

Abstract
Objective
To investigate the efficacy and safety of ixekizumab in patients with active radiographic axial spondyloarthritis (SpA) and prior inadequate response to or intolerance of 1 or 2 tumor necrosis factor inhibitors (TNFi).
Methods
In this phase III randomized, double‐blind, placebo‐controlled trial, adult patients with an inadequate response to or intolerance of 1 or 2 TNFi and an established diagnosis of axial SpA (according to the Assessment of SpondyloArthritis international Society [ASAS] criteria for radiographic axial SpA, with radiographic sacroiliitis defined according to the modified New York criteria and ≥1 feature of SpA) were recruited and randomized 1:1:1 to receive placebo or 80‐mg subcutaneous ixekizumab every 2 weeks (IXEQ2W) or 4 weeks (IXEQ4W), with an 80‐mg or 160‐mg starting dose. The primary end point was 40% improvement in disease activity according to the ASAS criteria (ASAS40) at week 16. Secondary outcomes and safety were also assessed.
Results
A total of 316 patients were randomized to receive placebo (n = 104), IXEQ2W (n = 98), or IXEQ4W (n = 114). At week 16, significantly higher proportions of IXEQ2W patients (n = 30 [30.6%]; P = 0.003) or IXEQ4W patients (n = 29 [25.4%]; P = 0.017) had achieved an ASAS40 response versus the placebo group (n = 13 [12.5%]), with statistically significant differences reported as early as week 1 with ixekizumab treatment. Statistically significant improvements in disease activity, function, quality of life, and spinal magnetic resonance imaging–evident inflammation were observed after 16 weeks of ixekizumab treatment versus placebo. Treatment‐emergent adverse events (AEs) with ixekizumab treatment were more frequent than with placebo. Serious AEs were similar across treatment arms. One death was reported (IXEQ2W group).
Conclusion
Ixekizumab treatment for 16 weeks in patients with active radiographic axial SpA and previous inadequate response to or intolerance of 1 or 2 TNFi yields rapid and significant improvements in the signs and symptoms of radiographic axial SpA versus placebo.
Introduction
Axial spondyloarthritis (SpA) is a chronic inflammatory disease that is estimated to affect 0.9–1.4% of adults in the US and encompasses both nonradiographic axial SpA and radiographic axial SpA 1, 2. Radiographic axial SpA is also referred to as ankylosing spondylitis (AS). The disease is typically characterized by inflammatory back pain and radiographically defined sacroiliac (SI) joint structural damage 2, 3. Patients with axial SpA may also exhibit peripheral musculoskeletal (inflammatory arthritis, enthesitis, and dactylitis) and extraarticular (uveitis, psoriasis, and inflammatory bowel disease [IBD]) involvement.
Currently, the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network, the Assessment of SpondyloArthritis international Society (ASAS)/European League Against Rheumatism, and the National Institute for Health and Care Excellence guidelines for the management of axial SpA recommend treatment with tumor necrosis factor inhibitors (TNFi) in patients with axial SpA who do not respond or tolerate nonsteroidal antiinflammatory drugs (NSAIDs) 4, 5, 6. Approximately 30–40% of patients with AS do not achieve adequate disease control or symptom relief according to clinical trials of TNFi 7, 8, 9, 10, 11, 12. In addition, some patients may not be eligible to receive TNFi due to relative contraindications 13.
The interleukin‐17 (IL‐17) axis has been linked to the immunopathology of axial SpA 14, 15. IL‐17 inhibition has demonstrated efficacy in patients with AS; however, an IL‐17 antagonist has not been evaluated in a population that exclusively consisted of patients with prior inadequate response to or intolerance of TNFi in a clinical trial setting 16. This is an important population on which to focus, given that it has been shown to be difficult to treat, with treatment responses lower in magnitude than observed in biologics‐naive populations 17, 18.
Ixekizumab is a high‐affinity monoclonal antibody that selectively targets IL‐17A 19. Here we present the 16‐week results of COAST‐W, a phase III clinical trial investigating the efficacy and safety of ixekizumab in patients with active radiographic axial SpA and previous inadequate response to or intolerance of 1 or 2 TNFi.
Patients and Methods
Trial design
COAST‐W is a multicenter, phase III, randomized, double‐blind, placebo‐controlled, parallel‐group, outpatient clinical trial of 1 year's duration, followed by an optional 2‐year extension trial (COAST‐Y) (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). Patient enrollment and data collection occurred at 106 sites located in 15 countries across North America, South America, Europe, and Asia (for a list of investigators and sites, see Supplementary Appendix A, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). This trial was conducted in accordance with the ethical principles of the Declaration of Helsinki and in compliance with local laws and regulations. All participants provided informed consent. COAST‐W protocol and consent forms were approved by each site's institutional review board or ethics committee. The trial was registered with ClinicalTrials.gov (NCT02696798) and the European Union Clinical Trials Register (2015‐003937‐84).
Trial participants
Complete inclusion and exclusion criteria are provided in Supplementary Appendix B (available at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). Eligible subjects were age ≥18 years, required to have an established diagnosis of axial SpA and fulfillment of ASAS classification criteria for radiographic axial SpA (i.e., radiographic evidence of sacroiliitis according to the modified New York criteria and having ≥1 SpA feature), and required to have a history of back pain for ≥3 months with an age at onset of <45 years 20, 21, 22. SI joint radiographs were scored by central readers. All patients fulfilling ASAS criteria for radiographic axial SpA (20) also fulfilled the modified New York criteria for AS (21).
Additional inclusion criteria included a baseline Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score (23) of ≥4, a baseline total back pain numeric rating scale score of ≥4, and a history of therapy for axial SpA of at least 12 weeks’ duration prior to screening. Patients were required to have discontinued at least 1 TNFi, but no more than 2 TNFi, either due to intolerance or due to an inadequate response (in the opinion of the investigator) to treatment with a single TNFi for at least 12 weeks at an adequate dose. The required TNFi washout periods are described in Supplementary Appendix B and were based upon the half‐lives of the respective TNFi.
The exclusion criteria included total spinal ankylosis (according to the site investigator's opinion), active or recent infections, current or history of lymphoproliferative or malignant disease (<5 years prior to baseline) or other medical conditions (e.g., systemic inflammatory diseases or chronic pain conditions such as fibromyalgia), other non‐TNF biologic or other immunomodulatory agent treatments, or surgical procedures that could pose an unacceptable risk to patients or that could confound interpretation of trial results. Patients with IBD (Crohn's disease or ulcerative colitis) were eligible if no disease exacerbations had occurred for ≥6 months (stable treatment allowed). Patients with anterior uveitis were eligible if no exacerbations had occurred for ≥4 weeks.
Patients could continue to receive the following medications at a stable dose: sulfasalazine (≤3 gm/day), methotrexate (≤25 mg/week), prednisone or equivalent (≤10 mg/day), and NSAIDs. No changes to these medications were allowed during the blinded treatment dosing period, except for safety reasons. Analgesics were also allowed according to the eligibility criteria (see Supplementary Appendix B, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract).
Randomization and blinding
Patients were randomly assigned (1:1:1) to receive subcutaneous administration of ixekizumab 80 mg every 2 weeks (IXEQ2W group), ixekizumab 80 mg every 4 weeks (IXEQ4W group), or matched placebo from week 0 to week 16. Patients randomized to the ixekizumab treatment regimens were randomized (1:1) to receive either an 80‐mg or 160‐mg starting dose of ixekizumab at week 0. The 2 starting doses were included in order to assess the impact of starting dose on week 16 responses, following regulatory agency feedback. All patients received the same frequency and number of injections regardless of treatment arm or assigned starting dose.
During the double‐blinded treatment period (weeks 0–16), site personnel, patients, and the sponsor trial team were blinded with regard to treatment. Randomization to treatment groups was determined using a computer‐generated, random‐sequence, interactive web‐response system with follow‐up confirmation by site personnel using the confirmation number present on the investigational product packaging. Randomization of treatment assignment (including starting dose) was stratified by country, high‐sensitivity C‐reactive protein (CRP) level (≤5 or >5 mg/liter) at screening, and the number of prior TNFi taken (1 or 2) to achieve between‐group comparability.
At week 16, patients entered the extended treatment period (weeks 16–52). Patients who were initially assigned to the placebo arm were, for the extended treatment period, randomly reassigned at week 16 to IXEQ4W or IXEQ2W with a 160‐mg starting dose. Patients already receiving ixekizumab remained on their assigned treatment regimens through week 52.
Procedures of product administration
The investigational product was supplied in prefilled manual syringes for subcutaneous administration, with trial‐specific labels. Ixekizumab and its matching placebo were visually indistinguishable from each other. To maintain blinding, all patients received 2 injections at week 0 and 1 injection every 2 weeks. Placebo patients received a placebo injection every 2 weeks and IXEQ4W patients received a placebo injection every other 2 weeks to maintain blinding. Patients assigned to an ixekizumab treatment regimen received their starting dose (either 80‐mg ixekizumab [1 80‐mg injection and 1 placebo injection] or ixekizumab 160 mg [2 80‐mg injections]) at week 0. Trial visits and data collection occurred at baseline (week 0) and at weeks 1, 2, 4, 8, 12, and 16 during the blinded treatment dosing period. The primary end point was assessed at week 16.
Efficacy and safety assessments
The primary end point of the trial was the proportion of patients achieving an ASAS 40% improvement in disease activity (ASAS40) 22 at week 16, with comparison of each ixekizumab dosing regimen to placebo. Major secondary end points assessed at week 16 were ASAS20, an Ankylosing Spondylitis Disease Activity Score (ASDAS) of <2.1 (inactive or low disease activity) (24), and changes from baseline in the ASDAS, BASDAI, Bath Ankylosing Spondylitis Functional Index (BASFI) 25, Medical Outcomes Study Short Form 36 (SF‐36) health survey physical component score (PCS) 26, ASAS Health Index (ASAS‐HI) 27, 28, and Spondyloarthritis Research Consortium of Canada (SPARCC) magnetic resonance imaging (MRI) index for the spine (MRI protocol addendum only) 29. These end points were assessed at every visit (i.e., screening, baseline, weeks 1, 2, 4, 8, 12, and 16) except for the SF‐36 PCS and ASAS‐HI (collected at baseline, weeks 4, 8, and 16) and MRI (collected at baseline and week 16). Additionally, CRP level was assessed at every visit. All listed end points are described in Supplementary Appendix C, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract.
Safety outcomes were assessed at every visit. Data on terms related to cerebrocardiovascular events and suspected IBD were adjudicated by external clinical event committees. Details on adjudication criteria are provided in Supplementary Appendix C.
Statistical analysis
The sample size of COAST‐W was estimated to have 96% power for testing the superiority of IXEQ2W to placebo for an ASAS40 response at week 16 (for details on power analysis assumptions, see Supplementary Appendix C).
Efficacy analyses for the blinded treatment dosing period included all randomized patients according to the treatment to which they were assigned. Analyses of the IXEQ2W and IXEQ4W treatment groups were performed without regard to the starting dose. Missing values (including for those patients who discontinued trial treatment) were imputed as nonresponders using nonresponder imputation for categorical variables; continuous variables were analyzed using a mixed‐effects model of repeated measures (MMRM) without imputation for missing values.
The primary analysis method for categorical outcome variables was logistic regression with treatment, geographic region, baseline CRP status (≤5 versus >5 mg/liter), and the number of prior TNFi taken included in the model. Secondary analysis of categorical outcomes was performed using Fisher's exact test when the logistic model did not converge due to sparse data.
The primary analysis method for continuous outcomes, except SPARCC MRI index scores, was MMRM with treatment, geographic region, baseline CRP level, the number of prior TNFi taken, baseline value, visit, baseline value–by‐visit, and treatment‐by‐visit interaction as fixed factors. The primary analysis method for SPARCC MRI index scores was analysis of covariance (ANCOVA) with observed case analysis, with inclusion only of patients with both baseline (between 42 days prior to and 14 days after the first injection) and week 16 (injection date at week 16 [±14 days]) SPARCC MRI index scores. The ANCOVA included treatment, geographic region, baseline CRP level, number of prior TNFi taken, and baseline value.
A graphical multiple testing strategy was implemented for primary and major secondary objectives to control the overall family‐wise Type I error rate at a 2‐sided alpha level of 0.05 (see Supplementary Figures 2–4, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). There was no adjustment for multiple comparisons for any other analyses. The analyses investigating efficacy prior to week 16 were not included in the multiple testing strategy.
Descriptive analyses (with no prespecified hypothesis testing) were performed using the safety population, which was defined as all randomized patients who received ≥1 dose of the trial drug, according to assigned treatments. Safety data were summarized as the frequency of events occurring in each treatment group during the blinded treatment dosing period.
Results
Patient disposition and baseline characteristics
The Consolidated Standards of Reporting Trials (CONSORT) diagram is provided in Figure 1. Of the 610 patients assessed for trial eligibility, 316 were randomly assigned to either placebo (n = 104), IXEQ2W (n = 98), or IXEQ4W (n =114), and 294 patients were designated as ineligible at screening or were discontinued prior to randomization. The predominant reason for ineligibility at screening was the lack of definitive sacroiliitis according to centrally read SI joint radiographs (n = 217 [35.6%]) (see Supplementary Table 1, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). The number of patients screened and enrolled for each country is shown in Supplementary Table 2 (available at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). The 16‐week blinded treatment dosing period was completed by 93 patients (89.4%) receiving placebo, 90 patients (91.8%) receiving IXEQ2W, and 99 patients (86.8%) receiving IXEQ4W. Overall, 282 patients (89.2%) completed week 16.
Figure 1.
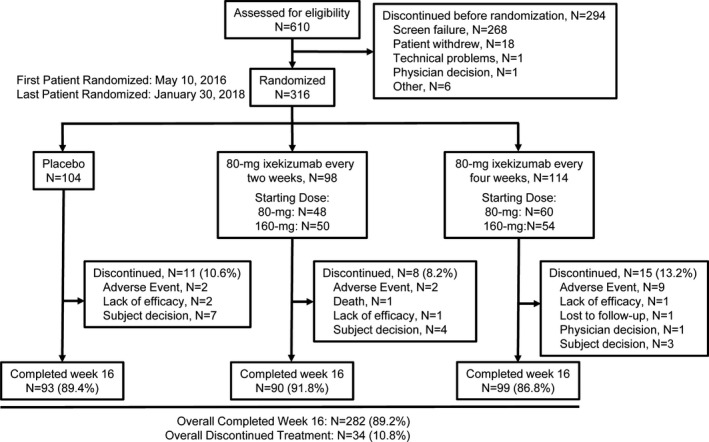
Disposition of the patients. Details are given according to the Consolidated Standards of Reporting Trials (CONSORT) statement for reporting randomized controlled trials.
Treatment arms were generally balanced for baseline demographic and clinical characteristics (Table 1). Baseline spondyloarthritis features are listed in Supplementary Table 3 (available at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). Overall, 315 patients had prior TNFi experience; 205 patients (65.1%) had an inadequate response to 1 TNFi, 78 patients (24.8%) had an inadequate response to 2 TNFi, and 32 patients (10.2%) were intolerant of TNFi. One patient was inadvertently enrolled without prior TNFi experience. Reasons for the discontinuation of previous biologic therapies are provided in Supplementary Table 4 (available at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). The median washout period for TNFi prior to study entry was 5 months (minimum 1 month, maximum 155 months). At baseline, 86 patients (27.2%) were receiving concomitant conventional synthetic disease‐modifying antirheumatic drugs (DMARDs; sulfasalazine or methotrexate).
Table 1.
Patient demographic and clinical characteristics for the intent‐to‐treat population in the COAST‐W studya
| Placebo group (n = 104) | IXEQ2W group (n = 98) | IXEQ4W group (n = 114) | |
|---|---|---|---|
| Age, mean ± SD years | 46.6 ± 12.7 | 44.2 ± 10.8 | 47.4 ± 13.4 |
| Male sex | 87 (83.7) | 75 (76.5) | 91 (79.8) |
| Race | |||
| White | 85 (81.7) | 78 (79.6) | 91 (80.5) |
| Asian | 13 (12.5) | 13 (13.3) | 14 (12.4) |
| Weight, mean ± SD kg | 84.3 ± 17.9 | 79.3 ± 17.3 | 85.5 ± 20.2 |
| <70 kg | 21 (20.2) | 25 (25.5) | 24 (21.1) |
| ≥70 kg | 83 (79.8) | 73 (74.5) | 90 (78.9) |
| Body mass index, mean ± SD kg/m2 | 28.9 ± 5.6 | 27.5 ± 5.4 | 29.4 ± 7.3 |
| Age at onset of axial SpA, mean ± SD years | 27.1 ± 8.8 | 28.1 ± 10 | 28.9 ± 9.6 |
| Duration of symptoms since axial SpA onset, mean ± SD years | 19.9 ± 11.6 | 16.5 ± 9.6 | 18.8 ± 11.6 |
| Duration of disease since axial SpA diagnosis, mean ± SD years | 13.0 ± 10.5 | 11.7 ± 8.8 | 10.1 ± 7.8 |
| Use of DMARDs | |||
| Methotrexate | 20 (19.2) | 9 (9.2) | 12 (10.5) |
| Sulfasalazine | 13 (12.5) | 16 (16.3) | 17 (14.9) |
| Use of oral corticosteroid | 14 (13.5) | 11 (11.2) | 11 (9.6) |
| Use of NSAIDs | 84 (80.8) | 71 (72.4) | 86 (75.4) |
| Prior TNFi experienceb | |||
| 1 prior TNFi | 62 (59.6) | 66 (68.0) | 70 (61.4) |
| 2 prior TNFi | 42 (40.4) | 31 (32.0) | 44 (38.6) |
| Reason for failing prior TNFic | |||
| Inadequate response to 1 TNFi | 64 (61.5) | 66 (68.0) | 75 (65.8) |
| Inadequate response to 2 TNFi | 32 (30.8) | 20 (20.6) | 26 (22.8) |
| Intolerance of TNFi | 8 (7.7) | 11 (11.3) | 13 (11.4) |
| TNFi washout period, median (minimum–maximum) daysd | 123.5 (31.0–4,053.0) | 143.0 (32.0–3,851.0) | 153.5 (29.0–4,639.0) |
| Baseline C‐reactive protein, mean ± SD mg/liter | 16.0 ± 22.3 | 16.9 ± 19.8 | 20.2 ± 34.3 |
| ≤5.00 mg/liter | 39 (37.5) | 26 (26.5) | 44 (38.6) |
| >5.00 mg/liter | 65 (62.6) | 72 (73.5) | 70 (61.4) |
| ASDAS, mean ± SD | 4.1 ± 0.8 | 4.2 ± 0.8 | 4.2 ± 0.9 |
| BASDAI score, mean ± SD | 7.3 ± 1.3 | 7.5 ± 1.3 | 7.5 ± 1.3 |
| BASFI score, mean ± SD | 7.0 ± 1.7 | 7.4 ± 1.4 | 7.4 ± 1.8 |
| ASAS‐HI score, mean ± SD | 9.0 ± 3.5 | 10.1 ± 3.6 | 10.0 ± 3.7 |
| SF‐36 PCS, mean ± SD | 30.6 ± 7.8 | 27.9 ± 7.3 | 27.5 ± 8.3 |
| SPARCC MRI spine score, mean ± SDe | 6.4 ± 10.2 | 11.1 ± 20.3 | 8.3 ± 16 |
| SPARCC MRI spine score ≥2e | 25 (49.0) | 24 (45.3) | 31 (53.4) |
Except where indicated otherwise, values are the number (%) of patients in the analysis population. IXEQ2W = 80‐mg subcutaneous ixekizumab every 2 weeks; IXEQ4W = 80‐mg subcutaneous ixekizumab every 4 weeks; SpA = spondyloarthritis; DMARDs = disease‐modifying antirheumatic drugs; NSAIDs = nonsteroidal antiinflammatory drugs; ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; ASAS‐HI = Assessment of Spondyloarthritis international Society Health Index; SF‐36 = Medical Outcomes Study Short Form 36 health survey; PCS = physical component score; SPARCC = Spondyloarthritis Research Consortium of Canada.
Patients were included regardless of whether they were inadequate responders to or intolerant of tumor necrosis factor inhibitors (TNFi).
If a patient had both an inadequate response to 1 TNFi and an intolerance of another TNFi, that patient was classified as having had an inadequate response to 1 TNFi. Patients in the intolerance category discontinued prior TNFi (1 or 2) due to intolerance only.
Washout period for the last TNFi taken. Data were available for all but 1 IXEQ2W patient.
Data were available for the magnetic resonance imaging (MRI) addendum population only (n = 51 for the placebo group, n = 58 for the IXEQ2W group, and n = 53 for the IXEQ4W group).
Clinical end points
Ixekizumab was found to be superior to placebo for the primary and all major secondary end points at week 16 with both ixekizumab treatment regimens, except for ASAS‐HI scores with IXEQ2W (Figures 2, 3, 4 and Supplementary Table 5, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). The proportion of patients who achieved the primary end point of an ASAS40 response at week 16 was significantly higher among patients treated with either IXEQ2W (n = 30 [30.6%]; P = 0.003) or IXEQ4W (n = 29 [25.4%]; P = 0.017) than among patients treated with placebo (n = 13 [12.5%]), with a significant response observed as early as week 1 for both ixekizumab regimens (Figure 2A). The proportion of patients who achieved an ASAS20 response at week 16 was also significantly higher among patients treated with either IXEQ2W (n = 46 [46.9%]) or IXEQ4W (n = 55 [48.2%]) than among patients treated with placebo (n = 31 [29.8%]), with a significant response as early as week 1 for both dosing regimens (Figure 2B). The starting dose of 160 mg versus 80 mg at week 0 did not lead to a significant improvement of the results observed at week 16 (Supplementary Figure 5, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). Further analyses are required in order to evaluate the effect of starting dose on speed of onset.
Figure 2.
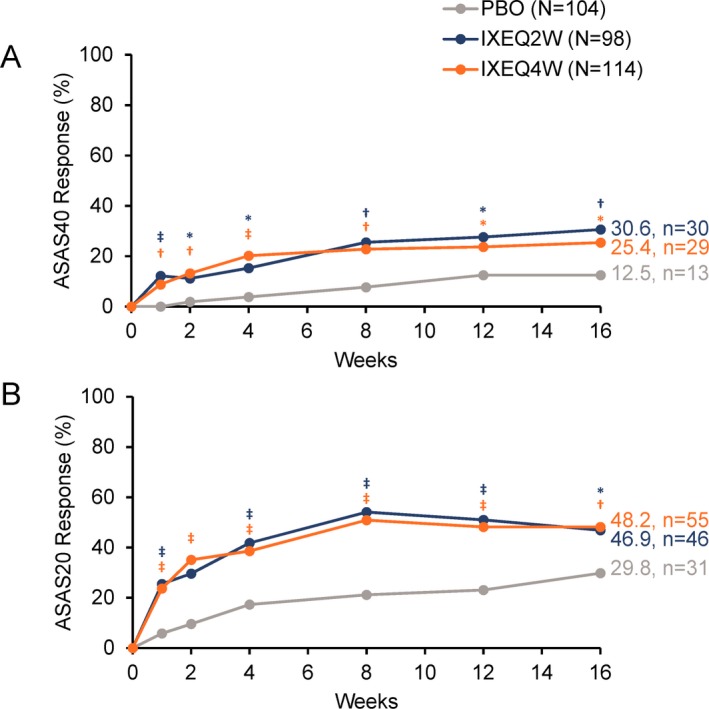
Proportion of patients achieving A, 40% improvement in disease activity according to the Assessment of SpondyloArthritis international Society criteria (ASAS40) or B, ASAS20 responses through week 16 when treated with placebo (PBO), ixekizumab every 2 weeks (IXEQ2W), or ixekizumab every 4 weeks (IXEQ4W). * = P < 0.05; † = P < 0.01; ‡ = P < 0.001, all versus placebo by logistic regression analysis, except for week 1 with ASAS40, for which Fisher's exact test was used due to model nonconvergence. Only analyses at week 16 were included in the prespecified multiple testing strategy. n = number of patients in the analysis category; N = number of patients in the analysis population.
Figure 3.
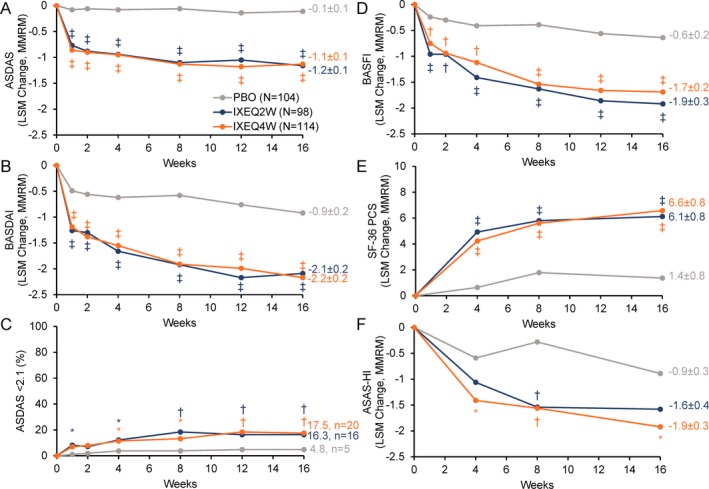
Major secondary outcomes investigating disease activity, function, and quality of life among patients treated with placebo (PBO), ixekizumab every 2 weeks (IXEQ2W), or ixekizumab every 4 weeks (IXEQ4W) for 16 weeks. A–D, Ankylosing Spondylitis Disease Activity Score (ASDAS) (A), Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) (B), ASDAS score <2.1 (C), and Bath Ankylosing Spondylitis Functional Index (BASFI) (D). E and F, Medical Outcomes Study Short Form 36 (SF‐36) health survey, physical component score (PCS) (E) and Assessment of Spondyloarthritis international Society Health Index (ASAS‐HI) (F). * = P < 0.05; † = P < 0.01; ‡ = P < 0.001, all versus placebo by mixed‐effects model of repeated measures (MMRM) or logistic regression analysis (C only). Only analyses at week 16 were included in the prespecified multiple testing strategy. Values shown at week 16 for all measures (except C) are the least squares mean (LSM) ± SE.
Figure 4.
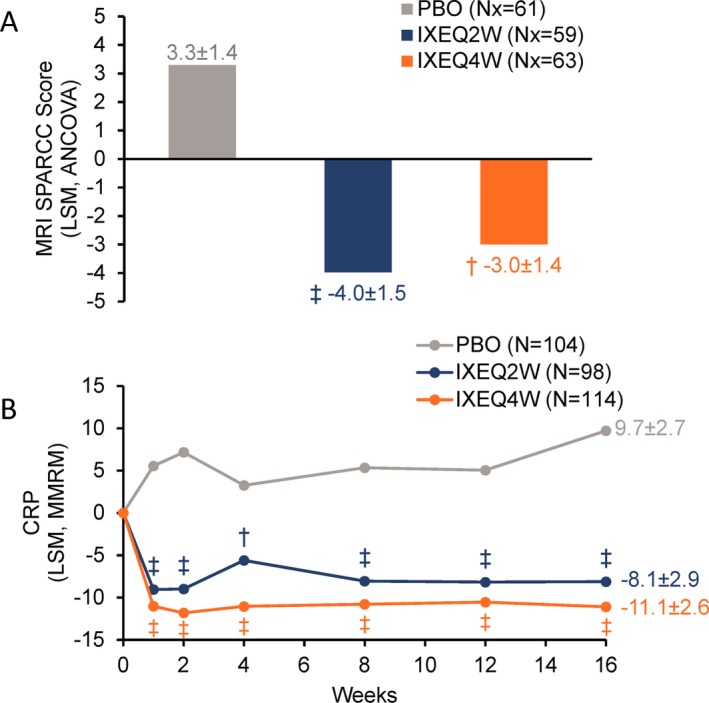
Spondyloarthritis Research Consortium of Canada (SPARCC) magnetic resonance imaging (MRI) index spine scores (A) and serum C‐reactive protein (CRP) levels (B) among patients treated with placebo (PBO), ixekizumab every 2 weeks (IXEQ2W), or ixekizumab every 4 weeks (IXEQ4W) through 16 weeks. † = P < 0.01; ‡ = P < 0.001, all versus placebo by analysis of covariance (ANCOVA) and least squares mean (LSM) (A) or mixed‐effects model of repeated measures (MMRM) and LSM (B). Only SPARCC MRI spine score analyses at week 16 were included in the prespecified multiple testing strategy. Values shown are the LSM ± SE. N = number of patients in the analysis population; Nx = number of patients in the MRI addendum population.
Statistically significant improvements in measures of disease activity were observed among patients treated with IXEQ2W or IXEQ4W relative to placebo at week 16, as measured by a change from baseline in ASDAS and BASDAI scores, as well as achievement of ASDAS <2.1 (inactive or low disease activity) (Figures 3A–C). Patient function was also significantly improved at week 16 in patients treated with IXEQ2W or IXEQ4W relative to placebo, as measured by a change from baseline in BASFI scores (Figure 3D).
Quality of life end points
Statistically significant improvements in quality of life, as measured by mean change from baseline in the SF‐36 PCS, were reported at week 16 for patients treated with IXEQ2W and IXEQ4W versus placebo (Figure 3E). Similarly, statistically significant improvements in health functioning, as measured by mean change from baseline in ASAS‐HI scores, were reported at week 16 among patients treated with IXEQ4W versus placebo (Figure 3F).
Spinal MRI and systemic inflammation
Spinal MRI and systemic inflammation significantly improved in patients treated with IXEQ2W or IXEQ4W, as demonstrated by the mean change from baseline in SPARCC MRI index spine scores and in mean change from serum baseline CRP levels, respectively, at week 16 versus placebo (Figures 4A and B).
Safety
Safety outcomes reported in COAST‐W during the blinded treatment dosing period are summarized in Table 2. Overall mean ± SD exposure was 109.1 ± 19.6 days or a total of 94.4 patient‐years. The proportions of patients in each ixekizumab treatment regimen who reported treatment‐emergent adverse events (TEAEs) were higher than those for placebo patients and were similar between ixekizumab treatment regimens. Most reported TEAEs were mild or moderate in severity. Severe TEAEs occurred in 7 placebo patients (6.7%), 4 IXEQ2W patients (4.1%), and 4 IXEQ4W patients (3.5%). The most frequently reported TEAEs (occurring in ≥5% of patients receiving ixekizumab overall) were upper respiratory tract infections and injection site reactions. AEs leading to discontinuation were reported for 2 placebo patients (1.9%), 3 IXEQ2W patients (3.1%), and 10 IXEQ4W patients (8.8%) (for specific AEs, see Supplementary Table 6, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40753/abstract). Few serious AEs were reported, with similar rates across treatment arms: n = 5 (4.8%) for placebo patients, n = 3 (3.1%) for IXEQ2W patients, and n = 4 (3.5%) for IXEQ4W patients. One death (suicide) occurred in the IXEQ2W treatment arm, in a patient with a documented prior history of depression of ~1 year (reported as mild at study entry), and was judged by the blinded principal investigator to be unrelated to the investigational product.
Table 2.
Adverse events (AEs) and treatment‐emergent adverse events (TEAEs) during the 16‐week blinded treatment dosing period of the COAST‐W studya
| Placebo group (n = 104) | IXEQ2W group (n = 98) | IXEQ4W group (n = 114) | |
|---|---|---|---|
| TEAE | 51 (49.0) | 59 (60.2) | 73 (64.0) |
| Mild | 18 (17.3) | 23 (23.5) | 34 (29.8) |
| Moderate | 26 (25.0) | 32 (32.7) | 35 (30.7) |
| Severe | 7 (6.7) | 4 (4.1) | 4 (3.5) |
| Discontinuation due to AE | 2 (1.9) | 3 (3.1) | 10 (8.8) |
| Serious AE | 5 (4.8) | 3 (3.1) | 4 (3.5) |
| Death | 0 | 1 (1.0) | 0 |
| Common TEAEsb | |||
| Upper respiratory tract infection | 3 (2.9) | 4 (4.1) | 9 (7.9) |
| Injection site reaction | 1 (1.0) | 8 (8.2) | 3 (2.6) |
| TEAEs of special interest | |||
| Hepatic | 2 (1.9) | 1 (1.0) | 5 (4.4) |
| Cytopenia | 0 | 2 (2.0) | 0 |
| Grade 1 neutropenia (≥1.5 to <2.0 × 109 cells/liter) | 1 (1.0) | 8 (8.2) | 10 (8.8) |
| Grade 2 neutropenia (≥1.0 to <1.5 × 109 cells/liter) | 0 | 1 (1.0) | 1 (0.9) |
| Grade 3 neutropenia (≥0.5 to <1.0 × 109 cells/liter) | 0 | 0 | 0 |
| Grade 4 neutropenia (<0.5 × 109 cells/liter) | 0 | 0 | 1 (0.9) |
| Infections | 10 (9.6) | 23 (23.5) | 34 (29.8) |
| Mild | 5 (4.8) | 14 (14.3) | 20 (17.5) |
| Moderate | 5 (4.8) | 9 (9.2) | 13 (11.4) |
| Severe | 0 | 0 | 1 (0.9) |
| Serious | 0 | 0 | 2 (1.8) |
| Candida (genital) | 0 | 1 (1.0) | 0 |
| Candida (esophageal) | 0 | 1 (1.0) | 0 |
| Herpes zoster | 0 | 0 | 1 (0.9) |
| Reactivated tuberculosis | 0 | 0 | 0 |
| Allergic reactions/hypersensitivities | 1 (1.0) | 6 (6.1) | 3 (2.6) |
| Potential anaphylaxis | 0 | 0 | 0 |
| Injection site reactionsc | 6 (5.8) | 16 (16.3) | 9 (7.9) |
| Cerebrocardiovascular eventsd | 1 (1.0) | 1 (1.0) | 0 |
| Malignancies | 0 | 0 | 1 (0.9) |
| Depression | 5 (4.8) | 2 (2.0) | 0 |
| Anterior uveitise | 0 | 3 (3.1) | 2 (1.8) |
| Inflammatory bowel disease | 1 (1.0) | 0 | 3 (2.6) |
| Interstitial lung disease | 0 | 0 | 0 |
Values are the number (%) of patients in the analysis population. IXEQ2W = 80‐mg subcutaneous ixekizumab every 2 weeks; IXEQ4W = 80‐mg subcutaneous ixekizumab every 4 weeks.
Common TEAEs are defined as those that occurred at a frequency of ≥5% for patients receiving ixekizumab (both treatment regimen populations combined).
Injection site reaction high‐level terms include injection site, pain, erythema, dermatitis, hypersensitivity, pruritus, bruising, rash, and paresthesia or reaction (unspecified).
Confirmed cerebrocardiovascular events only.
Anterior uveitis was not a prespecified AE of special interest but was included in the prespecified analyses.
The incidence of infections was higher for ixekizumab than for placebo, with most infections being mild or moderate (Table 2). Two Candida infections were reported in the IXEQ2W study arm (1 esophageal and 1 genital candidiasis), 1 herpes zoster infection was reported in the IXEQ4W study arm, and no infections were reported in the placebo group. Serious infections were reported in 2 patients in the IXEQ4W group (1 peritonitis and 1 pharyngitis; both patients continued the trial). No new cases of tuberculosis or reactivation were reported during the trial.
Reported injection site reactions were all mild or moderate in severity, occurred across all treatment arms (n = 6 [5.8%] for placebo, n = 16 [16.3%] for IXEQ2W, and n = 9 [7.9%] for IXEQ4W), and led to treatment discontinuation in only 2 patients (1 each in the IXEQ2W and IXEQ4W groups).
Mild neutropenia (grade 1) occurred more frequently in the ixekizumab arms than in the placebo arm, was transient in nature, and resolved spontaneously while ixekizumab treatment continued. A single case of grade 2 neutropenia was reported in each ixekizumab treatment arm; 1 of these cases existed before treatment began. One malignancy (acute promyelocytic leukemia [PML]) was reported 4 weeks postbaseline in the IXEQ4W treatment arm. The single report of grade 4 neutropenia was in this same patient. A post hoc analysis of a serum sample collected prior to investigational product exposure revealed that this patient had a genetic risk factor for acute PML (PML/retinoic acid receptor ɑ mutation) prior to trial entry. Treatment was discontinued in this patient.
Single adjudicated cerebrocardiovascular events were reported in the placebo and IXEQ2W treatment arms. The cerebrocardiovascular events were confirmed as stent placement (placebo group) and atrial fibrillation (IXEQ2W group), respectively. Both of these patients continued treatment through week 16. No major adverse cerebrocardiovascular events were reported during the blinded treatment dosing period. Depression was reported more often in patients taking placebo (n = 5 [4.8%]) than in patients taking IXEQ2W (n = 2 [2.0%]) and IXEQ4W (n = 0).
IBD was reported in 1 patient (1.0%) in the placebo arm (colitis), no patients in the IXEQ2W arm, and in 3 patients (2.6%) in the IXEQ4W arm (1 colitis, 1 ulcerative colitis, and 1 Crohn's disease). Two cases (1 in the placebo group and 1 in the IXEQ4W group) were flares in patients with a preexisting diagnosis of IBD. Of the other 2 IBD cases (IXEQ4W group), 1 case was in a current smoker (smoking since 1994) with a perianal cyst reported in 2010 and accompanying intermittent abdominal pain since that time. The other case was in a long‐term smoker (since 2009), who stopped smoking 3 months prior to trial entry, with a prior history of anemia (2012–2014) and intermittent diarrhea since 2011 (ongoing but mild at time of trial entry). All reported cases of IBD were adjudicated as “probable.” Anterior uveitis was reported in 3 IXEQ2W patients (3.1%) and 2 IXEQ4W patients (1.8%); 3 of these 5 patients (2 in the IXEQ2W group and 1 in the IXEQ4W group) were documented to have a preexisting history of anterior uveitis; 4 of the 5 patients were HLA–B27 positive, and all had longstanding disease (experiencing symptoms for at least 7 years and up to 28 years). Psoriasis was reported in 2 patients with a previous medical history of psoriasis (1 in the placebo group and 1 in the IXEQ4W group).
Treatment‐emergent antidrug antibodies were observed in 3 placebo patients (2.9%), 4 IXEQ2W patients (4.1%), and 8 IXEQ4W patients (7.1%). Most of the reported antidrug antibodies were classified as being low titer, 2 IXEQ2W patients (2.0%) and 1 IXEQ4W patient (0.9%) were reported as having moderate titers, and 1 IXEQ2W patient (0.9%) was reported with high titers. Neutralizing antidrug antibodies were detected in 3 IXEQ4W patients (2.7%). No associations were identified between treatment‐emergent antidrug antibody status and ASAS40 response, injection site reactions, or potential allergic/hypersensitivity events with either ixekizumab treatment regimen.
Discussion
The COAST‐W trial is the first large, randomized, controlled trial focusing exclusively on radiographic axial SpA patients with prior treatment failure (inadequate response to or intolerance of 1 or 2 TNFi). In that respect, this study differs from other reported phase III studies of radiographic axial SpA that enrolled exclusively or predominantly biologic DMARD (bDMARD)–naive patients. Ixekizumab was superior to placebo in reducing the signs and symptoms of radiographic axial SpA in this population of patients with longstanding disease and very high disease activity. For the primary end point (ASAS40 response), both IXEQ2W and IXEQ4W were superior to placebo at week 16. Statistically significant and clinically meaningful improvements over placebo were also observed at week 16 for all major secondary end points, including measures of disease activity, function, and quality of life for IXEQ4W and for all but 1 (ASAS‐HI) major secondary end points for IXEQ2W. Statistically significant improvements over placebo were reported early in the trial for all of these end points. In addition, and despite baseline levels of spinal inflammation being relatively low, thereby hampering the ability to show improvement, a statistically significant and objective effect on spinal inflammation was observed for IXEQ2W and IXEQ4W, as measured by the SPARCC MRI index score, which was supported by parallel improvements in CRP levels. The spinal MRI data are the first to be reported in a large subset of patients with prior intolerance of or inadequate response to TNFi in the context of a placebo‐controlled registration study.
Despite the greater exposure with the IXEQ2W regimen, IXEQ2W did not show a clinically meaningful incremental increase in observed efficacy relative to the IXEQ4W regimen. Similarly, the week 0 starting dose of ixekizumab 160 mg did not reveal a clinically meaningful incremental improvement in week 16 response rates relative to the 80‐mg starting dose for either ixekizumab regimen. The impact of starting dose on speed of onset requires further analysis.
Ixekizumab showed an acceptable safety profile in this patient population with longstanding and very active disease. The frequency of TEAEs in this population (patients with prior inadequate response to or intolerance of TNFi) was higher across all treatment arms, including placebo, than those reported in a bDMARD‐naive population with radiographic axial SpA from another phase III ixekizumab study (COAST‐V, NCT02696785) 30. TEAEs occurred more often with ixekizumab than with placebo, mostly driven by upper respiratory tract infections and injection site reactions. Serious AEs occurred at similar rates across ixekizumab and placebo treatment arms. The reported frequencies for total TEAEs and serious AEs are consistent with those reported among patients with psoriasis and psoriatic arthritis (PsA) treated with ixekizumab 31, 32, 33, 34. Discontinuations due to AEs were similar for IXEQ2W relative to placebo but higher for IXEQ4W relative to placebo, with no particular AE driving the difference. The explanation for this discrepancy is unclear, given that there were no discontinuations due to AEs in the IXEQ4W treatment arm in the study of ixekizumab treatment in bDMARD‐naive patients with radiographic axial SpA 30.
Infection frequencies were higher in patients treated with ixekizumab relative to placebo. The majority of infections were mild, with the most commonly reported being upper respiratory tract infections and nasopharyngitis. Serious infections were uncommon (n = 2). Reported infection frequencies were higher than those reported in COAST‐V, but consistent with or lower than those reported in trials of ixekizumab for PsA and psoriasis 30.
Injection site reactions (high‐level terms) were higher in patients treated with ixekizumab relative to placebo, and similar to or lower than previously observed in trials of ixekizumab for PsA and psoriasis 33, 34. One case of grade 4 neutropenia was reported in the IXEQ4W treatment arm in a patient with treatment‐emergent acute PML who had a genetic risk factor for this disease. In the IXEQ2W treatment arm, one patient with a documented prior history of depression committed suicide, which was judged by the blinded investigator to be unrelated to the investigational product.
The incidences of IBD and acute anterior uveitis in the current study involving patients with prior inadequate response to or intolerance of 1 or 2 TNFi, and longstanding (mean ± SD symptom duration 18.4 ± 11.1 years) and very active disease, were higher than reported among bDMARD‐naive patients with radiographic axial SpA treated with ixekizumab 30. Certain comorbidities in axial SpA, especially IBD and acute anterior uveitis, have been reported to be associated with longer disease duration 35. Integrated data from the ixekizumab axial SpA studies, as well as longer‐term data, are needed to better understand whether the IBD events observed in the current study reflect the recruited patient population, study treatment, other factors (such as discontinuation of prior TNFi), or a combination of the above 36.
The incidence of ixekizumab antidrug antibodies was low and consistent with previous reports in patients with PsA and prior inadequate response to or intolerance of 1 or 2 TNFi (34). Ixekizumab antidrug antibodies were not associated with immune reactions or reductions in efficacy.
TNFi and 1 IL‐17A antagonist (secukinumab) are the only approved biologic agents for treatment of AS. The ixekizumab results reported here for patients with inadequate response to or intolerance of 1 or 2 TNFi are consistent with those previously reported for secukinumab in patients with inadequate response to or intolerance of 1 TNFi (subset of 85 patients from the MEASURE‐2 study) 18. However, given that other baseline characteristics may also differ between these 2 studies, direct comparisons should not be made between the trial results. Both MEASURE‐2 and COAST‐W demonstrate that IL‐17A antagonists are efficacious and well‐tolerated in patients with prior intolerance of or inadequate response to TNFi.
The COAST‐W trial has several strengths. It is the first large, randomized, controlled trial to focus exclusively on patients with radiographic axial SpA with a prior inadequate response to or intolerance of 1 or 2 TNFi. This focus allowed for a comprehensive evaluation of the baseline characteristics, burden of disease, and treatment efficacy and safety in this population. COAST‐W also used the ASAS40 response as the primary end point, reflecting major improvement and representing a more stringent end point than the commonly used ASAS20. In addition, COAST‐W is the first placebo‐controlled trial to generate spinal MRI data in a TNFi‐intolerant or inadequate responder population. Conversely, the current data set is limited to a 16‐week treatment period. Longer‐term data, which are being collected through 1 year of treatment in the present trial, as well as during an optional 2‐year extension trial, will provide further information on the long‐term efficacy and safety of ixekizumab.
In conclusion, the findings of this study demonstrate that the ixekizumab treatment regimens yield rapid and significant improvements in the signs and symptoms of radiographic axial SpA, as well as a significant reduction in inflammation of the spine as measured by MRI, when compared to placebo. The current results support ixekizumab as a treatment option for patients with radiographic axial SpA and prior inadequate response to or intolerance of TNFi.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be submitted for publication. Dr. Deodhar had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Deodhar, Zhao, Carlier.
Acquisition of data
Deodhar, Poddubnyy, Pacheco‐Tena, Salvarani, Lespessailles, Rahman, Järvinen, Sanchez‐Burson, Gaffney, Lee, Santisteban, Li, Zhao, Carlier, Reveille.
Analysis and interpretation of data
Deodhar, Poddubnyy, Krishnan, Santisteban, Li, Zhao, Carlier.
Role of the Study Sponsor
An academic advisory committee was involved in the study design and data interpretation, together with authors from Eli Lilly and Company. Authors had full access to all the data in the study and had final responsibility for the decision to submit for publication. Eli Lilly and Company contributed to study design, data collection, data analysis, data interpretation, manuscript preparation, and publication decisions. Dr. Krishnan was responsible for the overall conduct of the study. Medical writing support was provided by Brian S. Comer, PhD. Editorial support was provided by P. Vidhyasagar Arya, PhD. Statistical analysis and programming support was provided by Emily Seem, MS, and Lingling Xie, MS. All are employees of Eli Lilly and Company. Publication of this article was not contingent upon approval by Eli Lilly and Company.
Supporting information
Appendix A. Members of the Coast‐W Study Group
Members of the COAST‐W Study Group are as follows: Christopher Antolini, Valderilio Azevedo, Magnus Barkham, Aaron Alejandro Barrera Rodriguez, Alberto Berman, Tomasz Blicharski, Jan Brzezicki, Gerd Burmester, Judith Carrio, Eduardo Collantes, Bernard Combe, Fidencio Cons‐Molina, Gregorio Cortes‐Maisonet, Anna Dudek, Sergio Duran Barragan, Ori Elkayam, Kathleen Flint, Mauro Galeazzi, Norman Gaylis, David Goddard, Carlos Gonzalez Fernandez, Philippe Goupille, Jordi Gratacos Masmitja, Maria Greenwald, Elisa Gremese, Seung Jae Hong, Mary Howell, Pawel Hrycaj, Akgun Ince, Ji Hyeon Ju, Jeffrey Kaine, Seong Wook Kang, Mauro Keiserman, Tae‐Hwan Kim, Alan Kivitz, Steven Klein, Joel Kremer, Chang Keun Lee, Sang Heon Lee, Sang‐Hoon Lee, Roger Lidman, James Loveless, Eleonora Lucero, Jose Maldonado Cocco, Flora Marcolino, Xavier Mariette, Daksha Mehta, Frederic Morin, Yolanda Moscovici, Eric Mueller, Eduardo Mysler, Francisco Navarro Blasco, Minh Nguyen, Carlos Pantojas, Min‐Chan Park, Amarilis Perez‐De Jesus, Eric Peters, Rafal Plebanski, Roel Querubin, Cesar Ramos Remus, Tatiana Reitblat, Tania Rivera, Juan Cruz Rizo Rodriguez, Michael Sayers, Antonio Scotton, Craig Scoville, David Shaw, Kichul Shin, Atul Singhal, Cassandra Skinner, Oscar Soto‐Raices, Martin Soubrier, Malgorzata Szymanska, Christine Thai, Marleen van de Sande, Alvin Wells, Rafal Wojciechowski, Ricardo Xavier, Antonio Ximenes, and Devy Zisman.
ClinicalTrials.gov identifier: NCT02696798.
Supported by Eli Lilly and Company.
Contributor Information
the COAST‐W Study Group:
Christopher Antolini, Valderilio Azevedo, Magnus Barkham, Aaron Alejandro Barrera Rodriguez, Alberto Berman, Tomasz Blicharski, Jan Brzezicki, Gerd Burmester, Judith Carrio, Eduardo Collantes, Bernard Combe, Fidencio Cons‐Molina, Gregorio Cortes‐Maisonet, Anna Dudek, Sergio Duran Barragan, Ori Elkayam, Kathleen Flint, Mauro Galeazzi, Norman Gaylis, David Goddard, Carlos Gonzalez Fernandez, Philippe Goupille, Jordi Gratacos Masmitja, Maria Greenwald, Elisa Gremese, Seung Jae Hong, Mary Howell, Pawel Hrycaj, Akgun Ince, Ji Hyeon Ju, Jeffrey Kaine, Seong Wook Kang, Mauro Keiserman, Tae‐Hwan Kim, Alan Kivitz, Steven Klein, Joel Kremer, Sang Heon Lee, Chang Keun Lee, Sang‐Hoon Lee, Roger Lidman, James Loveless, Eleonora Lucero, Jose Maldonado Cocco, Flora Marcolino, Xavier Mariette, Daksha Mehta, Frederic Morin, Yolanda Moscovici, Eric Mueller, Eduardo Mysler, Francisco Navarro Blasco, Minh Nguyen, Carlos Pantojas, Min‐Chan Park, Amarilis Perez‐De Jesus, Eric Peters, Rafal Plebanski, Roel Querubin, Cesar Ramos Remus, Tatiana Reitblat, Tania Rivera, Juan Cruz Rizo Rodriguez, Michael Sayers, Antonio Scotton, Craig Scoville, David Shaw, Kichul Shin, Atul Singhal, Cassandra Skinner, Oscar Soto‐Raices, Martin Soubrier, Malgorzata Szymanska, Christine Thai, Marleen van de Sande, Alvin Wells, Rafal Wojciechowski, Ricardo Xavier, Antonio Ximenes, and Devy Zisman
References
- 1. Reveille JD, Witter JP, Weisman MH. Prevalence of axial spondylarthritis in the United States: estimates from a cross‐sectional survey. Arthritis Care Res (Hoboken) 2012;64:905–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sieper J, Poddubnyy D. Axial spondyloarthritis. Lancet 2017;390:73–84. [DOI] [PubMed] [Google Scholar]
- 3. Braun J, Sieper J. Ankylosing spondylitis. Lancet 2007;369:1379–90. [DOI] [PubMed] [Google Scholar]
- 4. Van der Heijde D, Ramiro S, Landewe R, Baraliakos X, van den Bosch F, Sepriano A, et al. 2016 update of the ASAS‐EULAR management recommendations for axial spondyloarthritis. Ann Rheum Dis 2017;76:978–91. [DOI] [PubMed] [Google Scholar]
- 5. Ward MM, Deodhar A, Akl EA, Lui A, Ermann J, Gensler LS, et al. American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network 2015 recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheumatol 2016;68:282–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. National Institute for Health and Care Excellence . TA383: TNF‐α inhibitors for ankylosing spondylitis and non‐radiographic axial spondyloarthritis. 2016. URL: https://www.nice.org.uk/guidance/ta383.
- 7. Van der Heijde D, Kivitz A, Schiff MH, Sieper J, Dijkmans BA, Braun J, et al. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: results of a multicenter, randomized, double‐blind, placebo‐controlled trial. Arthritis Rheum 2006;54:2136–46. [DOI] [PubMed] [Google Scholar]
- 8. Inman RD, Davis JC Jr, van der Heijde D, Diekman L, Sieper J, Kim SI, et al. Efficacy and safety of golimumab in patients with ankylosing spondylitis: results of a randomized, double‐blind, placebo‐controlled, phase III trial. Arthritis Rheum 2008;58:3402–12. [DOI] [PubMed] [Google Scholar]
- 9. Van den Bosch F, Deodhar A. Treatment of spondyloarthritis beyond TNF‐α blockade. Best Pract Res Clin Rheumatol 2014;28:819–27. [DOI] [PubMed] [Google Scholar]
- 10. Deodhar A, Reveille JD, Harrison DD, Kim L, Lo KH, Leu JH, et al. Safety and efficacy of golimumab administered intravenously in adults with ankylosing spondylitis: results through week 28 of the GO‐ALIVE study. J Rheumatol 2018;45:341–8. [DOI] [PubMed] [Google Scholar]
- 11. Heiberg MS, Koldingsnes W, Mikkelsen K, Rodevand E, Kaufmann C, Mowinckel P, et al. The comparative one‐year performance of anti–tumor necrosis factor α drugs in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: results from a longitudinal, observational, multicenter study. Arthritis Rheum 2008;59:234–40. [DOI] [PubMed] [Google Scholar]
- 12. Glintborg B, Ostergaard M, Krogh NS, Dreyer L, Kristensen HL, Hetland ML. Predictors of treatment response and drug continuation in 842 patients with ankylosing spondylitis treated with anti‐tumour necrosis factor: results from 8 years’ surveillance in the Danish nationwide DANBIO registry. Ann Rheum Dis 2010;69:2002–8. [DOI] [PubMed] [Google Scholar]
- 13. D'Angelo S, Carriero A, Gilio M, Ursini F, Leccese P, Palazzi C. Safety of treatment options for spondyloarthritis: a narrative review. Expert Opin Drug Saf 2018;17:475–86. [DOI] [PubMed] [Google Scholar]
- 14. Kenna TJ, Davidson SI, Duan R, Bradbury LA, McFarlane J, Smith M, et al. Enrichment of circulating interleukin‐17–secreting interleukin‐23 receptor–positive γ/δ T cells in patients with active ankylosing spondylitis. Arthritis Rheum 2012;64:1420–9. [DOI] [PubMed] [Google Scholar]
- 15. Shen H, Goodall JC, Hill Gaston JS. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum 2009;60:1647–56. [DOI] [PubMed] [Google Scholar]
- 16. Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, et al. Secukinumab, an interleukin‐17A inhibitor, in ankylosing spondylitis. N Engl J Med 2015;373:2534–48. [DOI] [PubMed] [Google Scholar]
- 17. Pavelka K, Kivitz A, Dokoupilova E, Blanco R, Maradiaga M, Tahir H, et al. Efficacy, safety, and tolerability of secukinumab in patients with active ankylosing spondylitis: a randomized, double‐blind phase 3 study, MEASURE 3. Arthritis Res Ther 2017;19:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sieper J, Deodhar A, Marzo‐Ortega H, Aelion JA, Blanco R, Jui‐Cheng T, et al. Secukinumab efficacy in anti‐TNF‐naive and anti‐TNF‐experienced subjects with active ankylosing spondylitis: results from the MEASURE 2 Study. Ann Rheum Dis 2017;76:571–92. [DOI] [PubMed] [Google Scholar]
- 19. Liu L, Lu J, Allan BW, Tang Y, Tetreault J, Chow CK, et al. Generation and characterization of ixekizumab, a humanized monoclonal antibody that neutralizes interleukin‐17A. J Inflamm Res 2016;9:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rudwaleit M, van der Heijde D, Landewe R, Listing J, Akkoc N, Brandt J, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Ann Rheum Dis 2009;68:777–83. [DOI] [PubMed] [Google Scholar]
- 21. Van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis: a proposal for modification of the New York criteria. Arthritis Rheum 1984;27:361–8. [DOI] [PubMed] [Google Scholar]
- 22. Sieper J, Rudwaleit M, Baraliakos X, Brandt J, Braun J, Burgos‐Vargas R, et al. The Assessment of SpondyloArthritis international Society (ASAS) handbook: a guide to assess spondyloarthritis. Ann Rheum Dis 2009;68 Suppl 2:ii1–44. [DOI] [PubMed] [Google Scholar]
- 23. Garrett S, Jenkinson T, Kennedy LG, Whitelock H, Gaisford P, Calin A. A new approach to defining disease status in ankylosing spondylitis: the Bath Ankylosing Spondylitis Disease Activity Index. J Rheumatol 1994;21:2286–91. [PubMed] [Google Scholar]
- 24. Machado PM, Landewe RB, van der Heijde DM. Endorsement of definitions of disease activity states and improvement scores for the Ankylosing Spondylitis Disease Activity Score: results from OMERACT 10. J Rheumatol 2011;38:1502–6. [DOI] [PubMed] [Google Scholar]
- 25. Calin A, Garrett S, Whitelock H, Kennedy LG, O'Hea J, Mallorie P, et al. A new approach to defining functional ability in ankylosing spondylitis: the development of the Bath Ankylosing Spondylitis Functional Index. J Rheumatol 1994;21:2281–5. [PubMed] [Google Scholar]
- 26. Ware JE Jr, Snow KK, Kosinski M, Gandek B. SF‐36 health survey: manual and interpretation guide. 2nd ed Lincoln (RI): QualityMetric; 2000. [Google Scholar]
- 27. Kiltz U, van der Heijde D, Boonen A, Braun J. The ASAS Health Index (ASAS HI): a new tool to assess the health status of patients with spondyloarthritis. Clin Exp Rheumatol 2014;32 Suppl 85:S105–8. [PubMed] [Google Scholar]
- 28. Kiltz U, van der Heijde D, Boonen A, Cieza A, Stucki G, Khan MA, et al. Development of a health index in patients with ankylosing spondylitis (ASAS HI): final result of a global initiative based on the ICF guided by ASAS. Ann Rheum Dis 2015;74:830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maksymowych WP, Inman RD, Salonen D, Dhillon SS, Krishnananthan R, Stone M, et al. Spondyloarthritis Research Consortium of Canada magnetic resonance imaging index for assessment of spinal inflammation in ankylosing spondylitis. Arthritis Rheum 2005;53:502–9. [DOI] [PubMed] [Google Scholar]
- 30. Van der Heijde D, Wei J, Dougados M, Mease PJ, Deodhar A, Maksymowych W, et al. Ixekizumab significantly improves signs, symptoms, and spinal inflammation of active ankylosing spondylitis/radiographic axial spondyloarthritis: 16‐week results of a phase 3 randomized, active and placebo‐controlled trial [abstract]. Arthritis Rheumatol 2018;70 Suppl 10 URL: https://acrabstracts.org/abstract/ixekizumab-significantly-improves-signs-symptoms-and-spinal-inflammation-of-active-ankylosing-spondylitis-radiographic-axial-spondyloarthritis-16-week-results-of-a-phase-3-randomized-active-a/. [Google Scholar]
- 31. Gordon KB, Blauvelt A, Papp KA, Langley RG, Luger T, Ohtsuki M, et al. Phase 3 trials of ixekizumab in moderate‐to‐severe plaque psoriasis. N Engl J Med 2016;375:345–56. [DOI] [PubMed] [Google Scholar]
- 32. Griffiths CE, Reich K, Lebwohl M, van de Kerkhof P, Paul C, Menter A, et al. Comparison of ixekizumab with etanercept or placebo in moderate‐to‐severe psoriasis (UNCOVER‐2 and UNCOVER‐3): results from two phase 3 randomised trials. Lancet 2015;386:541–51. [DOI] [PubMed] [Google Scholar]
- 33. Mease PJ, van der Heijde D, Ritchlin CT, Okada M, Cuchacovich RS, Shuler CL, et al. Ixekizumab, an interleukin‐17A specific monoclonal antibody, for the treatment of biologic‐naive patients with active psoriatic arthritis: results from the 24‐week randomised, double‐blind, placebo‐controlled and active (adalimumab)‐controlled period of the phase III trial SPIRIT‐P1. Ann Rheum Dis 2017;76:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nash P, Kirkham B, Okada M, Rahman P, Combe B, Burmester GR, et al. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24‐week randomised, double‐blind, placebo‐controlled period of the SPIRIT‐P2 phase 3 trial. Lancet 2017;389:2317–27. [DOI] [PubMed] [Google Scholar]
- 35. Varkas G, Vastesaeger N, Cypers H, Colman R, Renson T, van Praet L, et al. Association of inflammatory bowel disease and acute anterior uveitis, but not psoriasis, with disease duration in axial spondyloarthritis: results from two Belgian nationwide axial spondyloarthritis cohorts. Arthritis Rheumatol 2018;70:1588–96. [DOI] [PubMed] [Google Scholar]
- 36. Kennedy NA, Warner B, Johnston EL, Flanders L, Hendy P, Ding NS, et al. Relapse after withdrawal from anti‐TNF therapy for inflammatory bowel disease: an observational study, plus systematic review and meta‐analysis. Aliment Pharmacol Ther 2016;43:910–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials