

Abstract
Convergent promoters exert transcriptional interference (TI) by several mechanisms including promoter occlusion, where elongating RNA polymerases (RNAPs) block access to a promoter. Here, we tested whether pausing of RNAPs by obstructive DNA‐bound proteins can enhance TI by promoter occlusion. Using the Lac repressor as a ‘roadblock’ to induce pausing over a target promoter, we found only a small increase in TI, with mathematical modelling suggesting that rapid termination of the stalled RNAP was limiting the occlusion effect. As predicted, the roadblock‐enhanced occlusion was significantly increased in the absence of the Mfd terminator protein. Thus, protein roadblocking of RNAP may cause pause‐enhanced occlusion throughout genomes, and the removal of stalled RNAP may be needed to minimize unwanted TI.
Keywords: bacteriophage, mathematical modelling, promoter occlusion, RNAP pausing, transcriptional interference, transcriptional roadblocking
Abbreviations
MM, minimal medium
RBS, ribosome‐binding sequence
REO, roadblock‐enhanced occlusion
RNAP, RNA polymerases
TI, transcriptional Interference
Transcriptional interference (TI) is ‘the suppressive influence of one transcriptional process, directly and in cis on a second transcriptional process’, and is the result of RNA polymerase (RNAP) encountering promoters, DNA‐bound transcriptional factors or other RNAPs in the process of transcription 1. In prokaryotes, TI can arise via five major mechanisms (Fig. 1) 2, 3. For overlapping promoters, promoter competition (Fig. 1A) is the predominant TI mechanism. Promoter competition occurs as a result of the steric hindrance between two initiation complexes such that only one of the overlapping promoters can be bound by an RNAP at any given time. Such promoter arrangements are common, being found for ~ 14% of annotated Escherichia coli promoters 4. The remaining TI mechanisms – promoter occlusion, collisions and dislodgement of promoter‐bound RNAPs or activators – apply for non‐overlapping promoters, with all four mechanisms possible when the promoters are convergent (Fig. 1B–E) 2. Convergent promoter arrangements are also common, with over 1000 antisense transcripts identified as starting within E. coli coding regions 5. TI between convergent promoters increases with promoter strength (i.e. the flux of elongating RNAPs) but the different mechanisms are affected by RNAP flux and various other factors in different ways 6. Thus, the overall impact of TI and the primary mechanisms involved varies for each case.
Figure 1.
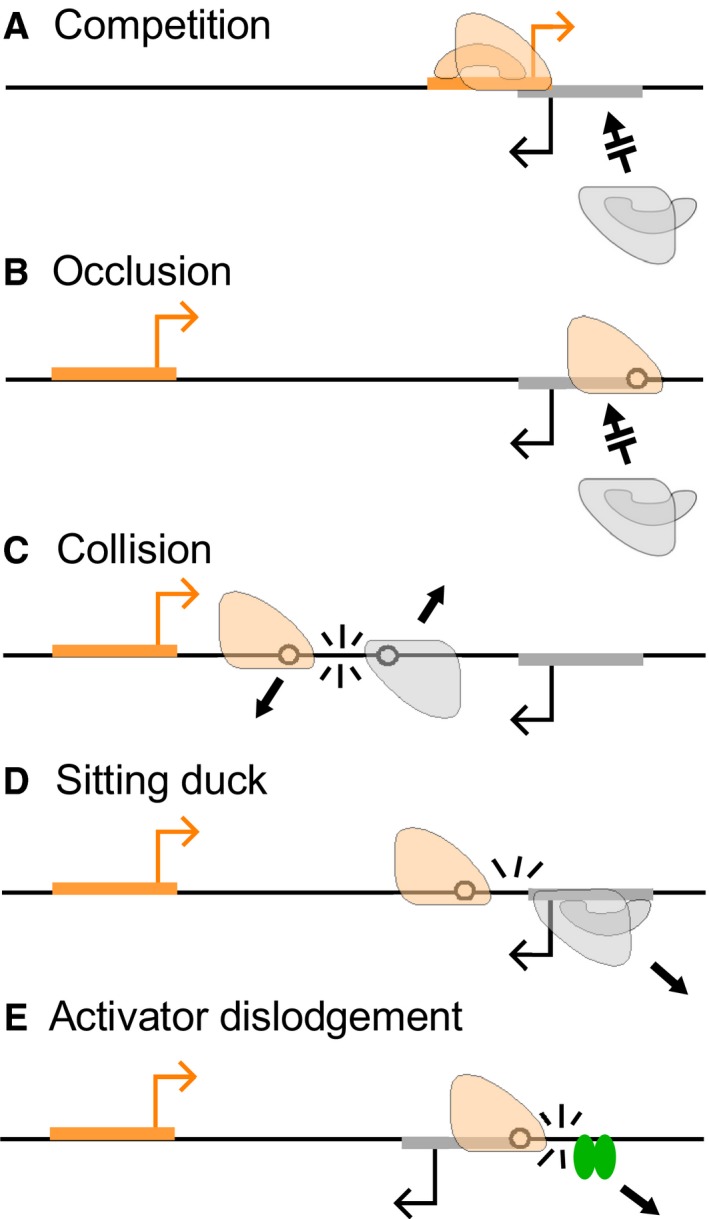
Five major mechanisms of transcriptional interference (TI) between convergent promoters.
Here, we focus on the promoter occlusion mechanism of TI, where elongating RNAPs positioned over a promoter prevent RNAPs in solution accessing the promoter (Fig. 1B). Because elongating RNAPs overlap a downstream promoter for only a short time, strong TI by occlusion requires either a very high flux of interfering RNAPs 6, or pausing of those RNAPs over the target promoter to increase occlusion time 7. Intrinsic RNAP pausing can be induced by specific sequences in the transcribed DNA or nascent RNA 8, 9, 10, 11 and occurs frequently in E. coli 12. We have shown that a strong pause site can significantly enhance occlusion 7. Since pausing can also be induced by DNA‐bound protein roadblocks 13, 14, we reasoned that a protein roadblock positioned such that the paused RNAP overlaps the target promoter would also enhance TI by promoter occlusion. Such roadblock‐induced pausing provides an alternative yet complementary avenue to study the pausing‐enhanced mechanism of TI, and may be a tool for manipulation of TI in regulatory circuits.
Collisional TI results from the termination of one or both elongating RNAPs when RNAPs from convergent promoters collide (Fig. 1C). Previous modelling of TI has suggested that collisions result in the removal of both RNAPs from the DNA 6, although imaging of convergent transcription in vitro has later suggested collisions might have diverse outcomes, with some RNAP stalling and remaining attached to the DNA and others being forced to backtrack 15. The magnitude of TI by collision depends on promoter separation; short promoter separations reduce the probability that an RNAP will be fired from the opposing promoter in the time it takes an RNAP to clear the region between the convergent promoters, and thus reduce collisional TI 6. Recent studies have indicated that RNAP loss after collisions can be asymmetric, favouring RNAPs whose transcripts are actively translated over RNAPs making RNA free of ribosomes 16, 17.
In sitting duck TI 18, an elongating RNAP from an opposing promoter removes RNAP at intermediate steps of initiation at the promoter (Fig. 1D), including stable closed complexes, open complexes and pre‐clearance initiation complexes 19. The magnitude of sitting duck interference felt by a promoter depends in part on its strength relative to that of the opposing promoter, and thus the overall amount of sitting duck TI experienced by a pair of convergent promoters is minimized when the promoters are of equal strength 6.
Dislodgement of activators by elongating RNAPs (Fig. 1E) has also been suggested as a form of TI 2, 7, as the consequent decrease in the occupancy of the activator's binding site should reduce activation of the target promoter. The magnitude of the loss of occupancy due to dislodgement is expected to be strongly affected by the DNA binding kinetics of the activator, with activators with slow binding and unbinding rates being more affected than activators with fast binding kinetics 2, 3.
These convergent promoter mechanisms combine to produce a case of strong TI in bacteriophage λ, in which the lytic promoter P R exerts ~ 6‐fold TI on the convergent lysogeny‐establishing P RE promoter 7 (Fig. 2A). P RE is activated by the λ CII protein and is necessary for production of sufficient CI immunity repressor to establish lysogeny after infection 20. Thus, inhibition of P RE by P R is likely to play an important role in the lysis–lysogeny decision of the phage. Pause‐enhanced occlusion is the major mechanism of TI in this case 7. P R and fully activated P RE are of similar strengths and are separated by just 320 bp, thus only moderate TI by the sitting duck and collision mechanisms would be expected. In addition, dislodgement of λ CII by RNAPs from P R was found to not impact on CII activity 7. However, the weak tR1 terminator induces pausing of P R RNAPs over P RE, significantly enhancing TI by occlusion. It was proposed that RNAP pausing may be a widespread mechanism to enhance TI; however, no other examples of this mechanism have been observed.
Figure 2.
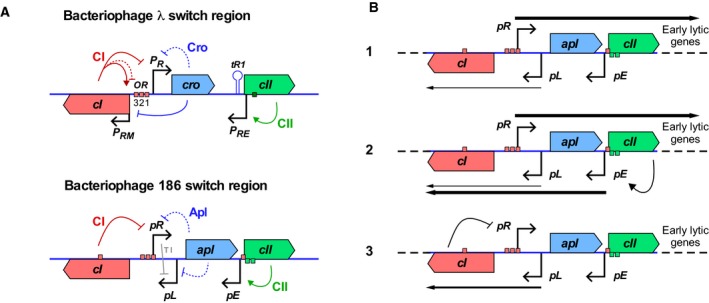
Regulation in the bacteriophage 186 pR‐pE region. (A) A comparison between phages 186 and λ lytic–lysogenic switch regions. The red and green boxes indicate the CI‐ and CII‐binding sites respectively. (B) Establishment of lysogeny in bacteriophage 186. (1) After infection, strong lytic transcription from pR inhibits pL by TI. (2) CII produced from pR activates pE, producing CI. (3) If sufficient CI is produced, pR is repressed, and the phage enters lysogeny. TI from pR at pL is alleviated, increasing pL activity and allowing pL to maintain CI production.
The P2‐family E. coli bacteriophage 186, though essentially unrelated to λ, has a remarkably similar arrangement of the lytic‐ and lysogeny‐establishing promoters (Fig. 2A), with the pR lytic promoter and the 186 CII‐activated pE promoter convergent and separated by 340 bp. As fully activated pE is of similar strength to pR, and with a lack of known RNAP pausing at pE, weak TI would be expected. Indeed, less than twofold mutual TI between pR and pE was observed in initial experiments 21. However, TI at physiologically relevant lower induction of pE was not examined. Here, we varied the expression level of CII and thus pE activity, and found that substantial TI of ~ 4‐fold by pR on pE occurred when pE was only partially activated. The results could be explained by a model lacking any pause‐enhanced occlusion but in which the 186 CII protein was sensitive to dislodgement by RNAPs from pR.
We next tested roadblock‐enhanced occlusion in a synthetic construct with convergent promoters in which Lac repressor was used to stall RNAP such that it overlapped one of the promoters and thereby interfered with its transcription. We saw strong roadblock‐enhanced TI in an E. coli mfd mutant, in which termination of stalled RNAPs is defective 22, 23, including at protein roadblocks 13. However, the TI enhancement in mfd wild‐type cells was modest, with modelling indicating that roadblock‐enhanced occlusion is limited by rapid removal of RNAPs by Mfd unless the interfering promoter is strong.
Materials and methods
Strains and reporters
All lacZ reporter constructs used in the TI experiments (Fig. S1) were integrated into the λ attB site of E. coli strain NK7049 (ΔlacIZYA)X74 galOP308 Str R Su −. The roadblock‐enhanced occlusion (REO) constructs (Fig. S1) were integrated into the λ attB site of E. coli MG1655 rph+ ΔlacIZYA, or a MG1655 derivative with an in‐frame deletion of the mfd gene 13. EC100D mcrA Δ(mrr‐hsdRMS‐mcrBC) φ80dlacZΔM15 ΔlacX74 recA1endA1 araD139 Δ (ara,leu)7697 galU galK λ− rpsL nupG pir + (DHFR) (Epicentre, Madison, WI, USA) was used for propagation of R6γK ori (pir‐dependent) plasmids.
DNA constructions used commercial DNA synthesis (Integrated DNA Technologies, Coralville, IA, USA), restriction enzyme‐based cloning and isothermal Gibson assembly.
The phage 186 pR and pE lacZ reporters for the TI experiments were based on the CRIM plasmid system 24. The pR to pE region was amplified from 186 phage DNA, and cloned into KpnI/SphI or KpnI/XbaI sites of placatt1‐∆lacY‐lacZ (Fig. S1). The pR and pE promoter mutants were generated by QuikChange mutagenesis. The activity of pR was suppressed by 186 CI expressed from pZC320 186 cI during the cloning process, as unrepressed multi‐copy P R ‐lacZ transcription led to cell lethality.
All REO reporters were derived from pIT3‐CL.lacZ* (Fig. S1). In this plasmid, the native ribosome‐binding sequence (RBS) of lacZ was weakened by mutagenesis, making it ~ 62 times weaker than that of the wild‐type lacZ RBS.
DNA constructions
CII was expressed from pZS15_pET_RBS_cII, a low copy number (pSC101 origin) plasmid derived from pZS15 25. The expression of CII was controlled by LacI, expressed from the medium copy (p15A origin) pUHA‐1 plasmid (H. Bujard, Heidelberg University, Germany), and induced by isopropyl β‐d‐1‐thiogalactopyranoside (IPTG).
LacZ assays
Microtiter plate‐based LacZ assays were carried out as previously described 13. Cultures were grown at 37 °C in microtiter plates until late log phase in either LB for TI experiments or M9 minimal medium (MM) for REO experiments. Twenty microlitres of culture was added to a combined lysis‐assay buffer, with each well of a microtiter plate containing: 30 μL culture medium (LB or MM), 150 μL of TZ8 (100 mm Tris/HCl, pH 8.0, 1 mm MgSO4, 10 mm KCl), 40 μL of ONPG (o‐nitrophenyl‐β‐d‐galactoside, 4 mg·mL−1 in TZ8), 1.9 μL of 2‐mercaptoethanol and 0.95 μL of polymyxin B (20 mg·mL−1). Assays were performed on cultures started from independent colonies and repeated on at least three different days. Error bars represent 95% confidence intervals.
Stochastic simulations
Programs for stochastic simulation were written in FORTRAN and were executed on a MacBook Pro. In a typical run, 108 time‐steps (~ 700 h) were simulated for each condition.
Rates are taken directly from previous simulations except where otherwise noted (Table S1). Promoter firing rates (k F) for the phage 186 pR and pE promoters used in the TI experiments were calibrated using the P Bla promoter, for which in vivo firing rates have been estimated 26. Under rich medium growth conditions (LB), P Bla fires approximately once every 55 s, 3.18 and 5.39 times slower than pR and CII‐activated pE, respectively, leading to k F estimates of 0.0582 s−1 for pR and 0.0989 s−1 for activated pE.
After promoter firing, an RNAP that has just fired can sterically block the promoter from access by another RNAP, until the first RNAP has transcribed a distance equal to its length. This process is referred to as self‐occlusion. To account for self‐occlusion, a further correction was made (Eqn (1)):
| (1) |
where k F* is the intrinsic firing rate, k F is the measured firing rate, l is the occlusion length of an elongating RNAP (30 bp) and ν is the elongation velocity (40 bp·s−1). After accounting for self‐occlusion, the final adjusted intrinsic strengths of pR and pE were calculated to be 0.0609 and 0.1072 s−1 respectively.
For REO experiments, cells were grown in M9 minimal medium instead of LB. It is known that promoter firing rates change with cellular growth rates 26. Thus, the promoter firing rates were recalibrated based on λ P L firing rates measured under the same growth conditions 26. In minimal medium, both 186 pR and P2 P e were about two times weaker than λ P L, estimated to fire approximately once every 10 s 26. After correcting for self‐occlusion, the final adjusted intrinsic firing rates for 186 pR and P2 P e were calculated to be 0.0554 and 0.0527 s−1 respectively.
Data transformation
The amount of LacZ expressed by very strong promoters with the native lacZ RBS can exceed the linear range of the LacZ assay. In a previous study 13, 11 promoter pairs of varying strengths, each expressing lacZ with either its native RBS or the weak RBS (lacZ*) were constructed and assayed, and a empirically derived rectangular hyperbola equation was used to transform the native RBS data to correct for the nonlinearity in the assay (Eqn (2)):
| (2) |
The LacZ data obtained for the phage 186 TI experiments were subjected to this transformation. The average LacZ produced by pR(pE‐).lacZ after background correction (against pR‐(pE‐).lacZ) over the IPTG concentration range was calculated to be ~ 23.44 transformed LacZ units, and the intrinsic firing rate for pR was 0.0609 s−1 or ~ 219.2 transcripts per hour in LB. One transformed LacZ unit was thus equivalent to 9.35 transcripts per hour. To align the experimental data with the stochastic simulations, the experimental LacZ data obtained for the 186 TI experiments were converted to the transcripts per hour units using this conversion factor.
For the REO experiments, a weak RBS version of lacZ reporter was used. We assumed that there was negligible nonlinearity in the observed LacZ activities assayed with the weak RBS and thus no data transformation was required to this data set. In minimal medium, the intrinsic firing rate for pR was 0.0554 s−1 or ~ 199.4 transcripts per hour, and the average LacZ produced by pR(P e ¯).lacZ* was 72.33 units. Thus, one LacZ unit was equivalent to 2.76 transcripts per hour in this system.
Statistical analysis
Measured lacZ values are presented as the mean ± the 95% confidence limits. The EC50 of pE induction curves with or without pR were calculated using the Hill function in graphpad prism (Graphpad Software, San Diego, CA, USA).
Results
Design and construction of a bacteriophage 186 pR‐pE reporter system
The bacteriophage 186 lysis–lysogeny decision is regulated by three promoters: pR, pE and pL, and two regulatory proteins: CI and CII (Fig. 2A). Unlike λ, where the P R lytic promoter and the P RM lysogenic promoter are arranged back‐to‐back, the 186 lytic promoter pR and the lysogenic promoter pL are convergent. This arrangement results in 5.6 fold TI at pL primarily by the sitting duck mechanism 6, 18. During lysogeny, CI repression of pR indirectly activates pL by removing this TI, allowing for expression of lysogenic genes including cI 27, 28 (Fig. 2B, panel 3). However, upon phage infection of a cell that contains no CI protein, pL transcription is low due to TI and thus establishment of lysogeny requires an alternative source of CI (Fig. 2B, panel 1).
Efficient establishment of lysogeny is dependent on the 186 CII protein 29, which activates transcription of cI from the pE promoter 30, 31, 32 (Fig. 2B, panel 2). CII contains a helix‐turn‐helix motif and is highly unstable in vivo, with an estimated half‐life of 2.6 min 32. CII binds an inverted repeat spaced two turns of the DNA helix apart, located at the −38 and −58 positions of pE, and contacts both the α and σ subunits of RNAP 32. Basal pE is of negligible strength, but is strong when induced by CII 30.
A previous study examined TI between pR and pE under conditions where pE was fully activated with a high level of CII expression. Weak TI was seen, with pR reducing the activity of pE 2.1‐fold and pE reducing the activity of pR 1.5‐fold 21, suggesting a lack of substantial pause‐enhanced occlusion in the 186 case. However, given the strong interference exerted by λ P R on P RE, we wished to test whether TI by 186 pR on pE might be stronger at lower pE induction levels.
In order to study the effect of pE induction level on the TI between pR and pE, a set of chromosomally integrated lacZ reporters was constructed (Fig. S1). The region of the 186 genome (NC_001317.1) used for these reporters spans from base 22 980, which is −81 from pR, to base 23 533, which is −132 from pE and ~ 70 bp from the end of the CII‐binding site (Fig. S2). This region of the 186 genome also contains the pL promoter and the full coding sequence of apl, both of which were mutationally inactivated by (a) altering the −10 site of pL 18 and (b) swapping residues E28 and R29 of Apl that lie within the recognition helix of Apl's helix‐turn‐helix motif. Together, these alternations are designed to retain the distance between pR and pE and to minimize changes to the native 186 DNA sequence. While the pL − mutation was designed to avoid affecting apl translation, we found that Apl expression was significantly reduced, consistent with disruption of the apl ribosome‐binding site (Fig. S3).
The CII protein was expressed in trans from a plasmid‐based LacI‐repressed system with IPTG induction (Fig. 3). The pZS15_pET_RBS_cII plasmid (Materials and methods) is capable of producing a gradient of CII expression, up to the levels necessary to maximally activate pE, while minimizing leaky CII expression and changes in cell growth due to a high level of CII.
Figure 3.
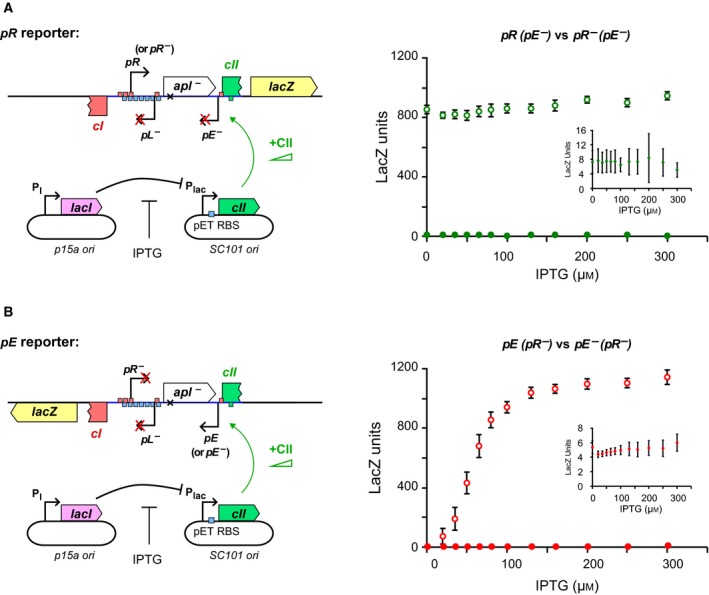
The effect of varying CII levels on pR and pE activity. (A) Left, a schematic representation of the chromosomally integrated pR(pE‐).lacZ and pR‐(pE‐).lacZ reporter constructs. CII was expressed from low copy number pZS15_pET_RBS_cII and was controlled by IPTG. Right, the pR activities were assayed across 12 IPTG concentrations from 0 to 300 μm. pR was constitutive and approximately constant, averaged across all IPTG concentrations at 862 ± 9 LacZ units (n = 238). Error bars represent 95% confidence intervals. Right insert, a zoomed in view of pR‐(pE‐).lacZ at different IPTG concentrations, showing that the mutation of pR almost completely abolished PR activity. Open circles: pR(pE‐).lacZ data; filled circles: pR‐(pE‐).lacZ data. (B) Left, a schematic representation of the chromosomally integrated pE(pR‐).lacZ and pE‐(pR‐).lacZ reporter constructs. Right, the PE activities across the IPTG range. pE was activated by the induction of CII expression with IPTG, from a basal activity of 5.1 ± 0.4 LacZ units to 1140 ± 50 lacZ units (n = 16). Right insert, a zoomed in view of pE‐(pR‐).lacZ. The PE promoter mutation is a single base pair change at the −10 site of pE, which almost completely knocks out CII‐dependent pE activation but does not alter CII binding at pE 31. Note that the binding half sites of CII are centred at −38 and −58 of pE (Fig. S2). Open circles: pE(pR‐).lacZ data; filled circles: pE‐(pR‐).lacZ data.
The pR and pE activities were first measured in the absence of the convergent promoter by constructing promoter null mutants (Fig. 3). The pR‐ mutant was made by altering the −10 and −35 sites of pR 18 (Fig. S2), while the pE‐ mutation is a single base substitution in the −10 site of pE (Fig. S2), which does not alter CII binding 31. In the absence of pE, transcription from pR was strong, constitutive, and not affected by the concentration of CII (Fig. 3A), indicating that DNA‐bound CII is not a transcriptional roadblock for elongating RNAPs from pR. In the absence of pR, pE had almost negligible basal activity but was strongly induced by IPTG‐regulated CII expression, reaching even greater activity than pR (Fig. 3B).
Assaying TI between pR and pE
Next we tested the activities of pR and pE when each promoter faced convergent transcription from the other (Fig. 4A). When pR was active, the activity of pE was reduced at all CII concentrations. The maximal pE activity was 1.63 (1.46–1.82) (95% confidence interval) fold less than that obtained when pR was mutationally inactivated (Fig. 4B). However, TI by pR on pE was stronger at lower CII induction levels, with 3.98 (2.96–5.50)‐fold TI at 50 μm IPTG (Fig. 4B). This effect substantially increased the IPTG concentration required to reach half‐maximal pE activation (the EC50), (from 72 μm without interference to 113 μm with interference), reflecting a requirement for higher CII expression levels (Fig. 4B). This change in EC50 is consistent with dislodgement of CII bound at pE by RNAP from pR. Furthermore, induction of pE by CII also caused a dose dependent reduction of pR, reflecting increased TI from a stronger pE (Fig. 4C). When pE was maximally activated, TI reduced pR about 2.27 (2.12–2.43)‐fold. The somewhat weaker 1.5‐fold inhibition of pR by pE seen previously 21 is likely to be due to non‐maximal pE activation.
Figure 4.
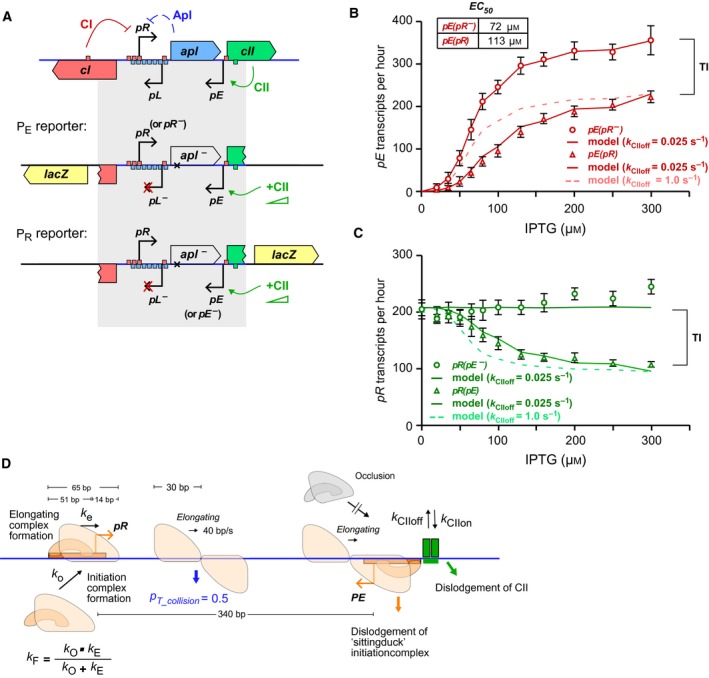
The convergent pR and pE promoters of bacteriophage 186 only weakly interfered with each other's transcription, even at high CII concentrations. (A) A schematic representation of the chromosomally integrated reporter constructs used to measure the activities of pR and pE with and without the convergent transcription from the other. (B) Data (symbols) and simulation (solid curve) for the activity of the pE in the pE(pR‐).lacZ and pE(pR).lacZ reporters with increasing concentration of CII. Data are mean ± 95% confidence intervals (n = 14). The dotted curve shows the simulation of pE(pR) with faster binding kinetics for CII (k CIIoff = 1 s−1). (C) Data and simulation (solid curve) for the activity of pR in the pR(pE‐).lacZ and pR(pE).lacZ reporters. The dotted curve showed the simulation of pR(pE) with faster CII binding kinetics, as in (B). For (B, C), the activity was expressed in units of transcripts per hour to allow direct comparison with the simulations. (D) Schematic of the stochastic model and associated parameters used to simulate the experimental data.
Modelling TI between pR and pE
To test if the observed TI between pR and pE can be explained by the known mechanisms of TI (Fig. 1), a mathematical model was developed based on our previous TI model, but altered to describe the convergent pR and pE promoters of phage 186 (Fig. 4D). A section of DNA from the −51 position of pR to the −51 position of pE was simulated. The model used the method of discrete fixed time‐steps to stochastically simulate the kinetics of RNAP and CII binding, as well as sitting duck, collision and occlusion TI on this DNA. In this simulation, each time‐step is set to 1/40 s, equivalent to the time taken for an elongating RNAP to advance one base pair 6. All possible events are assigned a specific rate k (Table S1). Whether or not a given event occurs during the next time‐step is decided by generating a random number between 0 and 1; if this number is less than 1 − e−k/40, then that event occurs. The simulation is updated to reflect this change, and the simulation proceeds to the next time‐step. Wherever possible, rates were taken directly from in vivo measurements from the literature. For the rates where no direct measurements are available, previously fitted rates were used, except where otherwise noted.
The key events in the simulation are as follows:
Promoter firing: Promoter firing is simulated as a two‐step process, consisting of loading of RNAP holoenzyme to form an open complex and firing of the open complex to form an elongating complex. Binding of an open complex to the promoter (defined as positions −51 to +14) is only possible when the promoter is not already overlapped by other RNAPs. A new open complex is loaded at pR with rate k O = 0.061 s−1 or at a CII‐bound pE with rate k O = 0.214 s−1. No open complex can be formed at pE if the CII‐binding site is unoccupied by CII. An open complex is converted to an elongating complex with rates k E, which are 61 and 0.214 s−1 respectively for pR and pE. Unlike k O, the k E for pE is CII independent, meaning that the k E for pE will not change even if CII dissociates after an open complex has formed at pE. After promoter firing, the open complex is converted to an elongating complex, and its footprint reduced from 65 bp for an open complex to 30 bp 33.
Binding and unbinding of CII at pE: Binding of CII at pE only occurs if the CII site is not already occupied by CII, and is not overlapped by RNAPs from pR. The binding rate of CII (k CIIon) depends on its concentration (Table S1), which can be extracted from the un‐interfered pE induction curve (Fig. 3B) by assuming that activity is proportional to CII occupation and that the CII site is fully occupied at the maximal pE activity. Since occupation equals the association rate k CIIon over the sum of k CIIon and the dissociation rate k CIIoff, these data alone give a fix on the ratio of k CIIon and k CIIoff at each CII concentration, but does not provide any fix on the exact values of k CIIon or k CIIoff. In the presence of pR, a DNA‐bound CII at pE either dissociates spontaneously with rate k CIIoff or is removed by RNAPs initiated from pR. Dislodgement of CII by RNAPs was assumed to be instantaneous as CII is not a transcriptional roadblock to RNAP from pR (Fig. 3A). Thus, the pR interfered pE induction curve (Fig. 4B) puts a further constraint on k CIIoff. If k CIIoff is fast, then dislodgement of CII by RNAPs will not have a large influence on the CII occupation; alternatively, if k CIIoff is slow, then dislodgement by RNAP will have a pronounced effect on the CII occupation.
Movement and termination of RNAPs: RNAP elongation is treated as occurring at a constant rate at 40 bp·s−1 6, consistent with measurements of the average speed of RNAP in vivo 34. RNAP velocity heterogeneity 35 was not simulated. In the model, collision of two convergent elongating RNAPs results in one (at random) being instantaneously removed, while the other remains (collision TI). Our results with synthetic TI constructs 16 indicated that RNAP loss after collisions is highly asymmetric between RNAPs whose RNAs are strongly translated and RNAPs whose RNAs are untranslated, with a 7% : 93% translated:untranslated removal ratio. However, while the pR transcripts would normally be translated over the apl coding sequence and the pE transcripts are untranslated, we did not include asymmetric removal of RNAPs in the model because the pL − mutation used in the constructs disrupts the apl ribosome‐binding site (Fig. S3). If the collision occurs between an elongating RNAP and an open complex, then the open complex is removed (sitting duck TI). Once the back of an elongating RNAP passes the end of the DNA, then that RNAP is eliminated from the DNA and a new transcript is counted.
The simulations use CII occupation as the independent variable versus transcripts per hour as the dependent variable, whereas the experimental measurements use IPTG concentration and lacZ activity respectively. To align the curves produced by simulation with the experimental values, the experimental LacZ activities were converted to transcripts per hour as described in the Materials and methods.
The model provided a reasonable fit to the data and reproduced the observed increase in EC50 as long as the k CIIoff of CII was set low at 0.025 s−1, so that CII dislodgement becomes a significant effect (Fig. 4B,C). Thus, modelling suggests that a DNA‐bound CII takes on average ~ 40 s to spontaneously leave the DNA or a half‐life of ∼28 s (= ln2/k CIIoff). If the dissociation rate of CII is increased 40‐fold to 1 s−1 (with a compensating increase in k CIIon to maintain occupation), inhibition of pE by pR is underestimated, especially at low CII concentrations (Fig. 4B, dotted line). In this case, the spontaneous rate of CII dissociation is high enough that the additional dissociation due to dislodgement by RNAPs from pR is minor and has little effect on CII occupation. The faster CII binding kinetics also produces too much inhibition of pR by pE (Fig. 4C, dotted line) as result of the higher pE activity.
Figure 5 shows how the measured TI between 186 pR and pE changes with induction of CII expression. The maximal observed TI by 186 pE on pR was ~ 2.3‐fold at the highest [IPTG] (Fig. 5A). The modelling allowed us to extract the relative proportions of the different TI mechanisms as CII concentration increased. This analysis (Fig. 5A) indicates that RNAP collisions were responsible for the majority (~ 75%) of the TI at all IPTG concentrations, and that promoter occlusion contributed to most of the remainder. The absence of sitting duck interference results from the very low aspect ratio (that is the rate of promoter binding over the rate of firing) assigned to pR (Table S1), which was necessary to fit the data.
Figure 5.
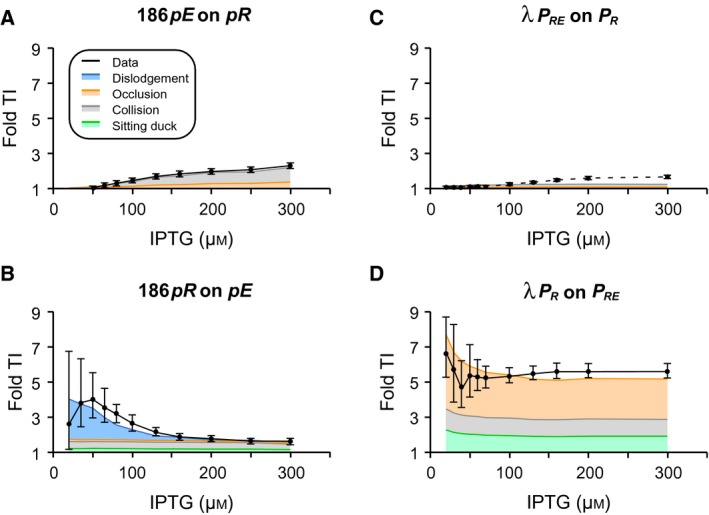
Bacteriophages 186 and lambda have evolved different mechanisms to induce TI between the convergent promoter pairs pR/pE and PR/PRE. (A) TI of 186 pE on pR is weak and predominantly via RNAP collision (B) TI of 186 pR on pE is stronger and peaks at low CII concentration as a result of CII dislodgement by transcription from pR (C) λ PR experiences very little TI from PRE. (D) In contrast, λ PRE experiences strong TI from PR, and is due to RNAP pausing at the tR1 terminator overlapping PR. The black solid curves show the fold TI calculated from experimental data, error bars represent 95% confidence intervals (n ≥ 6). The λ TI data were calculated from Palmer et al. 7. The different colour shadings indicate the contribution of the different TI mechanisms.
The measured TI of 186 pR on pE peaked at ~ 4‐fold at 20 μm IPTG when pE activity was weak, and then gradually reduced to ~ 1.6‐fold when pE started to gain strength (Fig. 5B). Modelling indicates that at low IPTG concentrations the TI was largely due to CII dislodgement by elongating RNAPs from pR, contributing up to 75% of observed TI at 20 μm IPTG. As expected, TI due to CII dislodgement became diminished at higher CII concentration, and the TI became predominately due to RNAP collision followed by sitting duck and promoter occlusion. Note that although the percentage contribution of each TI mechanism changed at different IPTG concentrations, the absolute TI due to RNAP collision, sitting duck and promoter occlusion mechanisms remained constant across the IPTG range.
For comparison, Fig. 5C,D shows how TI between λ P R and P RE changes with induction of λ CII 7. The separation of λ P R and P RE is 320 bp, similar to the 340 bp between 186 pR and pE. The establishment promoters are also of similar strength, with firing rates of once every 8.13 s for λ P RE compared to once per 9.3 s for 186 pE, while λ P R (once per 5.8 s) is ~ 2‐fold stronger than 186 pR at once per 14.5 s. However, a key difference is that RNAPs from P R pause for substantial periods at three sites within tR1 7. Interference by P RE on P R was at most 1.6‐fold (Fig. 5C), which is slightly lower than the 2.3‐fold for 186 pE on pR (Fig. 5A), primarily because of the higher strength of λ P R. However, in λ, interference at P RE was very strong, peaking at ~ 6.7‐fold at low λ CII concentration and staying at ~ 5.5‐fold TI even at high λ CII concentration, with pause‐enhanced occlusion the major TI mechanism 7 (Fig. 5D).
Thus, despite the similar promoter arrangements in 186 and λ, the two phages display different mechanisms and magnitudes of TI. While we have no direct measurements of transcriptional pausing in 186, our data and modelling indicate a lack of significant pause‐enhanced occlusion at 186 pE. The addition of substantial pause‐enhanced occlusion would increase TI of 186 pR on pE across all IPTG concentrations and would substantially worsen the match to the observed TI at high [IPTG]. Increased occlusion would also tend to dampen the observed EC50 shift, which is instead explained by 186 CII's sensitivity to dislodgement.
A roadblock‐enhanced occlusion circuit
While pause‐enhanced occlusion at λ P RE produces a substantial regulatory effect, the apparent lack of pausing‐enhanced occlusion in the 186 pR–pE system raises the question of whether this mechanism applies in other cases. There are three clustered pause sites at λ tR1 that produce an unusually long dwell time for RNAPs over P RE. Since the occlusion effect is strongly dependent on the RNAP pause time, it is possible that λ is a special case, and more typical pause sites may be unable to cause TI enhancement.
To test whether other pauses can enhance TI, we used a protein roadblock, specifically the Lac repressor (LacI), to pause RNAP at one of a pair of convergent promoters (Fig. 6A and Fig. S4). LacI is the best studied of a handful of DNA‐binding proteins that are known to block the progress of elongating RNAP in vitro and in vivo 13, 14, 34. We expected that LacI bound to its strong Oid operator located just upstream of one of the promoters would cause stalling of RNAPs from the second promoter such that they block access to the first promoter (Figs. 6A and Fig. S4). A potential advantage of using a protein‐induced pause is that controlling the availability or activity of the roadblocking protein should allow ready modulation of the occlusion effect.
Figure 6.
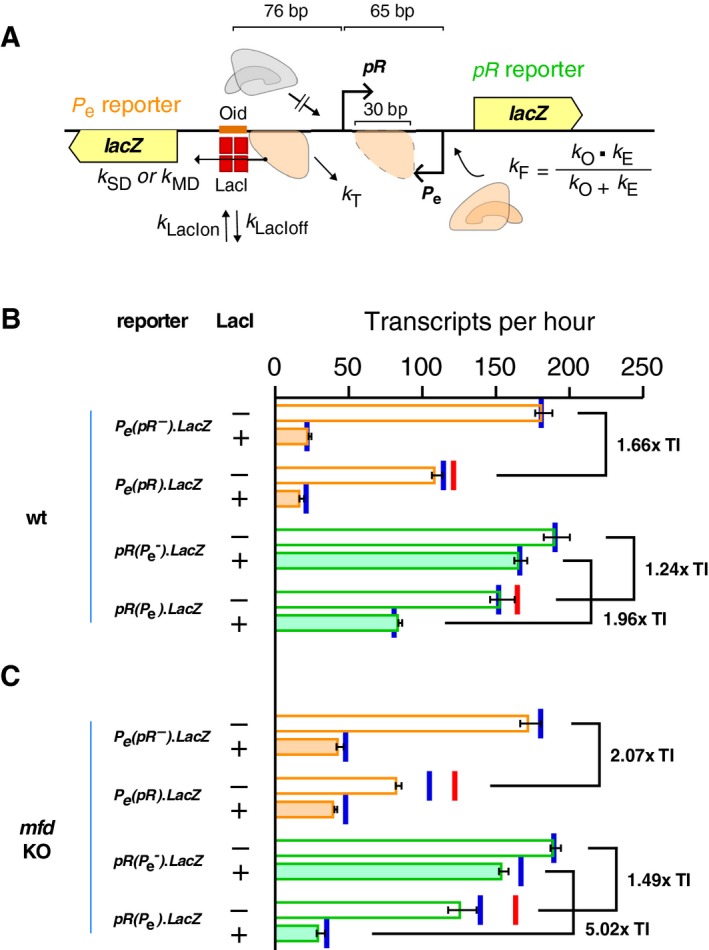
The roadblock‐enhanced occlusion (REO) circuit is a synthetic circuit with an engineered RNAP pause site at pR for the augmented induction of asymmetrical TI between convergent promoters pR and P e. (A) Schematic arrangement and parameters used for simulation of the REO circuit. The DNA region simulated in the modelling extends from the −85 position of 186 pR to the −51 position of P2Pe. (B) Data (horizontal bars) and simulations (red and blue vertical lines) for the TI between pR and P e promoters with (filled bars) or without (empty bars) a LacI roadblock in the mfd wild‐type strain (C) as for (B), but reporters integrated into a mfd knockout strain. To align the experimental data with the simulations, data were converted to transcripts per hour units (Materials and methods), error bars represent 95% confidence intervals (n = 9). Red bars represent the simulated TI when collision‐induced termination was considered instantaneous, while blue bars represent the TI when the rate of collision‐induced termination was set equal to the rate of termination at the lacI roadblock.
Our roadblock‐enhanced occlusion (REO) circuit system utilized the lytic promoters pR and P e of phages 186 and P2 as the convergent promoter pair. The use of two similar strength promoters (186 pR 0.0554 s−1 and P2 P e 0.0527 s−1) was designed to minimize sitting duck TI. The two promoters were also placed very close to each other (65 bp between the +1 of each promoter) to minimize TI by RNAP collision 6. The lacOid operator was centred at the −76 position of pR, a location where LacI binding had minor effects on pR activity. Four chromosomally integrated lacZ reporters were constructed (Fig. S1), two of which were used to report the activities of pR, either in the presence or absence of a convergent P e, and the other two reported the activities of P e with or without a convergent pR (Fig. 6B). The Lac repressor was expressed from a medium copy number plasmid (pUHA‐1), under the control of its native promoter P I 25. A ΔlacI version of the same plasmid was used as a no roadblock control 13.
In the absence of LacI, transcription from pR led to ~ 1.7‐fold TI on P e. Conversely, transcription from P e led to ~ 1.2‐fold TI on P R (Fig. 6B). This small difference in TI between P R and P e was probably due to the intrinsic kinetic parameters of the two promoters (Table S1). When LacI was present, the expression of lacZ from P e was reduced to only ~ 13% of its normal level due to transcriptional roadblocking by LacI (Fig. 6B). This drop in transcription occurs because RNAPs stalled at the LacI roadblock are subject to termination 13. The effect of LacI on transcription from the pR promoter in the absence of convergent transcription from P e was mild, reducing pR by ~ 10% (Fig. 6). However, in the presence of LacI, P e produced a ~ 2.0‐fold inhibition of pR (Fig. 6B). We attribute this P e‐dependent inhibition of pR by LacI to enhanced occlusion of pR due to RNAP pausing at the LacI roadblock.
The pausing time of RNAPs at a protein roadblock is affected by the transcription‐coupled repair protein Mfd. The Mfd translocase moves unidirectionally along DNA and upon encountering a stalled or backtracked RNAP either stimulates reinitiation of elongation by pushing RNAP forward, or, if a strong obstacle prevents this, stimulates termination of the RNAP 22, 23. Both outcomes should result in a reduction of the pause time. At strong protein roadblocks, such as LacI, increased termination due to Mfd leads to reduced transcription past the roadblock 13, 23. Thus, to test the idea that the P e‐dependent inhibition of pR by LacI is due to enhanced occlusion, we assayed the reporters in an Δmfd background (Fig. 6C). As expected, removal of Mfd increased P e readthrough of the LacI roadblock, from 13% to ~ 25%. Importantly, the LacI enhancement in TI by P e on pR increased dramatically from ~ 2‐fold to ~ 5‐fold in the mfd mutant (Fig. 6C), supporting the roadblock‐enhanced occlusion mechanism. Interestingly, the mutual TI between P R and P e in the absence of the LacI roadblock also increased slightly from ~ 1.2‐ to ~ 1.5‐fold and from ~ 1.7‐ to ~ 2.1‐fold, respectively, in the Δmfd strains (Fig. 6C).
Modelling the roadblock‐enhanced occlusion circuit
To test the roadblock‐enhanced occlusion mechanism further, we simulated the REO circuit, using a TI model modified to incorporate our previous transcriptional roadblocking model 13. In the model (Fig. 6A), an elongating RNAP becomes paused when it encounters a bound LacI roadblock. A paused RNAP stays paused unless it either spontaneously dissociates from DNA with rate k T, or dislodges the bound roadblock with rate k SD. If there is more than one RNAP paused behind a protein roadblock, then an increased dislodgement rate k MD is applied to account for RNAP cooperation 34. The same treatment was applied to both the wild‐type and the Δmfd strain, but with different values for k T, k SD, and k MD 13. All rates were as previously estimated except the k T in the Δmfd strain, which was increased ~ 4.4‐fold from 0.0045 to 0.02 s−1, a necessary adjustment required to fit the somewhat stronger than expected roadblocking of LacI on P e(pR‐)lacZ* (Fig. 6C).
Overall, the model was able to reproduce the wild‐type data reasonably well (red bars, Fig. 6B,C). However, we found that the model underestimated TI for both pR and P e in the Δmfd strains. This may be a result of how RNAP termination was simulated after a head‐on collision between RNAPs. As previously stated, the model treats the dissociation of collided RNAPs as an instantaneous process, a reasonable simplification for the wild‐type cells. However, it is possible that the removal of RNAPs after collision is delayed in the Δmfd cells, given the role Mfd plays in the resolution of RNAPs stalled at other obstacles 23. Indeed, when the rate of collision‐mediated termination was delayed by setting it equal to the k T used for RNAP termination at the LacI roadblock (0.02 s−1 for Δmfd cells and 0.66 s−1 for mfd + cells), the model was able to provide a better fit, particularly to the Δmfd data (blue bars, Fig. 6B,C). Because pR and P e are very close, this delay in termination causes promoter ‘clogging’, where queues of RNAPs extend back from a collision event to cover the promoters and prevent loading of new RNAPs 13. This additional inhibition of transcription due to collisions increases TI. The effect is minor in wild‐type cells because rapid termination by Mfd keeps RNAP queues to a minimum.
The modelling allowed us to assess the impact of system properties on the strength of roadblock‐enhanced occlusion in the REO circuit. Given a strong roadblock, the main factors are the strength of the promoter supplying RNAPs to the roadblock, and the rate of termination of RNAPs stalled at the roadblock. It is the balance between this gain and loss of paused RNAPs that determines the fractional occupancy of the occluding site. Figure 7A shows how the TI of P e on pR increases steeply in the presence of the LacI roadblock as the strength of P e increases beyond its actual firing rate of 0.053 s−1 (termination rate held constant). Figure 7B shows a strong but less steep effect of reducing the termination rate on the TI of P e on pR in the presence of the roadblock (P e intrinsic firing rate constant). The weaker effect of termination rate compared to promoter firing rate is also apparent when both factors are varied (Fig. 7C). This is because occluding RNAPs are lost not only through termination but also by passage through the roadblock. Even in the absence of termination, the rate of loss of occluding RNAPs at the LacI roadblock can be as high as 0.026 s−1 (k MD).
Figure 7.
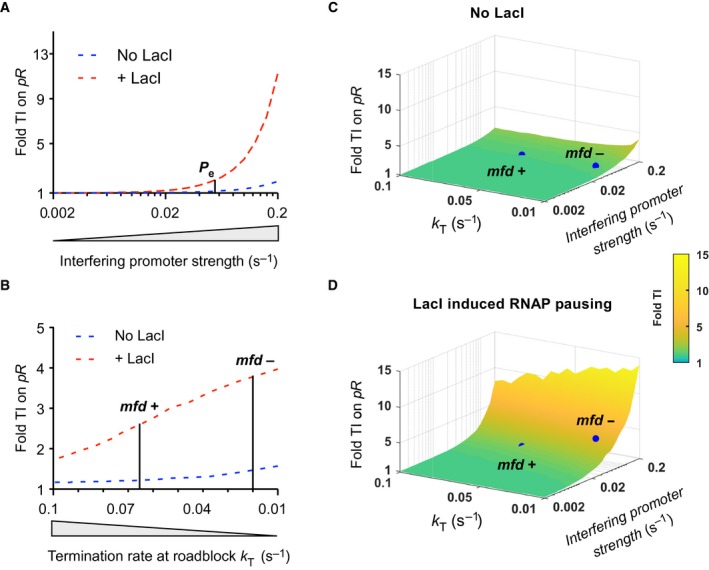
The strength of occlusion‐enhanced TI is dependent on both the strength of the promoter that controls RNAP flux at the roadblock site, and the rate of RNAP termination at the roadblock site. (A) Fold TI on pR increased with increasing P e strengths (i.e. increased firing of RNAP towards the roadblock). pR strength (0.055 s−1) and RNAP termination rate (0.066 s−1) were held constant. (B) Fold TI on pR increased as the RNAP termination rate at the roadblock site was lowered (i.e. reduced loss of RNAP at the roadblock). pR and P e strengths (0.055 and 0.053 s−1 respectively) were held fixed. (C) Roadblock‐enhanced occlusion on pR is maximized with a stronger P e and a reduced RNAP termination rate at the roadblock site. Blue dots show the experimentally determined TI on pR in the absence (C) or the presence (D) of the roadblock protein, LacI.
Discussion
The significance of these data and models are threefold; concerning how TI provides positive feedback in regulatory decision‐making, TI by dislodgement of transcriptional activators and mechanisms of promoter occlusion due to RNAP pausing.
The role of TI in the lysis–lysogeny decision of bacteriophages λ and 186
Phages λ and 186 both utilize TI in their developmental decisions but do so in different ways. The main similarity is that the lytic promoter (λ P R or 186 pR) is capable of substantially inhibiting the lysogeny‐establishing promoter (λ P RE or 186 pE). However, the major mechanism of this TI and its magnitude are different in the two phages, with pause‐enhanced occlusion contributing to ~ 6‐fold TI in λ, and CII dislodgment contributing to at most fourfold TI in 186 (Fig. 5). The different mechanisms also result in a different response to CII. In λ, the inhibition by P R is reasonably constant over a range of CII concentrations, while in 186, inhibition is maximal at low CII concentrations and decreases with increasing CII concentrations. Thus, in the presence of active pR, the level of expression of the lysogenic genes from 186 pE becomes highly sensitive to CII concentration, as increasing the CII concentration both directly activates pE and reduces its inhibition by pR (Fig. 4B). 186 pR also exerts TI on the convergent lysogenic promoter pL (Fig. 2). In this case TI is strong, ~ 6‐fold, and is primarily by the sitting duck mechanism 6, 18. In contrast, λ P R and P RM are back‐to‐back and cannot inhibit each other in vivo by any of the mechanisms shown in Fig. 1.
We note that our measurement of inhibition of pE by TI in 186 may be an underestimate because the decreased translation of the pR mRNA in our pL − constructs compared to the wild‐type case may have reduced the flux of RNAPs from pR at pE, due to the lack of ‘translation asymmetry’ in collisional TI. Our measurement of 2.3‐fold TI at maximal pE induction is similar to the 2.1‐fold value obtained for the same pL − mutant by 21. However, the TI value for the pL+ case at maximal pE induction was 3.5‐fold in 21, when an estimate of the pL contribution to leftward transcription is subtracted. The interaction of the three promoters makes TI difficult to analyse but the result suggests that, in the presence of full apl translation, the TI of pR on pE at sub‐maximal CII activation may be higher than the ~ 4‐fold we observed. On the other hand, the presence of Apl would likely reduce this TI because of its repressive effect on pR (Fig. S3).
We note also that our previous modelling of TI by P R on P RE in λ did not include translation asymmetry due to the translation of cro and lack of translation of the P RE RNA in the overlap region 7. Inclusion of translation asymmetry may allow the TI data to be explained with a weaker pause‐enhanced occlusion effect than we estimated.
Regardless of the mechanisms, the TI appears to provide for positive feedback by the lysogenic repressor CI, a feature that should sharpen the decision between lytic and lysogenic development. In λ, CI repression of P R relieves TI on P RE, increasing P RE transcription into the cI gene 7, 36. In 186, CI repression of pR relieves TI on pL, increasing lysogenic transcription 27, 28. In both cases, maximal relief of TI is possible because neither repressor presents a roadblock to RNAPs from the upstream promoter 27, 36. We expect that repression of pR by 186 CI will also relieve TI on pE. In addition, dislodgement of λ CI by the passing RNAPs does not impair repression of P R, implying fast binding kinetics for λ CI 36, while this has not been tested for 186 CI repression of pR. In λ, the relief of TI on P RE by CI should provide the first CI positive feedback mechanism operating after infection, with repression of Cro and direct activation of P RM being the second and third mechanisms 36. In 186, relief of TI is the only mechanism for positive feedback by CI at pL 27 and this appears to also be the case at pE. Thus, in these convergently evolved genetic switches, different mechanisms of TI are employed interchangeably but with a consistent function of establishing positive feedback.
TI by dislodgement
In our previous study of relief of TI in λ, we found that two different repressors of P R, the natural CI repressor and a Streptococcus pyogenes dCas protein targeted to P R, behaved quite differently in response to transcription from P RE 36. Both proteins strongly repressed P R, and neither protein acted as a roadblock to transcription from P RE (dCas was targeted to the P RE template strand), indicating that both were readily dislodged by elongating RNAPs from P RE. However, this dislodgement of dCas interfered with its repression of P R, but the dislodgement of λ CI did not 36. This difference was able to be explained by invoking a difference in binding kinetics, with slow binding kinetics by dCas and fast binding kinetics for λ CI.
The λ CII protein and the 186 CII protein provide a similar contrast in dislodgement sensitivity, but for transcription activators rather than transcription repressors. λ CII activation of P RE was not affected by passing RNAPs from λ P R, while we found here that 186 CII activation of pE was inhibited by RNAPs from 186 pR. We showed that this could be explained if the binding kinetics of 186 CII were slow, such that its dislodgement by RNAPs significantly increased its rate of dissociation from DNA. Our results thus further emphasize the importance of DNA binding kinetics in the interaction between transcription factors and elongating RNAPs 2. Even though transcription factors and RNAP cannot be simultaneously bound to the same nucleotide, when binding kinetics are sufficiently rapid these entities can effectively ‘pass through’ one another with little evidence of interaction.
Pausing‐enhanced occlusion
Our previous study of the interaction between λ P R and P RE provided evidence for pausing‐enhanced occlusion as a mechanism of TI 7. However, despite the similarities in promoter arrangement and regulatory requirements between the λ P R.P RE.cII system and the 186 pR.pE.cII system, the low TI exerted by 186 pR on pE at high CII levels indicates that 186 does not employ pause‐enhanced occlusion at pE. Instead, we were able to confirm the pause‐enhanced occlusion mechanism in a specifically designed synthetic (REO) construct by using a protein roadblock to pause RNAP, in contrast to the intrinsic pause mediated by λ tR1. In wild‐type cells, the LacI roadblock was responsible for a ~ 1.6‐fold enhancement of TI (1.96‐fold TI versus 1.24‐fold TI without the roadblock). As predicted, increasing the pausing time by using mfd − cells increased the enhancement of TI due to the roadblock, to ~ 3.4‐fold (5.02‐fold TI versus 1.49‐fold TI without the roadblock). Modelling indicated that the termination rate of paused RNAPs is a less critical factor in determining the magnitude of pause‐enhanced occlusion than the rate of firing of the promoter providing the occluding RNAPs. Thus, the weaker effect of the roadblock‐induced pause in the REO circuit compared to the tR1‐induced pause in the λ case is primarily due to the 3.3‐fold higher strength of λ pR compared to P2 P e. In general, strong pause‐enhanced occlusion, whatever the nature of the pause, requires a high rate of supply of RNAPs to the pause site to overcome their loss by termination and by passage through the roadblock. We expect the same principle to apply in other organisms.
Author contributions
IBD and KES conceived and supervised the study; NH, MTC, IBD and KES designed experiments; NH and MTC performed experiments; NH, MTC and ACP did the modelling analysis; NH, MTC, IBD and KES analysed data; NH, IBD and KES wrote the manuscript; NH, ACP, IBD and KES made manuscript revisions.
Funding
This work was supported by the Australian Research Council via a Discovery Early Career Researcher Award to NH (DE150100091) and Discovery Grants (DP150103009, DP160101450). NH was also supported in part by a Fellowship from Synthetic Biology Future Science Platform, Commonwealth Scientific and Industrial Research Organisation. ACP was supported by a James S. McDonnell Foundation Postdoctoral Fellowship.
Supporting information
Table S1. Modelling parameters.
Fig. S1. Chromosomally integrated 186 TI and REO reporter constructs.
Fig. S2. The sequence of the phage 186 switch region.
Fig. S3. The pL − mutation reduces Apl translation.
Fig. S4. Sequence of the REO construct.
Acknowledgements
We thank Dr Alexandra Ahlgren‐Berg for performing some of the initial REO experiments. We are also grateful to members of the Shearwin lab and Prof Kim Sneppen (University of Copenhagen) for discussions.
Nan Hao and Michael T. Crooks are joint First Authors
Edited by Claus Azzalin
References
- 1. Shearwin KE, Callen BP and Egan JB (2005) Transcriptional interference – a crash course. Trends Genet 21, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hao N, Palmer AC, Dodd IB and Shearwin KE (2017) Directing traffic on DNA‐How transcription factors relieve or induce transcriptional interference. Transcription 8, 120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Palmer AC, Egan JB and Shearwin KE (2011) Transcriptional interference by RNA polymerase pausing and dislodgement of transcription factors. Transcription 2, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bendtsen KM, Erdossy J, Csiszovszki Z, Svenningsen SL, Sneppen K, Krishna S and Semsey S (2011) Direct and indirect effects in the regulation of overlapping promoters. Nucleic Acids Res 39, 6879–6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dornenburg JE, Devita AM, Palumbo MJ and Wade JT (2010) Widespread antisense transcription in Escherichia coli . MBio 1, e00024–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sneppen K, Dodd IB, Shearwin KE, Palmer AC, Schubert RA, Callen BP and Egan JB (2005) A mathematical model for transcriptional interference by RNA polymerase traffic in Escherichia coli . J Mol Biol 346, 399–409. [DOI] [PubMed] [Google Scholar]
- 7. Palmer AC, Ahlgren‐Berg A, Egan JB, Dodd IB and Shearwin KE (2009) Potent transcriptional interference by pausing of RNA polymerases over a downstream promoter. Mol Cell 34, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang J and Landick R (2016) A two‐way street: regulatory interplay between RNA polymerase and nascent RNA structure. Trends Biochem Sci 41, 293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Toulme F, Mosrin‐Huaman C, Artsimovitch I and Rahmouni AR (2005) Transcriptional pausing in vivo: a nascent RNA hairpin restricts lateral movements of RNA polymerase in both forward and reverse directions. J Mol Biol 351, 39–51. [DOI] [PubMed] [Google Scholar]
- 10. Artsimovitch I and Landick R (2000) Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc Natl Acad Sci USA 97, 7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herbert KM, La Porta A, Wong BJ, Mooney RA, Neuman KC, Landick R and Block SM (2006) Sequence‐resolved detection of pausing by single RNA polymerase molecules. Cell 125, 1083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Larson MH, Mooney RA, Peters JM, Windgassen T, Nayak D, Gross CA, Block SM, Greenleaf WJ, Landick R and Weissman JS (2014) A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science 344, 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hao N, Krishna S, Ahlgren‐Berg A, Cutts EE, Shearwin KE and Dodd IB (2014) Road rules for traffic on DNA‐systematic analysis of transcriptional roadblocking in vivo. Nucleic Acids Res 42, 8861–8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deuschle U, Gentz R and Bujard H (1986) lac Repressor blocks transcribing RNA polymerase and terminates transcription. Proc Natl Acad Sci USA 83, 4134–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crampton N, Bonass WA, Kirkham J, Rivetti C and Thomson NH (2006) Collision events between RNA polymerases in convergent transcription studied by atomic force microscopy. Nucleic Acids Res 34, 5416–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoffmann S, Hao N, Shearwin KE and Arndt KM (2019) Characterizing transcriptional interference between converging genes in bacteria. ACS Synth Biol 8, 466–473. [DOI] [PubMed] [Google Scholar]
- 17. Brophy JA and Voigt CA (2016) Antisense transcription as a tool to tune gene expression. Mol Syst Biol 12, 854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Callen BP, Shearwin KE and Egan JB (2004) Transcriptional interference between convergent promoters caused by elongation over the promoter. Mol Cell 14, 647–656. [DOI] [PubMed] [Google Scholar]
- 19. Browning DF and Busby SJ (2016) Local and global regulation of transcription initiation in bacteria. Nat Rev Microbiol 14, 638–650. [DOI] [PubMed] [Google Scholar]
- 20. Ptashne M (2004) A Genetic Switch: Phage Lambda Revisited. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 21. Neufing PJ, Shearwin KE and Egan JB (2001) Establishing lysogenic transcription in the temperate coliphage 186. J Bacteriol 183, 2376–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park JS, Marr MT and Roberts JW (2002) E. coli transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell 109, 757–767. [DOI] [PubMed] [Google Scholar]
- 23. Le TT, Yang Y, Tan C, Suhanovsky MM, Fulbright RM Jr, Inman JT, Li M, Lee J, Perelman S, Roberts JW et al (2018) Mfd dynamically regulates transcription via a release and catch‐up mechanism. Cell 172, 344–357.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haldimann A and Wanner BL (2001) Conditional‐replication, integration, excision, and retrieval plasmid‐host systems for gene structure‐function studies of bacteria. J Bacteriol 183, 6384–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lutz R and Bujard H (1997) Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1‐I2 regulatory elements. Nucleic Acids Res 25, 1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang S, Bipatnath M, Xu Y, Chen S, Dennis P, Ehrenberg M and Bremer H (1999) Activities of constitutive promoters in Escherichia coli . J Mol Biol 292, 19–37. [DOI] [PubMed] [Google Scholar]
- 27. Dodd IB and Egan JB (2002) Action at a distance in CI repressor regulation of the bacteriophage 186 genetic switch. Mol Microbiol 45, 697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dodd IB, Shearwin KE and Sneppen K (2007) Modelling transcriptional interference and DNA looping in gene regulation. J Mol Biol 369, 1200–1213. [DOI] [PubMed] [Google Scholar]
- 29. Lamont I, Richardson H, Carter DR and Egan JB (1993) Genes for the establishment and maintenance of lysogeny by the temperate coliphage 186. J Bacteriol 175, 5286–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neufing PJ, Shearwin KE, Camerotto J and Egan JB (1996) The CII protein of bacteriophage 186 establishes lysogeny by activating a promoter upstream of the lysogenic promoter. Mol Microbiol 21, 751–761. [DOI] [PubMed] [Google Scholar]
- 31. Shearwin KE and Egan JB (2000) Establishment of lysogeny in bacteriophage 186. DNA binding and transcriptional activation by the CII protein. J Biol Chem 275, 29113–29122. [DOI] [PubMed] [Google Scholar]
- 32. Murchland I, Ahlgren‐Berg A, Priest DG, Dodd IB and Shearwin KE (2014) Promoter activation by CII, a potent transcriptional activator from bacteriophage 186. J Biol Chem 289, 32094–32108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vassylyev DG, Vassylyeva MN, Perederina A, Tahirov TH and Artsimovitch I (2007) Structural basis for transcription elongation by bacterial RNA polymerase. Nature 448, 157–162. [DOI] [PubMed] [Google Scholar]
- 34. Epshtein V and Nudler E (2003) Cooperation between RNA polymerase molecules in transcription elongation. Science 300, 801–805. [DOI] [PubMed] [Google Scholar]
- 35. Neuman KC, Abbondanzieri EA, Landick R, Gelles J and Block SM (2003) Ubiquitous transcriptional pausing is independent of RNA polymerase backtracking. Cell 115, 437–447. [DOI] [PubMed] [Google Scholar]
- 36. Hao N, Palmer AC, Ahlgren‐Berg A, Shearwin KE and Dodd IB (2016) The role of repressor kinetics in relief of transcriptional interference between convergent promoters. Nucleic Acids Res 44, 6625–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Modelling parameters.
Fig. S1. Chromosomally integrated 186 TI and REO reporter constructs.
Fig. S2. The sequence of the phage 186 switch region.
Fig. S3. The pL − mutation reduces Apl translation.
Fig. S4. Sequence of the REO construct.