

Abstract
A general approach for the efficient hydrogen‐isotope exchange of nucleobase derivatives is described. Catalyzed by ruthenium nanoparticles, using mild reaction conditions, and involving either D2 or T2 as isotopic sources, this reaction possesses a wide substrate scope and a high solvent tolerability. This novel method facilitates the access to essential diagnostic tools in drug discovery and development: tritiated pharmaceuticals with high specific activities and deuterated oligonucleotides suitable for use as internal standards during LC‐MS quantification.
Keywords: C−H activation, isotopes, isotopic labelling, nanoparticles, nucleotides
Nucleobases are one of the most important chemical building blocks of life. They can be divided into two groups: purines containing adenine and guanine, and pyrimidines including cytosine, thymine, and uracil. These nitrogen‐containing heterocycles are involved in many biological processes such as transmission of genetic information, energetic processes, or cell communication. Therefore, a myriad of nucleobase derivatives have been found to be potent drugs for the treatment of major public health concerns like HIV, asthma, or cancer.1 More recently, RNA interference has emerged as an experimental tool to inhibit the expression of genes and gave birth to, respectively, antisense oligonucleotides2 and small interfering RNA (siRNA),3 which are under intensive studies as high potential therapeutic agents.4 Because of their complex and particularly fragile nature, performing selective chemical transformations on these molecules represents an important challenge. Among those transformations, the development of efficient and selective hydrogen‐isotope exchange (HIE) reactions, allowing late‐stage deuterium and tritium labelling of nucleobase derivatives, is of paramount importance.5 Indeed, tritiated analogues of drug candidates, are essential tools for investigating the in vivo fate of substances within absorption, distribution, metabolism and excretion (ADME) studies.6 Deuterated molecules are also widely employed in various life‐science fields, such as metabolomics and proteomics, as stable isotopically labelled internal standards (SILS) for quantitative GC‐ or LC‐MS analyses.7 In the case of oligonucleotides, the synthetic access to SILS is particularly problematic because of their complexity and relative instability. Up to now, the nucleobase hydrogen‐isotope labelling is mainly achieved by multistep synthesis starting from precursors such as halogen, ketone, or aldehyde derivatives followed by, respectively, catalytic dehalogenation using D2/T2 gas8 or reduction with labelled reagents9 (NaBD4 or LiAlT4 for instance). Nevertheless, methods involving HIE have also been reported for such substructures based on the acido‐basic exchange of the position 8 of purine derivatives,10 or on Pd/C‐catalyzed HIE in deuterated water (Figure 1).11 In both cases, the high temperature required to obtain good isotopic enrichments drastically limits the application of such reactions for the labelling of complex and fragile nucleobase‐containing molecules.
Figure 1.
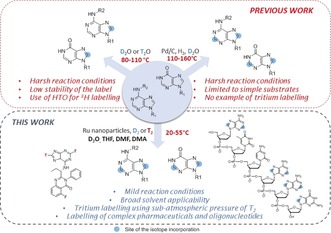
State‐of‐the‐art HIE for purine derivatives and their features compared to our new method using Ru nanocatalysts.
Moreover, methods previously described for tritium labelling are not particularly suited because use of hazardous tritiated water as an isotopic source is required. Herein, we present a wide‐spectrum method for the deuterium and tritium labelling of purine substructures using Ru nanoparticles (RuNp) as catalysts and deuterium and tritium gas, respectively, as isotopic sources (Figure 1). This new method allows the regioselective incorporation of hydrogen isotopes in complex biologically relevant substrates including pharmaceuticals and biomolecules under mild reaction conditions. Moreover, we show that our protocol can be used for the synthesis of radioactive tracers employing a sub‐atmospheric pressure of T2 gas, and of deuterated oligonucleotides suitable to be used as SILS for quantitative LC‐MS analyses.
At an early stage of this study, by screening different types of catalysts for HIE, we found that ruthenium nanoparticles embedded in a PVP polymer matrix (RuNp@PVP) may have great potential for the labelling of purine derivatives.12 Using optimized reaction conditions (5 mol % of catalyst, 2 bar of D2 and D2O as solvent at 55 °C), the purine (1d) was labelled, with an isotope incorporation at each available position α to a nitrogen atom (isotopic enrichments of 90 %), leading to a high deuterium uptake of 2.7 D without any secondary reactions otherwise observed (Figure 2; see the Supporting Information). Encouraged by this preliminary result we moved forward to the labelling of purine‐based natural substrates such as nucleosides, nucleotides, and analogues.
Figure 2.
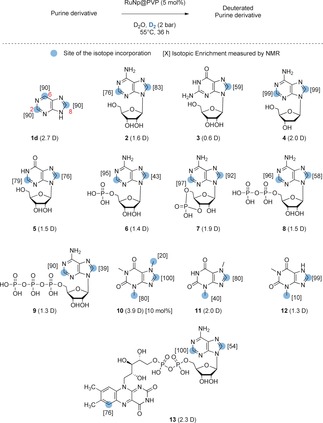
RuNp‐catalyzed deuterium labelling of representative purine derivatives.
Besides the numerous applications evoked in the introduction, deuterated nucleotides are also widely used in biochemistry and molecular biology to suppress non‐essential proton resonance in NMR structural studies and for atom‐transfer experiments with DNA damaging agents.9a, 13 Adenosine (2) and guanosine (3) were selectively deuterated on the purine core using the same reaction conditions described above (Figure 2). To ensure that the labelling was a result of the combined action of the Ru nanoparticles and D2 as an isotopic source, control experiments using only D2O were performed. As expected, a weak to average labelling of the most acidic position 8 was obtained without the presence of Ru nanoparticles and D2 gas. Interestingly, the attractive position 2, known to not be prone to back‐exchange during biodistribution studies, is efficiently labelled in the case of 2 using our method. Furthermore, the broad scope of this transformation was demonstrated by the labelling of deoxyadenosine (4), inosine (5), and nucleotides such as AMP (6), c‐AMP (7), ADP (8) and ATP (9). Remarkably, despite the coordination ability of phosphate groups for metallic nanoparticles, both selectivity and deuterium incorporation remained very high even in the case of ATP which contains a trisphosphate moiety. Interestingly, only slight changes in deuterium incorporation were observed depending on the pH of the solution for 9 (see Table S1 in the Supporting Information) highlighting the robustness of this new catalytic process. Analogues like the deuterated xanthines 10, 11, and 12, widely used as internal standards in qualitative or quantitative isotope dilution mass‐spectrometry for antidoping controls,14 were also obtained with a good deuterium incorporation and isotopic enrichment on the purine core (1.3 to 3.9 D incorporated). For those compounds, the additional labelling of the methyl group can be explained by a remote C−H activation process facilitated by the proximity of the sp3‐hybridized carbon center to the surface of the Ru nanocluster after the coordination of the nitrogen in position 9. Gratifyingly, the adoption of a slightly higher catalytic loading (10 mol %) allowed the synthesis of a highly deuterated caffeine (10), commonly used as SILS for quantitative LC‐MS analyses in antidoping tests.14 Flavin adenine dinucleotide (FAD; 13) was also labelled both at the purine and the flavin cores, thus demonstrating the usefulness of the method for the labelling of fragile and structurally complex substrates. Complex purine‐based drugs Doxofylline (14; bronchodilatator), Vidarabine (15; antiviral), Bucladesine (16; phosphodiesterase inhibitor), Didanosine (17; anti‐retroviral), Adefovir (18; reverse transcriptase inhibitor) and Idelalisib (19; kinase inhibitor; Figure 3), were successfully labelled using our HIE protocol, exemplifying its broad applicability relative to harsh labelling conditions described previously in the literature.15 Moreover, given the high solvent tolerability of this method, the best aprotic solvent could be chosen for each compound depending on its solubility.
Figure 3.
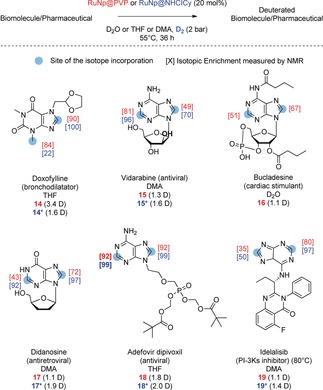
Deuterium labelling of pharmaceuticals. In red, isotopic enrichment obtained with RuNp@PVP (red) and with RuNp@NHCICy (blue).
For the compounds shown in Figure 3, higher deuterium uptakes were obtained using Ru nanoparticles stabilized by organosoluble carbenes (RuNp@NHCICy), highlighting the importance of nanoparticle engineering to enhance the activity for C−H activation processes.16 Moreover, their higher metal content (in weight) led to an easier work‐up and deuterated products purification. Only in the particular case of 14, a lower deuterium incorporation into the labelled methyl was obtained using RuNp@NHCICy instead of RuNp@PVP. This result may be explained by the higher steric hindrance at the surface of the catalyst induced by the carbene ligand. All in all, these complex deuterated compounds were obtained with high yields and good to excellent isotopic enrichments (from 50 to 99 % for position 2 and from 67 to 100 % for position 8). Each experiment was repeated at least twice using different batches of catalyst to ensure the reproducibility of the method.
We then optimized our reaction conditions to perform tritium labelling which provided compounds with high specific activities using a lower pressure of T2 gas (to limit the amount of radioactivity handled). Here, tritiated Didanosine (20) and Idelalisib (21) were synthesized in a single step using a sub‐atmospheric pressure of T2 gas and higher catalytic loadings (Figure 4). The high specific activities (>23 Ci mmol−1) obtained, exceeding the standards required for ADME studies (15 Ci mmol−1), highlight the potential of this novel HIE protocol to facilitate access to essential diagnostic tools in drug discovery and development.17
Figure 4.
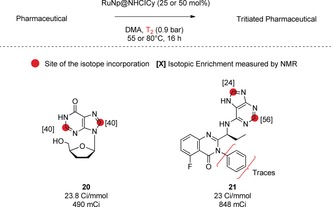
Tritium labelling of pharmaceuticals.
DNA‐ and RNA‐based therapeutics have a prominent place in the pharmaceutical industry, with at least 20 siRNA‐based drugs that have entered clinical trials for more than a dozen diseases.18 Regarding drug development process, quantitative analysis of oligonucleotide therapeutics in biological matrices is crucial for preclinical and clinical stages.19 Published bioanalytical methods devoted to the quantification of oligonucleotides by MS‐based approaches commonly overcome the lack of isotope‐labelled versions of oligonucleotides by the use of different sequences as internal standards (i.e. with slightly different physicochemical properties).20 There is no doubt that using D‐labelled oligonucleotides would greatly benefit to such quantitative analytical methods, thereby offering both high quantification accuracy and high precision with systematic errors arising from sample preparation and MS detection being largely eliminated. The method described in this paper can be applied to gain a rapid access to deuterated oligonucleotides as internal standards. Regarding such particularly complex macromolecules, optimization studies have demonstrated that the use of 1 equivalent of Ru nanoparticles was necessary to obtain adequate isotopic enrichments for SILS purpose. With regard to the value of the resulting labelled compounds, this stoichiometric use of Ru nanoparticles was not considered as a limiting preparative issue. In this case, the use of Ru nanoparticles stabilized by hydrosoluble carbenes (RuNp@NHCPriPr) has allowed a high deuterium incorporation to be obtained.16a This outcome can be explained by their higher water solubility and their better colloidal stability compared to RuNp@PVP. The best conditions developed were applied to the labelling of the model 6‐mer oligonucleotide 22 [RuNp@NHCPriPr (1 equiv) in D2O (80 mm) under D2 gas (2 bar), 36 h]. MALDI‐TOF MS data (see Figure 5 A) showed that an average of 5.5 D atoms can be introduced into the 6‐mer oligonucleotide backbone after 3 iterative runs. The location of up to 7 distinct D atoms was obtained by NMR spectroscopy, and demonstrated, as expected, preferential labelling of the purine core.
Figure 5.
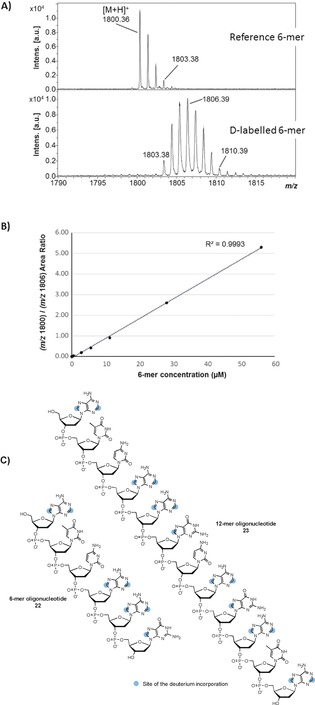
A) MALDI‐TOF mass spectra of native and deuterium‐labelled 6‐mer oligonucleotide. Non‐overlapping isotope massifs were observed after D‐labelling. B) Calibration curve obtained for the native 6‐mer from 0.56 μm to 56 μm, using a deuterium‐labelled 6‐mer concentration set at a constant value of 280 μm (overall concentration). Most intense isotopes were used for native and D‐labelled species (m/z 1800.36 and m/z 1806.39, respectively). C) Structures of the labelled oligonucleotides.
Of note, labelling was accompanied by a complete conservation of the structure while no reduction of the pyrimidine base was observed. Monitoring the MS intensity ratio between the ions of native and corresponding labelled species is a common practice in state‐of‐the‐art quantitative bioanalysis to correct for any particular experimental or analytical bias. The relationship between the 6‐mer concentration and the MS intensity ratio of unlabeled/labelled 6‐mer oligonucleotides proved linear over at least two orders of magnitude (0.56–56 μm) with a coefficient of determination R 2>0.99 (Figure 5 B), thus underlining the value of the labelled 6‐mer as a relevant internal standard. The implemented labelling method was also successfully applied to a more complex 12‐mer oligonucleotide (23) with an average deuterium atom uptake of 7.7 (see Figure S115) without any apparent compound degradation.
In summary, we have developed an efficient and convenient general approach for the hydrogen‐isotope labelling of nucleobase derivatives. Using gentle reaction conditions, this method exhibits broader substrate scope and solvent tolerability than previously published labelling procedures using direct HIE. We have also shown that nanoparticles can be judiciously engineered to significantly increase the efficacy of hydrogen isotope exchanges. Modifying the ligand at the nanoparticle's surface allows changes in the catalyst solubility, thus, leading to an increased efficiency of the labelling process. In terms of applications, this work represents an important advance, providing easy access to tritiated analogues of drug candidates which are essential tools in drug discovery and development processes, and to SILS for quantitative purposes notably for metabolomics.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to thank Dr. Bernard Rousseau for valuable discussions. A. P., V. P. and D. B. are participants in the EU Isotopics consortium. The ISOTOPICS project has received funding from the European Union Horizon2020 Research and Innovation Programme under the Marie Sklodowska‐Curie grant agreement 675071. This work was also supported by the MetaboHUB infrastructure (ANR‐11‐INBS‐0010 grant).
A. Palazzolo, S. Feuillastre, V. Pfeifer, S. Garcia-Argote, D. Bouzouita, S. Tricard, C. Chollet, E. Marcon, D.-A. Buisson, S. Cholet, F. Fenaille, G. Lippens, B. Chaudret, G. Pieters, Angew. Chem. Int. Ed. 2019, 58, 4891.
Contributor Information
Dr. Sophie Feuillastre, Email: sophie.feuillastre@cea.fr
Dr. Grégory Pieters, Email: gregory.pieters@cea.fr.
References
- 1. Nucleosides and Nucleotides as Antitumor and Antiviral Agents (Eds.: C. K. Chu, D. C. Baker), Plenum Press, New York, 1993. [Google Scholar]
- 2. Bennett C. F., Swayze E. E., Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [DOI] [PubMed] [Google Scholar]
- 3. Fire A., Xu S., Montgomery M. K., Kostas S. A., Driver S. E., Mello C. C., Nature 1998, 391, 806–811. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Wittrup A., Lieberman J., Nat. Rev. Genet. 2015, 16, 543–552; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Song E., Lee S.-K., Wang J., Ince N., Ouyang N., Min J., Chen J., Shankar P., Lieberman J., Nat. Med. 2003, 9, 347–351; [DOI] [PubMed] [Google Scholar]
- 4c. Davidson B. L., P. B. McCray, Jr. , Nat. Rev. Genet. 2011, 12, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Atzrodt J., Derdau V., Kerr W. J., Reid M., Angew. Chem. Int. Ed. 2018, 57, 3022–3047; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3074–3101. [Google Scholar]
- 6. Lockley W. J. S., McEwen A., Cooke R., J. Labelled Compd. Radiopharm. 2012, 55, 235–257. [Google Scholar]
- 7. Atzrodt J., Derdau V., Kerr W. J., Reid M., Angew. Chem. Int. Ed. 2018, 57, 1758–1784; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1774–1802. [Google Scholar]
- 8. Taylor G. F., Kepler J. A., J. Labelled Compd. Radiopharm. 1989, 27, 683–690. [Google Scholar]
- 9.
- 9a. Chen B., Jamieson E. R., Tullius T. D., Bioorg. Med. Chem. Lett. 2002, 12, 3093–3096; [DOI] [PubMed] [Google Scholar]
- 9b. Jardine I., Weidner M. M., J. Labelled Compd. Radiopharm. 1980, 17, 389–399; [Google Scholar]
- 9c. Guo X., Ashwell M., Sinnott M. L., Krenitsky T. A., Biochem. J. 1991, 278, 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shelton K. R., Clark J. M., Biochemistry 1967, 6, 2735–2739. [DOI] [PubMed] [Google Scholar]
- 11. Sajiki H., Esaki H., Aoki F., Maegawa T., Hirota K., Synlett 2005, 1385–1388. [Google Scholar]
- 12.
- 12a. Pieters G., Taglang C., Bonnefille E., Gutmann T., Puente C., Berthet J.-C., Dugave C., Chaudret B., Rousseau B., Angew. Chem. Int. Ed. 2014, 53, 230–234; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 234–238; [Google Scholar]
- 12b. Taglang C., Martinez-Prieto L. M., del Rosal I., Maron L., Poteau R., Philippot K., Chaudret B., Perato S., Sam Lone A., Puente C., Dugave C., Rousseau B., Pieters G., Angew. Chem. Int. Ed. 2015, 54, 10474–10477; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10620–10623; [Google Scholar]
- 12c. Gao L., Perato S., Garcia-Argote S., Taglang C., Martinez-Prieto L. M., Chollet C., Buisson D.-A., Dauvois V., Lesot P., Chaudret B., Rousseau B., Feuillastre S., Pieters G., Chem. Commun. 2018, 54, 2986–2989. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Chattopadhyaya J., Yamakage S.-I., Maltseva T. V., Nilson F. P., Foldesi A., Nucleic Acids Res. 1993, 21, 5005–5011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. De Voss J. J., Townsend C. A., Ding W. D., Morton G. O., Ellestad G. A., Zein N., Tabor A. B., Schreiber S. L., J. Am. Chem. Soc. 1990, 112, 9669–9670. [Google Scholar]
- 14.
- 14a. Balssa F., Bonnaire Y., J. Labelled Compd. Radiopharm. 2007, 50, 33–41; [Google Scholar]
- 14b.Regarding caffeine (10), when using 5 mol % of the catalyst, only the incorporation of deuterium atoms resulting from the preferential coordination site N9 is observed at positions 8 and 3 (N-CH3). When using a higher catalytic loading (10 mol % of the catalyst), the lifetime of a transient carbene in position 8 at the surface of the Ru catalyst may be increased, explaining thus the additional labelling at the 7-position N-CH3 (see Ref. [12a]).
- 15.For Vidarabine see: Baker D. C., Haskell T. H., Ann. N. Y. Acad. Sci. 1977, 284, 30–33; For Didanosine see Ref. [8]; For Idelalisib see:81638 [Google Scholar]; Harbeson S. L., Concert Pharmaceuticals, WO2015/179772A1, US, 2015.
- 16.
- 16a. Martínez-Prieto L. M., Baquero E. A. A., Pieters G., Flores J. C., de Jesús E., Nayral C., Delpech F., van Leeuwen P. W. N. M., Lippens G., Chaudret B., Chem. Commun. 2017, 53, 5850–5853; [DOI] [PubMed] [Google Scholar]
- 16b. Martínez-Prieto L. M., Chaudret B., Acc. Chem. Res. 2018, 51, 376–384. [DOI] [PubMed] [Google Scholar]
- 17.One example of tritiation of nucleobase derivative (Famciclovir, 1.9 Ci mmol−1) was described using Ni-catalyzed HIE: Yang H., Zarate C., Palmer W. N., Rivera N., Hesk D., Chirik P. J., ACS Catal. 2018, 8, 10210–10218. [Google Scholar]
- 18. Burnett J. C., Rossi J. J., Tiemann K., Biotechnol. J. 2011, 6, 1130–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Tafech A., Bassett T., Sparanese D., Lee C. H., Curr. Med. Chem. 2006, 13, 863–881; [DOI] [PubMed] [Google Scholar]
- 19b. Yu R. Z., Geary R. S., Monteith D. K., Matson J., Truong L., Fitchett J., Levin A. A., J. Pharm. Sci. 2004, 93, 48–59; [DOI] [PubMed] [Google Scholar]
- 19c. Marcucci G., Stock W., Dai G., Klisovic R. B., Liu S., Klisovic M. I., Blum W., Kefauver C., Sher D. A., Green M., Moran M., Maharry K., Novick S., Bloomfield C. D., Zwiebel J. A., Larson R. A., Grever M. R., Chan K. K., Byrd J. C., J. Clin. Oncol. 2005, 23, 3404–3411. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Lin Z. J., Li W., Dai G., J. Pharm. Biomed. Anal. 2007, 44, 330–341; [DOI] [PubMed] [Google Scholar]
- 20b. Basiri B., Bartlett M. G., Bioanalysis 2014, 6, 1525–1542; [DOI] [PubMed] [Google Scholar]
- 20c. McGinnis A. C., Chen B., Bartlett M. G., J. Chromatogr. B 2012, 883–884, 76–94; [DOI] [PubMed] [Google Scholar]
- 20d. O'Halloran S., Ilett K. F., Clin. Chem. 2008, 54, 1386–1389; [DOI] [PubMed] [Google Scholar]
- 20e. Hemsley M., Ewles M., Goodwin L., Bioanalysis 2012, 4, 1457–1469; [DOI] [PubMed] [Google Scholar]
- 20f. Ewles M., Goodwin L., Schneider A., Rothhammer-Hampl T., Bioanalysis 2014, 6, 447–464; [DOI] [PubMed] [Google Scholar]
- 20g. Franzoni S., Vezzelli A., Turtoro A., Solazzo L., Greco A., Tassone P., Di Martino M. T., Breda M., J. Pharm. Biomed. Anal. 2018, 150, 300–307; [DOI] [PubMed] [Google Scholar]
- 20h. Tozaki T., Gamo S., Takasu M., Kikuchi M., Kakoi H., Hirota K.-i., Kusano K., Nagata S.-i., BMC Res. Notes 2018, 11, 708–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary