c. Fedorov A., Batiste L., Bach A., Birney D. M., Chen P., J. Am. Chem. Soc. 2011, 133, 12162–12171; [DOI] [PubMed] [Google Scholar]
- 12d. Ringger D. H., Chen P., Angew. Chem. Int. Ed. 2013, 52, 4686–4689; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4784–4787; [Google Scholar]
- 12e. Batiste L., Chen P., J. Am. Chem. Soc. 2014, 136, 9296–9307; [DOI] [PubMed] [Google Scholar]
- 12f. Ringger D. H., Kobylianskii I. J., Serra D., Chen P., Chem. Eur. J. 2014, 20, 14270–14281. [DOI] [PubMed] [Google Scholar]
- 13. Swift C. A., Gronert S., Organometallics 2014, 33, 7135–7140. [Google Scholar]
- 14.CCDC 1887058, 1887068, 1887061, 1887064, 1887062, 1887059, 1887060, 1887067, 1887065, 1887063, and 1887066 (1 a, 1 b, 1 c, 1 d, 1 e, 7 a, 7 b, 10 a, 10 c, S1, S2) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 15.See Scheme S1 in the Supporting Information for alternative gold(I) carbenoids tested.
- 16.See the Supporting Information for spectroscopy characterization for 5 a–e.
- 17.See the Supporting Information: Optimized structures and structural discussion on 5 a–e can be found in Figure S17 and Table S15.
- 18.See Figure S32 in the Supporting Information: plot for the NLMO associated to lone pair at C2 (2pπ(C2)) for 5 c′.
- 19.Oxidation of gold(I) carbenes under air: Zhan H., Zhao L., Liao J., Li N., Chen Q., Qiu S., Cao H., Adv. Synth. Catal. 2015, 357, 46–50. [Google Scholar]
- 20.See the Supporting Information: see Scheme S1 for further information.
- 21. Homs A., Escofet I., Echavarren A. M., Org. Lett. 2013, 15, 5782–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.See the Supporting Information: a) See Figure S5 for a representative graph; b) See Figures S8–S13 for kinetic studies.
- 23.
- 23a. Merrifield J. H., Lin G. Y., Kiel W. A., Gladysz J. A., J. Am. Chem. Soc. 1983, 105, 5811–5819; [Google Scholar]
- 23b. Roger C., Bodner G. S., Hatton W. G., Gladysz J. A., Organometallics 1991, 10, 3266–3274. [Google Scholar]
- 24. Heinekey D. M., Radzewich C. E., Organometallics 1998, 17, 51–58. [Google Scholar]
- 25. Bailey G. A., Foscato M., Higman C. S., Day C. S., Jensen V. R., Fogg D. E., J. Am. Chem. Soc. 2018, 140, 6931–6944. [DOI] [PubMed] [Google Scholar]
- 26. Graban E., Lemke F. R., Organometallics 2002, 21, 3823–3826. [Google Scholar]
- 27.
- 27a. Casey C. P., Anderson R. L., J. Chem. Soc. Chem. Commun. 1975, 895–896; [Google Scholar]
- 27b. Masters C., Van der Woude C., Van Doorn J. A., J. Am. Chem. Soc. 1979, 101, 1633–1634. [Google Scholar]
- 28.
- 28a. Schrock R. R., Sharp P. R., J. Am. Chem. Soc. 1978, 100, 2389–2399; [Google Scholar]
- 28b. Berry D. H., Koloski T. S., Carroll P. J., Organometallics 1990, 9, 2952–2962; [Google Scholar]
- 28c. Schrock R. R., Copeŕet C., Organometallics 2017, 36, 1884–1892. [Google Scholar]
- 29.The cyclopropane 3 b undergoes partial cis to trans isomerization in the presence of GaCl3, and therefore, the diastereoselectivity varies along the experiments.
- 30.
- 30a. Wang Y., Muratore M. E., Rong Z., Echavarren A. M., Angew. Chem. Int. Ed. 2014, 53, 14022–14026; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14246–14250; [Google Scholar]
- 30b. Leboeuf D., Gaydou M., Wang Y., Echavarren A. M., Org. Chem. Front. 2014, 1, 759–764; [Google Scholar]
- 30c. Wang Y., McGonigal P. R., Herlé B., Besora M., Echavarren A. M., J. Am. Chem. Soc. 2014, 136, 801–809; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30d. Yin X., Mato M., Echavarren A. M., Angew. Chem. Int. Ed. 2017, 56, 14591–14595; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14783–14787. [Google Scholar]
- 31.The diastereomeric ratio can vary from along the experiment because small amounts of GaCl3 can catalyze the isomerization of cis-3 c into trans-3 c (see Table S3 in the Supporting Information).
- 32.See the Supporting Information: see Figures S31 and S33 for further DFT details.
- 33.
- 33a. Pérez-Galán P., Herrero-Gómez E., Hog D. T., Martin N. J. A., Maseras F., Echavarren A. M., Chem. Sci. 2011, 2, 141–149; [Google Scholar]
- 33b. López-Carrillo V., Huguet N., Mosquera Á., Echavarren A. M., Chem. Eur. J. 2011, 17, 10972–10978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary