

Abstract
Objective
A minipig model was employed to explore the changes in endogenous leptin transport into the central nervous system and in hypothalamic sensitivity to exogenous leptin when individuals are placed on high‐fat diet (HFD) compared with standard diet.
Methods
Serum and cerebrospinal fluid (CSF) leptin concentrations during 10 weeks of HFD versus standard diet and exogenous leptin‐induced STAT3 phosphorylation in the hypothalamus of minipigs were assessed, and the hypothalamic leptin‐sensitive cells were characterized by immunofluorescence.
Results
The efficiency of the passage of endogenous blood‐borne leptin into the CSF (measured as the log [CSF:serum leptin ratio]) decreased over time in minipigs fed a HFD (β = –0.04 ± 0.005 per kilogram of weight gain in HFD; P < 0.0001), while it remained stable in minipigs fed a standard diet. However, the ability of peripherally administered leptin to activate its receptor in hypothalamic neurons was preserved in obese minipigs at 10 weeks of HFD.
Conclusions
Together, these data are consistent with the existence of an early‐onset tranport deficiency for endogenous circulating leptin into the brain in individuals developing obesity, preceding the acquisition of hypothalamic leptin resistance. Although additional studies are required to identify the underlying mechanisms, our study paves the way for the development of new preclinical pharmacological models targeting the restoration of the shuttling of peripheral leptin into the central nervous system to manage obesity.
Introduction
Leptin is a peptide hormone mainly derived from adipocytes, and it is present in serum at levels directly proportional to fat mass and adipocyte size and, thus, to peripheral energy stores 1. It targets brain networks, notably leptin receptor (LepR)‐expressing neurons in the hypothalamus, to decrease food intake, increase energy expenditure, and control glucose homeostasis 2, 3. Thus, leptin plays a crucial role in modulating the pathways that maintain energy homeostasis, and a low leptin level represents a powerful signal to promote the anabolic response to negative energy balance 4.
Human studies have shown that serum leptin enters the central nervous system (CNS) in proportion to its circulating levels and that the passage of circulating leptin from the blood to the cerebrospinal fluid (CSF) is dramatically impaired in patients with obesity 5, 6. As highlighted by Jeffrey M. Friedman and Christos S. Mantzoros in a retrospective about the discovery of leptin 7 20 years after the pioneering studies, how leptin is transported into the CNS still remains an enigma. Previous studies have suggested that leptin enters the brain by transport across the vascular endothelium 8. However, evidence has indicated that leptin first enters a circumventricular organ, the median eminence, where it is taken up by processes of specialized glial cells called tanycytes and transcytosed into the third ventricle; from there, leptin then circulates through the ventricular system and accesses deeper brain structures 9, 10, 11. This tanycytic transport of leptin is dependent on LepR signaling in these cells. These rodent studies have also suggested that in animals with diet‐induced obesity, tanycytes are among the first cells of the CNS to be affected by resistance to peripheral leptin caused by an early impairment of LepR signaling, which likely occurs when serum leptin levels exceed the capacity of tanycytes to shuttle leptin into the CSF 9. Early impairment of LepR signaling in neuronal circuits controlling feeding in the hypothalamus has also been proposed to explain the limited response to exogenous leptin in animals with diet‐induced obesity 4.
However, the kinetics of the alteration of leptin transport across the blood‐CSF barrier in individuals who develop obesity because of diet, as well as whether the alteration is causal or adaptive to the hypothalamic leptin resistance, remains unknown. Here, we explored the hypothesis that the alteration of leptin transport into the CSF is a gradual process that heralds hypothalamic resistance to peripheral leptin in minipigs fed a high‐fat diet (HFD), an animal model that is predictive of human biology 12 and that permits the direct assessment of leptin uptake by the CNS for preclinical pharmacology purposes.
Methods
Animals and diets
Seventeen 1‐year‐old male minipigs (weight 33.2 ± 0.9 kg) derived from Göttingen minipigs were selected from Elevage Denis breeding (Templeuve, France). All animals were housed individually with free access to water. Eight animals were fed a standard diet with normal fat content (normal‐fat diet [NFD]) of 2,463 kcal/kg (including 2% fat, 33% barley, 25% wheat bran, 12% soy shell, 10% wheat, 10% sunflower meal, 6% soy meal). Each animal received a ratio of 41.5 g/kg0.75 body weight once a day at 9:00 am for 10 weeks, calculated weekly to maintain a lean phenotype. In the HFD group, nine animals were fed with a Western diet of 3,473 kcal/kg (including 23% fat from saturated fatty acids, 10% barley, 12% wheat bran, 33% wheat, 15% soy meal, and 5% sugar). Each animal received a ratio of 41.5 g/kg0.75 body weight twice a day for 10 weeks, calculated weekly to develop an obesity phenotype, as previously described by others 13. Animal studies were approved by the Institutional Ethics Committees for the Care and Use of Experimental Animals of the University of Lille (CEEA Nord Pas‐de‐Calais, protocol n°032012); all experiments were performed in accordance with the guidelines for animal use specified by the European Union Council Directive of September 22, 2010 (2010/63/EU).
Body weight and adipose tissue volume assessment
Body weight was assessed weekly after an overnight fasting period with an electronic scale (Marechalle Pesage, Chauny, France). Volumetric adipose tissue analysis was performed by magnetic resonance imaging (MRI) to estimate visceral and subcutaneous adipose tissue. The study was carried out on an Airis Mate 0.2‐Tesla MRI machine (maximum gradient strength: 15 milliTesla/meter; gradient slew rate: 30 milliTesla/meter/millisecond, with a panoramic, open gantry design and permanent magnet; Hitachi Medical Co., Tokyo, Japan). The measurements were realized at the level of T13 and L2, two locations corresponding to the greatest abdominal girth in minipigs 14, at the end of the 10 weeks of experimentation in both groups. To perform these experiments, animals were lightly anesthetized using a combination of ketamine (1 mL/10 kg) and xylazine (1 mL/10 kg).
Serum and CSF leptin assessment
Blood and CSF samples were collected at the same time as body weight measurements after an overnight fasting period and in lightly anesthetized animals. Blood was collected by auricular venipuncture in 5‐mL sterile tubes (Vacutainer Systems Europe, BD Diagnostics‐Preanalytical Systems, Le Pont de Claix, France). Immediately after being drawn, the blood sample was centrifuged at 4°C and 1,500g for 15 minutes, and the serum was separated and stored at −20°C until assayed. CSF was collected at the same time by lumbar puncture in lateral decubitus in 1.5‐mL Eppendorf tubes (Sigma Aldrich, St. Quentin Fallavier, France) and immediately stored at −20°C. Serum and CSF leptin concentrations were measured using a Millipore Multi‐Species Leptin RIA kit (XL‐85K; MilliporeSigma, Burlington, Massachusetts), with interassay variability of 6.5% to 8.7% and intra‐assay variability of 2.8% to 3.6%. The standard curve was affine, adding a point at 0.78 ng/mL. CSF samples were concentrated before leptin measurement by lyophilization. Dry extracts were resuspended in one‐third of initial volume buffer, and leptin was measured using the kit.
Subcutaneous leptin or saline injections
At the end of the nutritional challenge, animals randomly received, after an overnight fasting period, a subcutaneous injection of 0.025 mg/kg of recombinant leptin (PeproTech, Rocky Hill, New Jersey) or control solution of 1 mL of sodium citrate (5nM, pH 4).
Euthanasia and brain extraction
Forty‐five minutes after the leptin or saline injection, animals were deeply anesthetized with a combination of ketamine (1 mL/10 kg), xylazine (1 mL/10 kg), and inhaled isoflurane (Aerrane; Baxter, Deerfield, Illinois) and were intubated with a laryngoscope. Animals underwent a median sternotomy with bone shears. A small incision was made in the pericardial sac. The cardiac structures were identified, especially the aorta and the superior vena cava. First, an exsanguination‐perfusion was performed as follows: a perfusion cannula was inserted into the aorta, pointing toward the brachiocephalic trunk and left common carotid artery, to infuse the solution; then another cannula was inserted into the superior vena cava for blood draining 15. The vascular system was rinsed with 6 L (200 mL/kg) of isotonic saline solution (VIAFLO, Baxter; NaCl 0.9%; 4°C) coupled with a single injection of 5,000‐IU/mL Choay heparin (Sanofi‐Aventis, Paris, France) until the discharge was clear. Next, 5 L (160 mL/kg) of paraformaldehyde 2% (pH 7.4; 4°C) was perfused directly through the aortic cannula continuously for 15 minutes with a Duotronic III pump (Hygeco, Paris, France). Lastly, the scalp was removed by a longitudinal incision with the animal in prone position. A craniotomy was performed following the coronal and lambdoid bone sutures with an electric jigsaw. The brains, and especially the hypothalamus, were then removed with special care taken to preserve the median eminence.
Tissue processing
Blocks of tissue containing the hypothalamic nuclei were dissected, postfixed in the same fixative for 2 hours, and subsequently treated with cryoprotectant 20% sucrose for 4 days prior to freezing in OCT Compound (Tissue‐tek; Sakura, Alphen aan den Rijn, The Netherlands) with isopentane and liquid nitrogen. Six series of 30‐µm‐thick coronal sections from the preoptic area to the mammillary bodies were collected with a Leica 3050SM cryostat (Leica Biosystems, Wetzlar, Germany), mounted directly onto Superfrost Plus slides (VWR International, Radnor, Pennsylvania), and stored at −80°C until further use. We localized the different structures of the hypothalamus based on previous neuroanatomical studies in the minipig 16.
Immunofluorescence, image acquisition, and quantification
The detection of phosphorylation of signal transducer and activator of transcription 3 (STAT3) was carried out using protocols we have previously described 9.
Phosphorylated STAT3 (pSTAT3) immunohistochemistry
Immunohistochemistry was adapted from a protocol previously described in mice 9. Sections were pretreated for 20 minutes with 0.05% NaOH and 0.5% H2O2 in potassium‐phosphate‐buffered saline (KPBS), followed by immersion in 0.03% glycine for 10 minutes. Sections were then placed in 0.03% sodium dodecyl sulfate (SDS) for 10 minutes and incubated in 4% normal goat serum (NGS) + 0.4% Triton (Sigma Aldrich) + 1% bovine serum albumin (BSA) (fraction V) for 20 minutes. Sections were then incubated with the primary antibody, rabbit anti‐pSTAT3 (Tyr705; 9131; 1/1000 in 0.3% Triton X‐100, 1% NGS, and 1% BSA; Cell Signaling Technology, Danvers, Massachusetts) for 48 hours at 4°C. After washing, sections were incubated for 1 hour at room temperature with the secondary antibody, goat antirabbit conjugated to Alexa Fluor 568 (1:200 in KPBS with 0.3% Triton X‐100 and 1% NGS; Molecular Probes, Eugene, Oregon).
ß‐endorphin and HuC/D immunohistochemistry
As both ß‐endorphin and pSTAT3 antibodies were raised in rabbit, sections previously subjected to pSTAT3 immunostaining were rinsed in 0.02M KPBS and then immersed in paraformaldehyde 4% for 30 minutes prior to incubation in NGS diluted 1:1,500 in 0.02M KPBS containing 0.3% Triton X‐100 for 30 minutes. They were then incubated with the primary antibodies, rabbit anti‐ß‐endorphin 1:750 (homemade) 9 and mouse anti‐HuC/D (1:200, A21271, Invitrogen, Carlsbad, California) in 0.02M KPBS containing 0.3% Triton X‐100 at 4°C for 48 hours. Sections were then incubated with a biotinylated goat antirabbit antibody (1:500; Vector Laboratories, Burlingame, California) in 0.02M KPBS for 1 hour, followed by the ABC kit (Vectastain, Vector Laboratories, Peterborough, UK) for 30 minutes. After washing, they were incubated with tyramide 0.5% (Perkin Elmer, Waltham, Massachusetts) for 20 minutes and rinsed in 0.02M KPBS. Sections were incubated with Alexa Fluor® 488 streptavidin (1:500) and the secondary antibody Alexa Fluor‐conjugated goat antimouse (1:500; Abcam, Cambridge, UK) for 1 hour. The sections were then counterstained with Hoechst (Invitrogen) for 3 minutes at room temperature. The sections were finally put under cover slips slightly wet with buffered Mowiol mounting medium (Sigma Aldrich).
Image processing
Tissue sections were analyzed and digitally photographed using a Zeiss Axio Imager Z2 apotome microscope mounted with an AxioCamMRm digital color camera (Carl Zeiss, Oberkochen, Germany) and AxioVision software (Carl Zeiss). To create photomontages, single‐plane images were captured using the MosaiX module of the AxioVision 4.6 system (Carl Zeiss) and a Zeiss 20 X objective (N.A.0.8) for each fluorophore sequentially. Images were subsequently adjusted for intensity and contrast using Photoshop software (Adobe, San Jose, California).
Immunolabeling quantification
Cell counts were performed using the count tool of Photoshop (CS5.0) software; pSTAT3‐positive‐, ß‐endorphin‐positive, and double‐labeled cells were manually counted on digitized images. For each animal, two hemisections of the tuberal region of the hypothalamus were counted by an investigator blind to the experimental condition, and the mean of the two counts was used for analysis. Counting was performed on a homogenous area on the 24 slices of interest (i.e., two slices per animal for six HFD and six NFD minipigs, of which half had received leptin injection in each group). The arcuate nucleus (ARH), ventromedial nucleus, and dorsomedial nucleus of the hypothalamus were delineated using global morphology on Hoechst counterstaining and the distribution of ß‐endorphin‐immunoreactive cells. The number of pSTAT3‐positive cells was counted in each nucleus, followed by ß‐endorphin‐positive and double‐labeled cells.
Statistical analysis
Values are expressed as means ± SEM. For comparisons of body weight, serum leptin, and CSF leptin between HFD and NFD minipigs, we used a two‐way ANOVA with repeated measures (diet × time), followed by Bonferroni multiple comparison test at each time point. The ratio of CSF to serum leptin was log‐transformed (as recommended elsewhere) 6 before performing two‐way repeated‐measures ANOVA (diet × time) followed by Bonferroni multiple comparison test at each time point. Adipose tissue volumes were compared between animal groups by using two‐way ANOVA. Immunolabeled cell counts were compared between NFD and HFD groups and between treatment conditions using two‐way ANOVA (diet × treatment) followed by Bonferroni multiple comparison test in each diet group. Finally, to estimate the longitudinal relationships between serum leptin, CSF leptin, ratio of CSF to serum leptin, and weight, we used a linear regression model. P values were considered to be significant below a threshold of 0.05. Analyses were performed using R (version 3.4.2; The R Foundation, Vienna, Austria), and graphs were generated by GraphPad Prism 5 software (GraphPad, San Diego, California).
Results
Evolution of body weight and body fat distribution in minipigs fed HFD vs. NFD
Seventeen 1‐year old (young adult) male minipigs of comparable initial weights were fed NFD for 10 weeks (n = 8; weight at week 0: 34.9 ± 1.1 kg) or an HFD (n = 9; weight at week 0: 31.8 ± 1.2 kg; the difference in initial weight between the two groups was not significant). Under this regimen, minipigs presented different weight evolutions (P diet < 0.0001, P time < 0.0001, P diet×time < 0.0001), notably from week 5 (P < 0.001, HFD vs. NFD) (Figure 1A). At week 10, while minipigs on NFD showed stable body weights (37.7 ± 3.2 kg), minipigs on HFD had doubled their body weights (66.9 ± 2.1 kg; P < 0.0001 vs. baseline) (Figure 1A). Volumetric analyses of adipose tissue performed at the 10th week by MRI showed a 3.2‐fold increase in subcutaneous fat and a 3.7‐fold increase in visceral fat in minipigs fed HFD compared with NFD‐fed animals (P < 0.0001 for both fat location, HFD vs. NFD) (Figure 1B).
Figure 1.
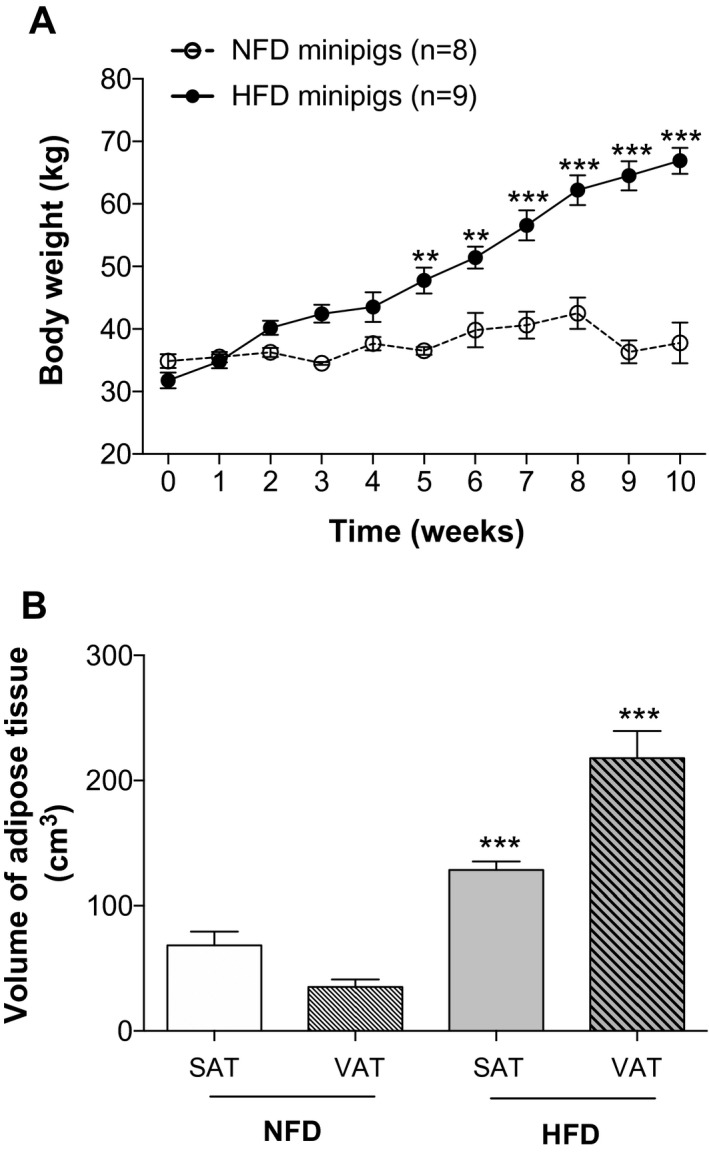
Diet‐induced obesity in minipigs. (A) Body weight curves in minipigs fed normal‐fat diet (NFD) and high‐fat diet (HFD), revealing diet‐induced obesity in minipigs. (B) Adiposity measurement by MRI in minipigs. Graphs represent the volume of each fat compartment. Values shown are means ± SEM. **P < 0.001, ***P < 0.0001, HFD vs. NFD. VAT, visceral adipose tissue; SAT, subcutaneous adipose tissue.
Relationships of CSF and serum leptin with body weight in minipigs fed HFD vs. NFD
In agreement with rodent and human studies 1, 5, 6, as well as previous data gathered in pigs 17, circulating serum leptin levels were significantly higher in obese than in lean minipigs at the 10th week (0.9 ± 1.2 ng/mL in NFD vs. 8.0 ± 0.9 ng/mL in HFD, P < 0.0001). While serum leptin levels became significantly different between groups only at the seventh week of the regimen (P diet < 0.0001, P time < 0.03, P diet×time = 0.004) (Figure 2A) and CSF leptin levels remained steady over time (P diet < 0.0001, P time = 0.82, P diet×time = 0.99) (Figure 2B), the relationship between CSF and serum leptin became significantly different between the NFD and HFD groups as early as the fourth week of the regimen (P diet < 0.0001, P time < 0.0001, P diet×time < 0.0001) (Figure 2C). At the 10th week, the CSF:serum leptin ratio in lean individuals (0.266 ± 0.039) was five‐times higher than that in individuals with obesity (0.053 ± 0.007, P < 0.0001). Moreover, serum leptin and CSF leptin levels were linearly correlated in the NFD group (β = 0.10 ± 0.05; P = 0.04) but not in the HFD group (P = 0.68).
Figure 2.
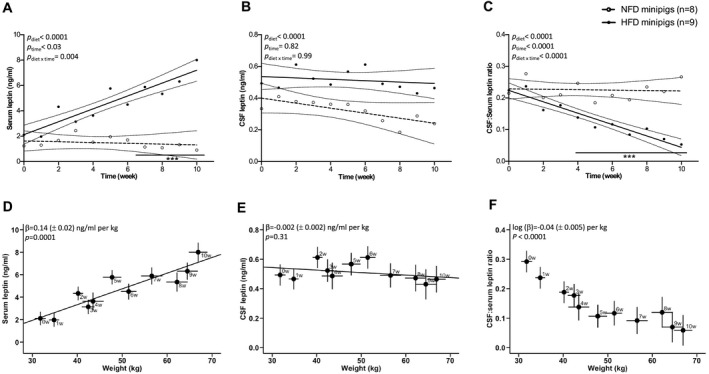
Longitudinal relationships of serum leptin, cerebrospinal fluid (CSF) leptin, CSF:serum leptin ratio, and body weight in minipigs fed normal‐fat diet (NFD) and high‐fat diet (HFD). (A‐C) Evolution of serum and CSF leptin and CSF:serum leptin ratios with time. Values are means and 95% CI. Bonferroni post hoc analysis, ***P < 0.0001, HFD vs. NFD. Evolution of (D) serum leptin, (E) CSF leptin, and (F) CSF:serum leptin ratio according to body weight in HFD minipigs. Values are means ± SEM at each week.
Consistent with the fact that leptin circulates in proportion to body adiposity, serum leptin levels significantly increased with weight gain in HFD minipigs by 0.14 (± 0.02) ng/mL per kilogram (P < 0.0001) (Figure 2D). CSF leptin levels were not correlated with body weight (P = 0.31) (Figure 2E). Consequently, there was a negative correlation between the CSF:serum leptin ratio and body weight in minipigs fed HFD (log [β] = –0.04 ± 0.005/kg, P < 0.0001) (Figure 2F). In the NFD minipigs, no correlations were found between weight and serum leptin (P = 0.16) or log (CSF:serum leptin ratio) (P = 0.93). However, a slight negative correlation between weight and CSF leptin (β = −0.02 [± 0.006] ng/mL per kilogram, P = 0.006) was observed in the NFD minipigs, which could be explained by a slight negative energy balance as access to food was restricted to maintain weight stability during the experiment.
Leptin‐induced STAT3 phosphorylation in the hypothalamus of minipigs
To determine whether 10 weeks of HFD hampered the ability of peripheral leptin to promote LepR signaling in the hypothalamus, we used immunohistochemistry to assess pSTAT3, commonly used as a marker of LepR activation 18, 90 minutes after subcutaneous leptin administration (0.025 mg/kg). Immunoreactivity for pSTAT3 was readily detected in the hypothalamus of both lean and obese minipigs after leptin treatment (n = 3 per group), whereas only a few cells exhibited pSTAT3 immunoreactivity in vehicle‐injected animals (n = 3 per group) (Figure 3A).
Figure 3.
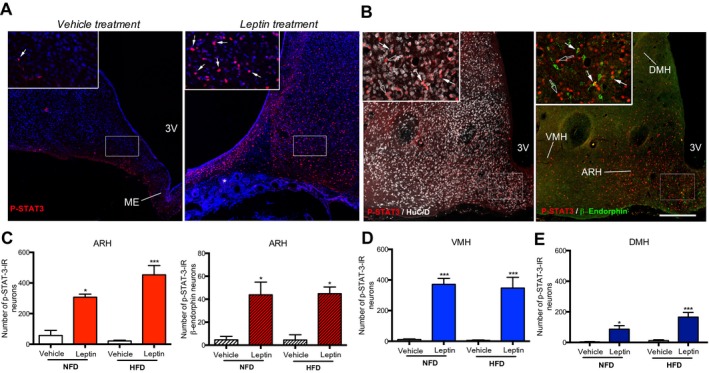
Peripheral leptin activates leptin receptor signaling in hypothalamic neurons of minipigs even after 10 weeks of high‐fat diet (HFD). (A,B) Distribution of pSTAT3 immunoreactivity (arrows) in coronal sections of the ARH in minipigs after subcutaneous administration of leptin (right images in panels A and B) or vehicle (left image in panel A) (n = 3 per group). Insets show higher magnification of the area delineated by a white rectangle in the main image. In panel B, the white (left panel) and green (right panel) signals show HuC/D‐ and β‐endorphin‐immunoreactive (IR) neurons, respectively, some of which express pSTAT3 (arrows). Empty arrows point to single‐labeled pSTAT3‐immunoreactive cells. Scale bars: 300 μm (100 μm in insets). Number of pSTAT3‐immunoreactive neurons in the (C) ARH, (D) VMH, and (E) DMH in minipigs fed normal‐fat diet (NFD) (n = 6) and HFD (n = 6) for 10 weeks and subjected to vehicle (n = 3 per group) or leptin treatment (n = 3 per group). *P < 0.05, ***P < 0.0001, vehicle vs. leptin. 3V, third ventricle; ME, median eminence; ARH, arcuate nucleus of the hypothalamus; VMH, ventromedial nucleus of the hypothalamus; DMH, dorsomedial nucleus of the hypothalamus. [Colour figure can be viewed at wileyonlinelibrary.com]
Chemical characterization of leptin‐sensitive cells in the hypothalamus of minipigs
Cellular analysis of double‐labeled material clearly revealed that pSTAT3 immunoreactivity following leptin administration was almost exclusively restricted to HuC/D‐immunoreactive cells (Figure 3B), indicating that the majority of these cells were neurons. In the ARH of minipigs, pSTAT3 was found in proopiomelanocortin (POMC)‐producing anorexigenic neurons, as revealed by immunoreactivity for β‐endorphin (Figure 3B). Quantitative analyses showed that leptin administrated at a pharmacological dose was equally potent in stimulating STAT3 phosphorylation in the ARH and dorsomedial and ventromedial hypothalamic nuclei of minipigs fed NFD or HFD for 10 weeks (Figure 3C‐3E) (Supporting Information Table S1).
Discussion
Here, we studied the evolution of the transport of endogenous leptin during 10 weeks of HFD versus NFD as well as the hypothalamic sensitivity to exogenous leptin. First, our data show that blood‐borne leptin, which is correlated with adiposity, is transported into the CNS in proportion to its circulating levels in lean minipigs fed a NFD. In animals fed a HFD, which develop obesity, the CSF leptin levels remain stable over time, while the serum leptin levels increase. This corroborates the hypothesis of an early‐onset saturation of the leptin transport into the CNS.
Studies have suggested that circulating leptin 9, and possibly other metabolic hormones 19, 20, enter the brain through the hypothalamic median eminence and that tanycytes, which are specialized but highly plastic ependymo‐glial cells 21, 22 that capture leptin from the bloodstream 9, act as a checkpoint along this route 10. It was shown that leptin‐neutralizing antibodies infused into the third ventricle impeded hypothalamic neuron activation, indicating that tanycyte‐mediated leptin transport from the periphery to the CSF is necessary for its downstream effects 9. CSF‐borne leptin is then likely redistributed to its sites of action in the CNS by the beating of ciliated ependymal cells lining the third ventricle 23, 24, which generate CSF currents that flow alongside the median eminence and the ARH 25. These findings have mostly been demonstrated in rodents, whereas our study is the first to observe this phenomenon in minipigs, which is a better model than rodents to extrapolate animal findings to human biology 13. Consistently, saturation of leptin transport through the blood‐CSF barrier has been reported in humans to partially explain the mechanism of leptin resistance in obesity 6.
Second, our quantitative analyses show that leptin sensitivity is preserved in the neuronal circuits controlling feeding in the hypothalamus of minipigs fed a HFD. Our findings in that model corroborates with previous studies in rodents, supporting the fact that exogenous leptin is still relevantly signaling in the early phase of diet‐induced obesity 26 despite the fact that the ARH has been considered to be the major site affected early by leptin resistance 27.
Our neuroanatomical results, together with the fact that 10 weeks of HFD did not cause any major alteration of the blood metabolic profile of minipigs (Supporting Information Figure S1), indicate that the decline of the transport of circulating leptin into the CNS that is likely to worsen over time precedes the onset of hypothalamic resistance to peripheral leptin 28, 29. Our results stand in contrast with recent reports in rodents suggesting that impaired transport of leptin in the hypothalamus does not precede diet‐induced obesity 30 and that leptin receptor‐mediated transport across the blood‐brain barrier at the level of cerebral vessels and the blood‐CSF barrier at the choroid plexus is not critical for homeostatic feeding 31. However, the study of Kleinert et al. 30 focused on leptin actions in response to the administration of pharmacological doses of exogenous leptin and could not conclude about the transport dynamics of endogenous blood‐borne leptin across the blood‐CSF barrier that may involve tanycytic shuttles 9, which remained intact in the study by Di Spiezio et al. 31.
Our study has notable methodological advantages that strengthen our findings. First, we used an original protocol to investigate both the endogenous leptin uptake by the CNS and the central leptin sensitivity on exogenous leptin. Second, the minipig has closer physiological parameters to humans compared with rodents 12. We cannot exclude that some of the discrepancies of our results with previous rodent studies may be linked to natural physiological differences existing between mice and minipigs. Our study also has certain limitations. First, we did not investigate the functional aspect of exogenous leptin injection on food intake and body weight in minipigs and thus cannot depict a complete biology system model. Second, CSF leptin levels were not assessed after exogenous leptin injections and therefore this study cannot provide evidence for the relative access of exogenous leptin to the CNS in our model. Third, our study was not designed to test the assumption of a linear relationships between plasma and CSF leptin, regardless of leptin concentrations or nutritional status (lean versus obese conditions), and thus could not provide evidence that CSF:serum ratios would remain constant in lean minipigs following an experimental rise in plasma leptin to levels similar to the obese ones.
From a translational point of view, the phenotype of the minipigs fed a HFD for 10 weeks resembles that of “metabolically healthy” subjects with obesity, who, given more time or additional weight gain, are likely to experience functional deterioration leading to metabolically unhealthy obesity 32, with a significantly greater risk of death and cardiovascular events than individuals with normal weight 33. Identifying key pathways to be targeted pharmacologically for early intervention to hamper the onset of metabolically unhealthy obesity and associated comorbidities in the population with obesity is of high importance. For example, the LepR‐dependent tanycytic regulation of hypothalamic leptin uptake that is impaired in diet‐induced obesity can be rescued by pharmacological treatment 9. Although we did not investigate whether an intervention increasing the amount of leptin able to bind to its receptor in the hypothalamus would lead to an additional effect than the ceiling effect observed in our study, it has been suggested that leptin sensitizers (e.g., metformin) coadministered with leptin improve the exogenous leptin action by restoring both the leptin transport and the leptin receptor signaling 34. Future antiobesity pharmacotherapy would clearly benefit from simultaneously targeting different causal mechanisms involved in obesity 34.
To conclude, our minipig model is consistent with the conjecture that the alteration of leptin transport into the CNS precedes the occurrence of any metabolic dysfunction despite the progression to diet‐induced obesity, although additional studies are still required to confirm this mechanism. Assessment of leptin in CSF and serum is an easy method for preclinical pharmacology studies, which would target the restoration of leptin transport in obesity. In this translational application, the minipig represents a relevant preclinical model to investigate the pharmacological effect of such novel leptin sensitizers.
Supporting information
Acknowledgments
The authors thank Dr. S. Rasika for the editing of the manuscript and Julie Kerr‐Conte and her whole team from the laboratory of biotherapies for diabetes, Inserm U1190, for their support and advice; Michel Pottier, Arnold Dive, Audrey Quenon, Martin Fourdrinier, and Nicolas Durieux for MRI assessment; and Maurice de Meulaere from the laboratory of anatomy and Martine Michiels from the Centre de Biologie et Pathologie, Centre Hospitalo‐Universitaire de Lille, for their technical help.
Funding agencies: This work was supported by grants from the Fond Français de l’Alimentation (FFA), the Société Française de Nutrition (SFN), the Institut Benjamin Delessert, the Diamenord Association, and the Agence Nationale de la Recherche (ANR‐15‐CE14‐0025, Glioshuttles4metabolism; Distalz).
Disclosure: The authors declared no conflicts of interest.
Author contributions: MP, VP, and FP designed research studies; AC, AD, TH, DM, MD, VP, and MP conducted experiments, acquired data, and analyzed data; AC, AD, TH, PC, CF, and RC provided reagents and discussed data; and AC, AD, MP, and VP wrote the manuscript.
Contributor Information
Vincent Prevot, Email: vincent.prevot@inserm.fr.
Marie Pigeyre, Email: marie.pigeyre@univ-lille.fr.
References
- 1. Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet‐induced resistance to leptin action. Nat Med 1995;1:1311‐1314. [DOI] [PubMed] [Google Scholar]
- 2. de Luca C, Kowalski TJ, Zhang Y, et al. Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron‐specific LEPR‐B transgenes. J Clin Invest 2005;115:3484‐3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coppari R, Ichinose M, Lee CE, et al. The hypothalamic arcuate nucleus: a key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab 2005;1:63‐72. [DOI] [PubMed] [Google Scholar]
- 4. Pan WW, Myers MG. Leptin and the maintenance of elevated body weight. Nat Rev Neurosci 2018;19:95‐105. [DOI] [PubMed] [Google Scholar]
- 5. Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D. Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med 1996;2:589‐593. [DOI] [PubMed] [Google Scholar]
- 6. Caro JF, Kolaczynski JW, Nyce MR, et al. Decreased cerebrospinal‐fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet 1996;348:159‐161. [DOI] [PubMed] [Google Scholar]
- 7. Friedman JM, Mantzoros CS. 20 years of leptin: from the discovery of the leptin gene to leptin in our therapeutic armamentarium. Metabolism 2015;64:1‐4. [DOI] [PubMed] [Google Scholar]
- 8. Banks WA, Farrell CL. Impaired transport of leptin across the blood‐brain barrier in obesity is acquired and reversible. Am J Physiol Endocrinol Metab 2003;285:E10‐E15. [DOI] [PubMed] [Google Scholar]
- 9. Balland E, Dam J, Langlet F, et al. Hypothalamic tanycytes are an ERK‐gated conduit for leptin into the brain. Cell Metab 2014;19:293‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prevot V, Dehouck B, Sharif A, Ciofi P, Giacobini P, Clasadonte J. The versatile tanycyte: a hypothalamic integrator of reproduction and energy metabolism. Endocr Rev 2018;39:333‐368. [DOI] [PubMed] [Google Scholar]
- 11. García‐Cáceres C, Balland E, Prevot V, et al. Role of astrocytes, microglia, and tanycytes in brain control of systemic metabolism. Nat Neurosci 2019;22:7‐14. [DOI] [PubMed] [Google Scholar]
- 12. Dolgin E. Minipig, minipig, let me in. Nat Med 2010;16:1349.21135823 [Google Scholar]
- 13. Val‐Laillet D, Guerin S, Malbert CH. Slower eating rate is independent to gastric emptying in obese minipigs. Physiol Behav 2010;101:462‐468. [DOI] [PubMed] [Google Scholar]
- 14. Val‐Laillet D, Blat S, Louveau I, Malbert CH. A computed tomography scan application to evaluate adiposity in a minipig model of human obesity. Br J Nutr 2010;104:1719‐1728. [DOI] [PubMed] [Google Scholar]
- 15. Ettrup KS, Glud AN, Orlowski D, et al. Basic surgical techniques in the Gottingen minipig: intubation, bladder catheterization, femoral vessel catheterization, and transcardial perfusion. J Vis Exp 2011;52:2652. doi: 10.3791/2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ettrup KS, Sorensen JC, Bjarkam CR. The anatomy of the Gottingen minipig hypothalamus. J Chem Neuroanat 2010;39:151‐165. [DOI] [PubMed] [Google Scholar]
- 17. Lee L, Alloosh M, Saxena R, et al. Nutritional model of steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Hepatology 2009;50:56‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caron E, Sachot C, Prevot V, Bouret SG. Distribution of leptin‐sensitive cells in the postnatal and adult mouse brain. J Comp Neurol 2010;518:459‐476. [DOI] [PubMed] [Google Scholar]
- 19. Collden G, Balland E, Parkash J, et al. Neonatal overnutrition causes early alterations in the central response to peripheral ghrelin. Mol Metab 2015;4:15‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Secher A, Jelsing J, Baquero AF, et al. The arcuate nucleus mediates GLP‐1 receptor agonist liraglutide‐dependent weight loss. J Clin Invest 2014;124:4473‐4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parkash J, Messina A, Langlet F, et al. Semaphorin7A regulates neuroglial plasticity in the adult hypothalamic median eminence. Nat Commun 2015;6:6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Langlet F, Levin BE, Luquet S, et al. Tanycytic VEGF‐A boosts blood‐hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell Metab 2013;17:607‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sawamoto K, Wichterle H, Gonzalez‐Perez O, et al. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science 2006;311:629‐632. [DOI] [PubMed] [Google Scholar]
- 24. Conductier G, Brau F, Viola A, et al. Melanin‐concentrating hormone regulates beat frequency of ependymal cilia and ventricular volume. Nat Neurosci 2013;16:845‐847. [DOI] [PubMed] [Google Scholar]
- 25. Faubel R, Westendorf C, Bodenschatz E, Eichele G. Cilia‐based flow network in the brain ventricles. Science 2016;353:176‐178. [DOI] [PubMed] [Google Scholar]
- 26. Ottaway N, Mahbod P, Rivero B, et al. Diet‐induced obese mice retain endogenous leptin action. Cell Metab 2015;21:877‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Munzberg H, Flier JS, Bjorbaek C. Region‐specific leptin resistance within the hypothalamus of diet‐induced obese mice. Endocrinology 2004;145:4880‐4889. [DOI] [PubMed] [Google Scholar]
- 28. Van Heek M, Compton DS, France CF, et al. Diet‐induced obese mice develop peripheral, but not central, resistance to leptin. J Clin Invest 1997;99:385‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. El‐Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet‐induced obesity. J Clin Invest 2000;105:1827‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kleinert M, Kotzbeck P, Altendorfer‐Kroath T, Birngruber T, Tschöp MH, Clemmensen C. Time‐resolved hypothalamic open flow micro‐perfusion reveals normal leptin transport across the blood‐brain barrier in leptin resistant mice. Mol Metab 2018;13:77‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di Spiezio A, Sandin ES, Dore R, et al. The LepR‐mediated leptin transport across brain barriers controls food reward. Mol Metab 2018;8:13‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cefalu WT, Bray GA, Home PD, et al. Advances in the science, treatment, and prevention of the disease of obesity: reflections from a Diabetes Care Editors’ Expert Forum. Diabetes Care 2015;38:1567‐1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kramer CK, Zinman B, Retnakaran R. Are metabolically healthy overweight and obesity benign conditions?: A systematic review and meta‐analysis. Ann Intern Med 2013;159:758‐769. [DOI] [PubMed] [Google Scholar]
- 34. Quarta C, Sánchez‐Garrido MA, Tschöp MH, Clemmensen C. Renaissance of leptin for obesity therapy. Diabetologia 2016;59:920‐927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials