

Abstract
Boron has been employed in materials science as a marker for imaging specific structures by electron energy loss spectroscopy (EELS) or secondary ion mass spectrometry (SIMS). It has a strong potential in biological analyses as well; however, the specific coupling of a sufficient number of boron atoms to a biological structure has proven challenging. Herein, we synthesize tags containing closo‐1,2‐dicarbadodecaborane, coupled to soluble peptides, which were integrated in specific proteins by click chemistry in mammalian cells and were also coupled to nanobodies for use in immunocytochemistry experiments. The tags were fully functional in biological samples, as demonstrated by nanoSIMS imaging of cell cultures. The boron signal revealed the protein of interest, while other SIMS channels were used for imaging different positive ions, such as the cellular metal ions. This allows, for the first time, the simultaneous imaging of such ions with a protein of interest and will enable new biological applications in the SIMS field.
Keywords: carboranes, click chemistry, nanobodies, secondary-ion mass spectrometry, specific labeling
Optical imaging is extremely powerful in biological sciences because it is based on a variety of fluorescent tags that are easily modified and thus enable a large variety of experiments.1, 2 However, many types of information cannot be obtained by light microscopy, including extremely small structures, which are typically addressed by electron microscopy (EM), or the sample composition, in terms of atoms or molecules, which can be studied by secondary ion mass spectrometry imaging (SIMS).2, 3 These non‐optical imaging approaches also need to employ labeling tags to reveal specific biological structures. However, the labeling probes for EM, and especially for SIMS, lag substantially behind the potential of the fluorescence tags in terms of flexibility, specificity, and amount of signal generated. EM typically relies on probes containing heavy metals, such as gold particles, which are dense enough to be observed in the resulting images.4 Such probes have been optimized for several decades and perform satisfactorily. Probes for SIMS have been less researched. They need to consist of elements that are revealed by ionization,5 and such ions need to be rare in the sample of interest and present on the target molecule in sufficient density to be detectable (as one fluorophore is detectable in light microscopy, but one atom is not sufficient for SIMS). One could in principle, use gold‐labeled antibodies as in EM. They have been used successfully in a few publications,6, 7 and gold or platinum deposits have also been analyzed,8, 9 especially in tumors, in relation to tumor therapy approaches. However, all of the SIMS analyses of gold have been based on Au− ions since Au+ is difficult to measure owing to its very poor ionization characteristics.10, 11, 12 At the same time, it has been noted that gold particles appear to modify the sample ionization in their surroundings, for example for CN− ions.7 Therefore, efforts are needed for generating new probes, especially for the positive ion domain. Lanthanides have been employed in several technologies (SIMS; EELS) but typically require individual metal particles to be either linked to antibodies (which lowers the particle density that can be used, owing to the large antibody size) or to be precipitated on the cells.13, 14
A simple and flexible solution to this could be to use boron, which typically has a strong signal in techniques such as nanoSIMS or EELS.15 The main advantage of boron over gold is that the former can be coupled to small, flexible molecular scaffolds. These could, in turn, be linked directly to target proteins or could be covalently attached to immunolabeling reagents, including antibodies or even camelid single domain antibodies (sdAbs, also known as nanobodies).16 This approach, in which high amounts of boron would be integrated either in a protein or in a small nanobody that is directly bound to the protein, would provide high labeling densities, as well as high spatial precision owing to the small probe size (ca. 2–4 nm), which places the label far closer to the protein of interest than do the conventional antibodies (ca. 15 nm) coupled to metal particles.
However, boron is chemically intractable and is difficult to couple to specific proteins or other biological elements. In this study, we solved this issue by targeting boron‐rich polyhedral clusters, especially the ortho‐carboranes. Their pseudoaromatic characteristics enables a substitution reaction on carbon without cage decomposition,17 thus facilitating their coupling to peptides. We applied carboranyl peptides to several biological labeling procedures, followed by nanoSIMS imaging.
For this purpose, specific incorporation of carborane derivatives was expanded to different strategies (Figure 1 A). The first strategy is based on co‐translational incorporation of an unnatural amino acid (UAA) in cellular proteins followed by a bioorthogonal and chemoselective reaction between the UAA and the carborane probe.18, 19 The alkyne‐modified proteins are coupled directly with azide‐functionalized carborane probes by the copper(I)‐catalyzed Huisgen‐cycloaddition,20 also known as click chemistry. The second strategy relies on first coupling the reactive carborane probe to an ectopic cysteine of a nanobody through a thiol–maleimide reaction, followed by the immunostaining of cells.21
Figure 1.

A) Strategies of specific incorporation of boron‐enriched compounds into cellular proteins. BorEncode and BorLink, shown in detail in (B), consist of hydrophobic aminobutyl‐ortho‐carboranes, linked to soluble short peptides bearing additional functional groups, azide or maleimide, respectively, and the fluorophore Star635.
Two molecules were synthesized with the same boron precursor, but with different reactive functional groups, to fulfill the requirements of both strategies. We call them BorEncode and BorLink for the click chemistry and nanobody strategies, respectively (Figure 1 B). We selected aminobutyl‐ortho‐carborane22 to attach it to peptides, similar to the coupling of amino acids. Each synthesis started from a Sieber amide resin to generate nonapeptides, by following the automatic or manual microwave‐assisted Fmoc protocol (Supporting Information, Section 3). Afterwards, the N α,N ϵ‐unprotected l‐lysine in the resin‐bound peptides was linked with succinic acid (SA) by a short treatment with succinic anhydride, N,N‐diisopropylethyamine (DIEA) in N,N‐dimethylformamide (DMF). Then, the N‐terminally obtained carboxylic groups were converted in situ to active esters by using O‐(7‐azabenzotriazol‐1‐yl)‐N,N,N′,N′‐tetramethyl‐uronium‐hexafluoro‐phosphate (HATU) in the presence of DIEA and accompanied by coupling of amino‐ortho‐carborane (o‐CB). After acidic cleavage of the carboranyl peptides from the resin, the fluorophore Star635 was attached, either to a lysine by an amine‐NHS ester reaction (for BorEncode, see the Supporting Information, Section 3.1) or to a cysteine by a thiol–maleimide reaction (for BorLink, see the Supporting Information, Section 3.2). The fluorophore was implemented to enable us to quickly verify the cellular incorporation of the probes by fluorescence microscopy, before the more time‐consuming SIMS analysis, and also to facilitate the selection of areas of interest in the sample. In the case of BorLink, an additional step was required to activate the probe for nanobody coupling. For this, we have chosen succinimidyl 4‐(N‐maleimidomethyl)‐cyclohexane‐1‐carboxylate (SMCC) to functionalize the lysine at position 2. The maleimide in SMCC is known to be more stable towards hydrolysis under physiological conditions, owing to the steric hindrance caused by the cyclohexane ring.23 Each reaction, performed after cleavage from solid support, was analyzed by HPLC. The peaks related to the desired products could be confirmed by HR‐ESI‐MS measurements. After identification, the products were cleaned by HPLC to reach purities of ≥95 %. BorEncode was ready to be used in click‐based approaches, after testing its function in a reaction with propargyl‐l‐lysine (PRK) catalyzed by copper(I) in aqueous ethanol (Supporting Information, Section 3.1). BorLink had to be coupled first to a nanobody, then purified by size exclusion chromatography (SEC), and was analyzed by SDS‐PAGE (Supporting Information, Section 3.3). As a simple proof of principle, two commercially available nanobodies against green fluorescent protein (GFP) were used, which were able to recognize simultaneously two independent epitopes on each GFP molecule. BorEncode and BorLink bear 20 boron atoms each. Therefore, by using both anti‐GFP BorLink‐nanobodies (BorLink‐GFP‐Nbs), the number of boron atoms is doubled per GFP.
To test the boron contents of the probes, we turned to nanoscale secondary ion mass spectrometry (nanoSIMS), which enables isotopic element analysis at high spatial resolution by detecting secondary ions sputtered from the surface of biological samples. In the past, we have generated two probes containing elements (19F, 15N) that generate negative ions under nanoSIMS, which enabled the analysis of the proteins of interest together with other negative ions that are highly abundant in biological samples (e.g., C and CN).24, 25 However, probes that enable the analysis of specific proteins or structures in the positive ion mode, together with positive ions, such as the cellular metal ions, have been more difficult to obtain. We therefore tested our probes in the positive ion mode to detect 11B ions and to visualize specific structures with subcellular resolution.
We first tested the BorEncode in cells having alkyne‐modified proteins after co‐translational incorporation. Then we tested the BorLink‐GFP‐Nbs by immunostaining cells expressing a specific protein fused to a GFP variant (Supporting Information, Section 4). For the first strategy, baby hamster kidney cells (BHK) were cultured in a medium lacking methionine but containing homopropargyl‐l‐glycine (HPG), which resulted in the integration of this unnatural amino acid in all newly synthesized proteins at the methionine location.19 Afterwards the cultured cells were fixed and permeabilized, and their alkyne‐modified proteins were stained with the azide‐bearing BorEncode in the presence of copper(I). After plastic embedding of the sample, sections of 200 nm were prepared, and nanoSIMS images were recorded on a NanoSIMS 50 L instrument using the Hyperion II dual polarity oxygen source as a primary ion source. The signal of 11B species was collected in a m/z detector to visualize the abundance of probe‐labeled samples (Figure 2 A). In addition, 23Na and 40Ca signals were simultaneously collected to show the natural occurrences of these isotopes. Both isotopes ensured cell detection and acted as controls. 23Na was concentrated in the nucleus region, whereas 40Ca was more widely distributed throughout the cell. 11B could be clearly visualized inside the probe‐containing cells, while non‐labeled cells failed to give 11B signals. Moreover, 11B was distributed almost homogenously across the cell as expected owing to the incorporation of HPG in all newly produced proteins. Statistical data analysis showed a highly significant difference of 11B intensity between boron‐labeled and non‐labeled cells.
Figure 2.

BHK cells that incorporated HPG in all newly synthesized proteins were labeled with BorEncode following strategy I. A) 11B nanoSIMS image of a representative labeled cell (top row) and a non‐labeled cell (bottom row). The other images show 23Na and 40Ca. The overlay shows the co‐localization of 11B (green), 23Na (blue), and 40Ca (red) in the cells. Scale bar=5 μm. B) Images of the labeled (top row) and non‐labeled cells (bottom row) obtained with an epifluorescence microscope before the nanoSIMS measurement. The fluorescence signal of the BorEncode is shown in magenta. The autofluorescence of the cells is shown in gray. The overlay image compares the signal of 40Ca to the autofluorescence (gray) in cells, to indicate that indeed the same cell has been imaged. The fluorescence and 11B nanoSIMS images show good correlation. Scale bar=5 μm. C) Plot of the normalized boron signal intensity. Significantly higher levels of 11B are detected in labeled cells than in the negative control. The difference was highly significant (p<0.0001), as determined by Wilcoxon rank sum test. Analyzed number of circular cellular regions of interest (ROIs): 60. The middle line indicates the median, the box edges the 25th percentiles, the error bars the 75th percentiles, and the dots indicate the 90th percentile.
Based on these encouraging results, we proceeded to tag one specific protein from the entire cell. Therefore, BHK cells were transfected with a plasmid encoding for the transmembrane protein syntaxin 1, fused to yellow fluorescent protein (YFP) and including an Amber stop codon in between syntaxin 1 and YFP. Using an expanded genetic code system,18, 24, 25 cells incorporated propargyl‐l‐lysine (PRK) on a defined position, at the Amber codon in syntaxin 1–YFP. Afterwards, cells were chemically fixed and stained with BorEncode in the same fashion as described above. The labeling of syntaxin 1‐YFP with BorEncode was verified by fluorescence microscopy through overlapping of the YFP and the BorEncode signals, before embedding. After embedding and sectioning, the fluorescence of YFP was lost. However, faint fluorescence signals from the Star635 fluorophore were observed, which helped to locate the click‐labeled cells (magenta in Figure 3 B), while all other cells were detected by their autofluorescence (gray in Figure 3 B). These Star635 positive areas were then imaged using nanoSIMS. The signal measured by nanoSIMS was in good agreement with the fluorescence image, in that high signals from 11B were observed in transfected cells but not in the non‐transfected ones. The 11B signal from cells expressing syntaxin 1‐YFP was, as expected, lower in comparison to the signal obtained from total protein labeling in Figure 2, but still sufficient to reveal this protein specifically in the cellular context of other naturally present metal ions such as 23Na and 40Ca (Figure 3).
Figure 3.

BHK cells expressing PRK‐modified syntaxin 1 were labeled with BorEncode following strategy I. A) NanoSIMS images of 11B signal (green) in transfected (top row) and non‐transfected cells (bottom row). 23Na (blue) and 40Ca (red) are also shown, as well as overlay images of 11B, 23Na, and 40Ca. Scale bar=5 μm. B) Images of the transfected (top row) and non‐transfected cells (bottom row) obtained with an epifluorescence microscope before the nanoSIMS measurement. Although the different pixel sizes and imaging depth of the two procedures makes it difficult to overlay the two images perfectly, the fluorescence and 11B signals are in agreement in the transfected cells. Scale bar=5 μm. The less “crisp” appearance of the images, when compared to Figure 2, is due to the difference in the image sizes; the actual resolution is similar in both figures. C) Plot of the normalized boron signal intensity (*** p<0.0001). Analyzed number of ROIs: 120.
For the second strategy, cell lines derived from monkey kidney (COS‐7) or human embryonic kidney (HEK293) were first transfected with a plasmid encoding for the mitochondrial protein TOM70 fused to GFP. After 18 hours of expressing this fusion protein, cells were chemically fixed and were immunostained with both BorLink‐GFP‐Nbs (Figure 4; see Methods in the Supporting Information for details). By mixing both nanobodies, a density of up to 40 boron atoms per GFP protein can be achieved. The embedding and sectioning was performed similarly to the first strategy. The immunostaining with anti‐GFP nanobodies conjugated to BorLink on COS‐7 cells was also examined by fluorescence microscopy. The results demonstrate a highly specific labeling for the protein of interest TOM70‐GFP (Supporting Information, Section 6). The labeling strategy using nanobody immunostaining thus provided sufficient sensitivity for specific target proteins imaged with nanoSIMS.
Figure 4.
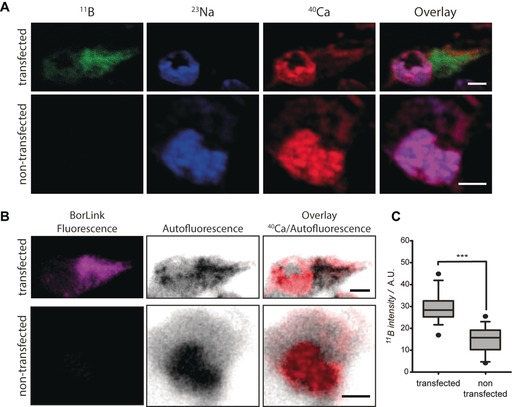
COS‐7 cells transfected with the protein TOM70‐GFP and labeled afterwards with BorLink‐GFP‐Nbs. A) NanoSIMS images of 11B signal (green) in transfected and non‐transfected cells. 23Na (blue) and 40Ca (red) are also shown, as well as overlay images of 11B, 23Na, and 40Ca. Scale bar=5 μm. B) Images of the transfected and non‐transfected cells obtained with an epifluorescence microscope before the nanoSIMS measurement. Although the different pixel sizes and imaging depth of the two procedures makes it difficult to overlay the two images perfectly, the fluorescence and 11B signals are in agreement in the transfected cells. Scale bar=5 μm. C) Plot of the normalized boron signal intensity for transfected cells compared to non‐transfected cells (*** p<0.0001). Analyzed number of ROIs: 150.
To image the molecules of interest with nanoSIMS, it is necessary to label them with specific isotopes. Other SIMS approaches, such as cluster ion SIMS, also work in a label‐free fashion, at least for small metabolites and lipids up to m/z 2000,26, 27, 28 albeit not for larger molecules such as proteins and peptides. For these technologies, the boron probes we developed could be used for labeling and imaging specific proteins. This would enable cluster ion SIMS to investigate the metabolite and lipid composition of a sample, while also knowing exactly where specific proteins of interest are located,29 which would open new avenues in tissue and metabolite analysis.
We have demonstrated that these boron probes are potential labeling probes for total and specific molecular imaging using non‐optical nanoSIMS. The probes work as small tags, which can be directly coupled to their targets, either by co‐translational incorporation of alkyne‐modified amino acids followed by click chemistry or by immunostaining with nanobodies. This should also enable their use in a more general way, by coupling similar probes to secondary nanobodies30 (directed against mouse or rabbit antibodies), which should render the probes immediately applicable in any type of antibody‐based imaging and in any type of sample.
Boron probes are more advantageous than previously available probes for positive ion mode SIMS imaging, such as the metal‐containing tags, functionalized by polymerized ligand backbones. These probes are very large, which results in low spatial precision (not ideal for nanoSIMS) and a low signal density.14 Nevertheless, further improvements in the sensitivity of these probes for nanoSIMS analysis should be made by increasing the amount of elements (boron atoms) without reducing the solubility of the boron probes and without affecting the coupling reactions. Such improved probes should also be applicable for other non‐optical techniques, even in the EM domain, through electron energy loss spectroscopy (EELS).31 Finally, the boron probe could be in the future combined with two other similarly flexible probes we developed in the past24, 25 for the analysis of negative ions. This would allow multi‐protein detection with nanoSIMS or cluster ion SIMS and would thereby enable a plethora of new experiments. At the same time, this should encourage future work on developing further specific SIMS probes, containing other elements that are rare in biological samples, such as I or Si.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Prof. Dr. Ulf Diederichsen and Dr. Franziska Thomas (Institute of Organic and Biomolecular Chemistry, University of Göttingen, Germany) for the generous support by using their infrastructure. We also acknowledge the service department of the faculty of chemistry (University Göttingen, Germany), especially Dr. Holm Frauendorf, Dr. Michael John, and their teams for mass and NMR measurements. The work was supported by grants to S.O.R. from the European Research Council (ERC‐2013‐CoG NeuroMolAnatomy) and from the Deutsche Forschungsgemeinschaft (DFG) 1967/7‐1 and by grants from DFG (SFB1286/B1) and VR (Swedish Research Council) to N.T.N.P.
S. Kabatas, P. Agüi-Gonzalez, K.-A. Saal, S. Jähne, F. Opazo, S. O. Rizzoli, N. T. N. Phan, Angew. Chem. Int. Ed. 2019, 58, 3438.
Contributor Information
Dr. Selda Kabatas, Email: skabata@gwdg.de.
Prof. Dr. Silvio O. Rizzoli, Email: srizzol@gwdg.de.
Dr. Nhu T. N. Phan, Email: thi.phan@med.uni-goettingen.de.
References
- 1. Drummen G. P. C., Molecules 2012, 17, 14067–14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fornasiero E. F., Opazo F., Bioessays 2015, 37, 436–451. [DOI] [PubMed] [Google Scholar]
- 3. da Cunha M. M. L., Trepout S., Messaoudi C., Wu T.-D., Ortega R., Guerquin-Kern J.-L., Marco S., Micron 2016, 84, 26–36. [DOI] [PubMed] [Google Scholar]
- 4. Watson M. L., J. Biophys. Biochem. Cytol. 1958, 4, 475–478; [DOI] [PMC free article] [PubMed] [Google Scholar]; Chatterjee S., Roy A., Laskar A., Adv. Sci. Technol. 2012, 891–902. [Google Scholar]
- 5. Pól J., Strohalm M., Havlíček V., Volný M., Histochem. Cell Biol. 2010, 134, 423–443. [DOI] [PubMed] [Google Scholar]
- 6. Thiery-Lavenant G., Guillermier C., Wang M., Lechene C., Surf. Interface Anal. 2014, 46, 147–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pett-Ridge J., Weber P. K., Methods Mol. Biol. 2012, 881, 375–408. [DOI] [PubMed] [Google Scholar]
- 8. Wedlock L. E., Kilburn M. R., Cliff J. B., Filgueira L., Saunders M., Berners-Price S. J., Metallomics 2011, 3, 917–925. [DOI] [PubMed] [Google Scholar]
- 9. Wedlock L. E., Kilburn M. R., Liu R., Shaw J. A., Berners-Price S. J., Farrell N. P., Chem. Commun. 2013, 49, 6944–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nuñez J., Renslow R., Cliff J. B., Anderton C. R., Biointerphases 2018, 13, 03B301. [DOI] [PubMed] [Google Scholar]
- 11. Clegg J. B., Surf. Interface Anal. 1991, 17, 221–221. [Google Scholar]
- 12. Newbury D. E. in Characterization of Particles, (Ed.: K. F. J. Heinrich), NBS special publication 533, Michigan, 1980, pp. 139–151. [Google Scholar]
- 13. Adams S. R., Mackey M. R., Ramachandra R., Lemieux S. F. P., Steinbach P., Bushong E. A., Butko M. T., Glepmans B. N. G., Ellisman M. H., Tsien R. Y., Cell Chem. Biol. 2016, 23, 1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Angelo M., Bendall S. C., Finck R., Hale M. B., Hitzman C., Borowsky A. D., Levenson R. M., Lowe J. B., Liu S. D., Zhao S., Natkunam Y., Nolan G. P., Nat. Med. 2014, 20, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wirtz T., Philipp P., Audinot J. N., Dowsett D., Eswara S., Nanotechnology 2015, 26, 434001. [DOI] [PubMed] [Google Scholar]
- 16. Beghein E., Gettemans J., Front. Immunol. 2017, 8, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leśnikowski Z. J., J. Med. Chem. 2016, 59, 7738–7758. [DOI] [PubMed] [Google Scholar]
- 18. Liu C. C., Schultz P. G., Annu. Rev. Biochem. 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- 19. Beatty K. E., Liu J. C., Xie F., Dieterich D. C., Schuman E. M., Wang Q., Tirell D. A., Angew. Chem. Int. Ed. 2006, 45, 7364–7367; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7524–7527. [Google Scholar]
- 20. Wang Q., Chan T. R., Hilgraf R., Fokin V. V., Sharpless K. B., Finn M. G., J. Am. Chem. Soc. 2003, 125, 3192–3193. [DOI] [PubMed] [Google Scholar]
- 21. Massa S., Xavier C., De Vos J., Caveliers V., Lahoutte T., Muyldermans S., Devoogdt N., Bioconjugate Chem. 2014, 25, 979–988. [DOI] [PubMed] [Google Scholar]
- 22. Lai C.-H-, Lin Y.-C., Chou F.-I., Liang C.-F., Lin E.-W., Chuang Y.-J., Lin C.-C., Chem. Commun. 2012, 48, 612–614. [DOI] [PubMed] [Google Scholar]
- 23. Hashida S., Imagawa M., Inoue S., Ruan K. H., Ishikawa E., J. Appl. Biochem. 1984, 6, 56–63. [PubMed] [Google Scholar]
- 24. Vreja I. C., Kabatas S., Saka S. K., Kröhnert K., Höschen C., Opazo F., Diederichsen U., Rizzoli S. O., Angew. Chem. Int. Ed. 2015, 54, 5784–5788; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5876–5880. [Google Scholar]
- 25. Kabatas S., Vreja I. C., Saka S. K., Höschen C., Kröhnert K., Opazo F., Rizzoli S. O., Diederichsen U., Chem. Commun. 2015, 51, 13221–13224. [DOI] [PubMed] [Google Scholar]
- 26. Tian H., Sparvero L. J., Amoscato A. A., Bloom A., Bayır H., Kagan V. E., Winograd N., Anal. Chem. 2017, 89, 4611–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fletcher J. S., Analyst 2009, 134, 2204–2215. [DOI] [PubMed] [Google Scholar]
- 28. Passarelli M. K., Ewing A. G., Winograd N., Anal. Chem. 2013, 85, 2231–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sheng L., Cai L., Wang J., Li Z., Mo Y., Zhang S., Xu J.-J., Zhang X., Chen H.-Y., Int. J. Mass Spectrom. 2017, 421, 238–244. [Google Scholar]
- 30. Pleiner T., Bates M., Görlich D., J. Cell Biol. 2018, 217, 1143–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Michel J., Sauerwein W., Wittig A., Balossier G., Zierold K., J. Microsc. 2003, 210, 25–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary