

Abstract
Objective
Plasminogen activator inhibitor‐1 (PAI‐1) is the key endogenous inhibitor of fibrinolysis, and enhances clot formation after injury. In traumatic brain injury, dysregulation of fibrinolysis may lead to sustained microthrombosis and accelerated lesion expansion. In the present study, we hypothesized that PAI‐1 mediates post‐traumatic malfunction of coagulation, with inhibition or genetic depletion of PAI‐1 attenuating clot formation and lesion expansion after brain trauma.
Methods
We evaluated PAI‐1 as a possible new target in a mouse controlled cortical impact (CCI) model of traumatic brain injury. We performed the pharmacological inhibition of PAI‐1 with PAI‐039 and stimulation by tranexamic acid, and we confirmed our results in PAI‐1–deficient animals.
Results
PAI‐1 mRNA was time‐dependently upregulated, with a 305‐fold peak 12 hours after CCI, which effectively counteracted the 2‐ to 3‐fold increase in cerebral tissue‐type/urokinase plasminogen activator expression. PAI‐039 reduced brain lesion volume by 26% at 24 hours and 43% at 5 days after insult. This treatment also attenuated neuronal apoptosis and improved neurofunctional outcome. Moreover, intravital microscopy demonstrated reduced post‐traumatic thrombus formation in the pericontusional cortical microvasculature. In PAI‐1–deficient mice, the therapeutic effect of PAI‐039 was absent. These mice also displayed 13% reduced brain damage compared with wild type. In contrast, inhibition of fibrinolysis with tranexamic acid increased lesion volume by 25% compared with vehicle.
Interpretation
This study identifies impaired fibrinolysis as a critical process in post‐traumatic secondary brain damage and suggests that PAI‐1 may be a central endogenous inhibitor of the fibrinolytic pathway, promoting a procoagulatory state and clot formation in the cerebral microvasculature. Ann Neurol 2019;85:667–680
Traumatic brain injury (TBI) is a serious health care burden, representing the most common cause of trauma‐related death and disability in industrialized countries, with an annual incidence of 295/100,000 people.1 Mechanical force to the brain not only causes tissue damage, but is also a trigger for delayed neuronal cell loss.2, 3 Multiple mechanisms have been investigated to limit secondary brain damage; however, little is known regarding the role of clot formation and resolution.
The formation of microclots and vascular occlusion is a frequent event after brain injury.4 In vivo studies have demonstrated pericontusional microclot formation as early as 1 hour after insult.5, 6 To prevent the overshooting of clot formation, proper fibrinolysis by plasminogen activators is required. Recent data obtained from urokinase plasminogen activator (uPA)‐deficient mice have shown an impairment of thrombolysis, with enhanced post‐traumatic motor dysfunction and tissue damage.7 In addition, tissue‐type plasminogen activator (tPA) knockout mice show impaired long‐term recovery of white matter and neurological function after experimental TBI,8 whereas the activation of tissue‐type plasminogen activator (tPA) and uPA causes intracerebral hemorrhage after experimental brain trauma.9
The key regulator of plasmin‐mediated fibrinolysis is plasminogen activator inhibitor‐1 (PAI‐1), which is a member of the serine protease inhibitor (serpin) superfamily. PAI‐1 facilitates local homeostasis by maintaining a tight balance between clot formation and clot lysis. The mechanical destruction of brain parenchyma causes vascular rupture, the release and presentation of tissue factor, and the initiation of the clot formation process.10 At the same time, tPA is released from the injured endothelium11 to limit the overshooting of clot formation and to prevent vascular occlusion. The binding partners of PAI‐1 are the active centers of tPA and uPA,12 which form a reversible Michaelis complex. Upon binding, structural changes and the irreversible inactivation of PAI‐1 occur by folding the reactive center loop into the β‐sheet A.13 PAI‐1 has been found to be present in plasma and various tissues and cell types, including the vascular endothelium, liver, adipose tissue, neutrophils, astrocytes, and platelets.14, 15, 16, 17 PAI‐1 is released upon inflammatory stimuli via interleukin (IL)‐118 as well as tumor necrosis factor (TNF)‐α.19
The clinical importance of PAI‐1 has been highlighted in pathologies associated with ischemia, inflammation, and vascular dysfunction.20, 21 High PAI‐1 plasma levels are both a risk factor22 and a negative predictor of survival23 after ischemic stroke. In a previous study, we recently showed that genetic PAI‐1 deficiency reduces brain damage after experimental ischemic stroke.21 Increased levels of PAI‐1 are also present in patients after head injury,24 where it reaches 80% of PAI‐1 plasma activity in the cerebrospinal fluid and is associated with poor outcome.25 The tPA:PAI‐1 complex enhances post‐traumatic damage due to matrix metalloproteinase‐3–dependent proteolytic degradation of the neurovascular unit.26 However, PAI‐1 is also considered to mediate neuroprotective effects in brain tissue through the activation of the MAPK/ERK (mitogen‐activated protein kinases/extracellular signal‐regulated kinases) pathway.27 Accordingly, the intracerebral application of PAI‐1 has been shown to be protective in a model of neonatal cerebral hypoxia.28
Knowledge regarding the role of fibrinolysis after head injury and specifically how PAI‐1 acts as a modulator of coagulopathy after TBI is scarce. Therefore, the present study was designed to address this issue through a genetic approach and by utilizing pharmacological stimulation and inhibition of PAI‐1 in an experimental murine model of TBI.
Materials and Methods
Animals
Male C57Bl/6 N (Charles River Laboratory, Sulzfeld, Germany), PAI‐1 deficient mice (JAX Mice and Services; Jackson Laboratory, Bar Harbor, ME)29 and corresponding wild‐type C57Bl/6J mice (Charles River Laboratory) were used in this study. The care of animals before and during the experiments was performed in compliance with institutional and national guidelines. The animal ethics committee of the Landesuntersuchungsamt Rheinland‐Pfalz approved all the experiments (protocol number 23177‐07/G10‐1‐024).
Traumatic Brain Injury
TBI was performed as described previously.30 A pneumatic brain trauma was induced on the right parietal cortex on the dura‐covered brain via controlled cortical impact (CCI; 1.5 mm displacement). After the injury, mice recovered in their individual cages for 2 hours in a humidity‐ and temperature‐controlled incubator (IC8000; Draeger, Lübeck, Germany).
Drug Preparation
PAI‐039 (Tiplaxtinin; Axon Medchem, Groningen, the Netherlands) was dissolved in dimethylsulfoxide (DMSO; Sigma‐Aldrich, St Louis, MO) and diluted with normal saline to 0.5% DMSO. The vehicle solution (0.5% DMSO) or PAI‐039 (1 mg/kg) was injected subcutaneously twice at 30 minutes and 6 hours after trauma to ensure a sufficient concentration of PAI‐039.31
Tranexamic acid (Fagron, Barsbüttel, Germany) was freshly dissolved before use. The vehicle solution (0.5% DMSO) or tranexamic acid (0.5 g/kg) was injected subcutaneously 30 minutes after trauma.
Neurological Severity Score
Before and after CCI, an investigator blinded to the experimental groups determined the functional outcome using a 15‐point neurologic severity score ranging from 0 (healthy, successful in all tasks) to 15 (severely impaired) points.32
Histological Evaluation
Brains were carefully removed from isoflurane‐anesthetized animals (Abbott, Wiesbaden, Germany), were immediately frozen in powdered dry ice, and were stored at −20°C. The brain contusion area was determined in 10 μm‐thick sections stained with Cresyl violet as described.33
Gene Expression Analysis
Between histologic slices of 500 μm intervals, tissue samples were collected of the right upper quadrant containing the lesion core and the perilesional tissue, were snap frozen in liquid nitrogen, and were stored at −80°C. RNA isolation (RNeasy Lipid Tissue Mini Kit; Qiagen, Hilden, Germany), DNAse treatment, reverse transcription (QuantiTect Reverse Transcription Kit; Qiagen), and real‐time polymerase chain reaction (PCR) were performed as described.34 Absolute copy numbers of target gene mRNA expression were normalized by the absolute number of housekeeping gene copy numbers (cyclophilin A, PPIA) as described previously.35
Isolation of Brain Endothelial Cells
The isolation of brain endothelial cells (EC) was performed using Dynabeads (Invitrogen, Darmstadt, Germany) as described previously.36 Pericontusional brain tissues (total weight = 0.2–0.3 g) were pooled (2 brains) from naive animals and at 6, 24, and 72 hours after CCI. Before isolation, pellets were resuspended in 1 ml ice‐cold Hank balanced salt solution, and one 100 μl sample was taken for quantitative PCR (qPCR) analysis of the total brain (TB) sample. From the residual 900 μl homogenate, endothelial cells were isolated using PECAM‐1 antibody (ab32457‐100, GR18226‐1; Abcam, Cambridge, UK)‐bound Dynabeads (122.03D, M280, goat, Invitrogen). TB samples and brain endothelial fractions were lysed in mRNA‐lysis buffer and were stored at −80°C before mRNA isolation, cDNA transcription, and qPCR analysis.
Immunostaining of Claudin‐5, CD41, and Iba‐1
Immunostaining was performed in frozen brain sections as described previously using the following primary antibodies37: monoclonal rat anti‐CD41 (LS‐C44479; Biozol, Eching, Germany; dilution 1:100), polyclonal rabbit anti–claudin‐5 (34‐1600, Zymed/Invitrogen, dilution 1:100), and anti–Iba‐1 (1:500; Wako Chemicals, Neuss, Germany). The secondary Alexa Fluor–conjugated antibodies were as follows: Alexa Fluor 594 goat antirat IgG (A11007, Invitrogen, 1:200) and Alexa Fluor 488 donkey antirabbit IgG (A21206, Invitrogen, 1:200). Images of fluorescent immunohistochemical staining were captured using equal filter and acquisition parameters to assure comparable conditions. Claudin‐5 and CD41 images were captured with a confocal laser‐scanning microscope (TCS SP5; Leica, Wetzlar, Germany). The number of Iba‐1 microglia/macrophages were determined within the perilesional brain tissue. Images were acquired by conventional fluorescence (AxioVert200; Carl Zeiss, Oberkochen, Germany) and were processed for quantification using ImageJ software (NIH Image, Bethesda, MD) with the appropriate threshold settings for background subtraction and particle count plugin (Iba‐1, 20–1,000 pixel units).
TUNEL Staining
TdT‐mediated dUTP‐biotin nick end labeling (TUNEL) staining was performed in cryostat sections according to the manufacturer's instructions (in situ cell death detection kit; Roche Molecular Biochemicals, Indianapolis, IN; Cat # 1168480991) in combination with 4′,6‐diamidino‐2‐phenylindole solution (0.5 μg/ml in phosphate‐buffered saline). Images of the ipsilesional dentate gyrus were taken from 1 section per brain at bregma −2.36 mm according to the Mouse Brain Library Atlas using a × 20 objective. The number of TUNEL‐positive nuclei was counted by an investigator blinded to group allocation in the region of interest (ROI), encompassing the upper and lower blades of the granule cell layer and the hilus of the hippocampal dentate gyrus.
Immunoblotting of αII‐Spectrin Fragments
Immunoblotting was performed as described previously38 using the anti–αII‐spectrin antibody (1:500 dilution; Enzo Life Sciences, Farmingdale, NY) and IRDye 800CW goat antimouse antibody (1:15,000 dilution; LI‐COR Biosciences, Lincoln, NE). All results were normalized to glyceraldehyde‐3‐phosphate dehydrogenase expression. The Odyssey system (LI‐COR Biosciences) was used to perform detection and quantification.
PAI‐1 Activity Assay
Intracardiac blood samples were collected from isoflurane‐anesthetized animals. The blood samples were collected in 0.5 ml citrate microtubes (0.106 mol/l trisodium citrate solution; Sarstedt, Nümbrecht, Germany), centrifuged at 1,500 × g for 10 minutes at 4°C, and the plasma was stored at −20°C. PAI‐1 activity assays were performed according to the manufacturer's instructions (Cat # 400055 MPAIKT; Loxo, Dossenheim, Germany).
Intravital Microscopy of Microclots
Intravital microscopy (IVM) and the analysis of microcirculatory parameters were performed as described previously.6 The intravenous injection of the fluorescent plasma marker fluorescein isothiocyanate–dextran (100 μl of a 0.5% solution, Sigma‐Aldrich) illustrated the microvessels and platelets, which were visualized by intravenous injection of 50 μl of a 0.02% solution of the fluorescent dye rhodamine 6G (Merck, Darmstadt, Germany) before each measurement. IVM images of the overall 20 vessels per group were analyzed for the number of microthrombi by frame‐by‐frame analysis at a total magnification of ×625 with a computer‐assisted microcirculation analysis system (Capimage; Dr Heinrich Zeintl Ingenieurbüro, Heidelberg, Germany). The occurrence of microthrombi in the ROI was categorized as none, moving, and fixed at 15 minutes pre‐CCI, 15 minutes post‐CCI, and 120 minutes post‐CCI. In addition, cerebral blood flow velocity in the perilesional cortical brain vasculature was quantified by categories (0 = no or reversed flow, 1 = slow flow, 2 = reduced flow, 3 = normal flow).6
Statistics
All experiments were randomized and performed by blinded investigators (computer‐based randomization). To determine the required sample size, an a priori power analysis using G*Power was performed using lesion volume data from previously published studies.39 This analysis was performed to determine an effect size of 0.7, standard statistical power (1 − β) of 0.95, and a significance level (α) of 0.05. Prism 8 statistical software (GraphPad, La Jolla, CA) was used to perform statistical analysis. Prior to analysis, we checked the test assumptions. Due to the limited power in small samples, we did not perform formal goodness‐of‐fit tests prior to the t test or analysis of variance (ANOVA), but instead relied on the graphical assessment of distribution characteristics.40 Normality was checked by inspecting the unimodality and symmetry of histograms, as well as by Q‐Q plots. The equality of variances was checked by inspecting histograms and standard deviations. For comparison of multiple independent groups, 1‐way ANOVA with post hoc Holm–Šidák comparisons test (comparisons between all groups) or Dunnett multiple comparisons test (comparisons to group of naive animals only) was employed. To evaluate group differences in repeated‐measurements from the same animals (neurological function, clot formation, cerebral perfusion), repeated measures 2‐way ANOVA was applied (factors: treatment and time), followed by Šidák multiple comparisons test. Comparisons between 2 independent groups were carried out by the Welch t test. Supplementary Table 2 shows details on the applied tests for each set of experiments. Values of p < 0.05 were considered significant. Data are presented as the mean ± standard deviation.
Results
TBI‐Induced Expression of the Plasminogen Activators tPA and uPA Is Followed by the Induction of PAI‐1 Expression in Brain EC and Increased Plasma Activity
The mRNA expressions of tPA, uPA, and their endogenous blocker PAI‐1 were determined in naive animals and pericontusional tissue at 15 minutes and 3, 6, 12, and 24 hours postinjury (n = 6 each; 12 and 24 hours, n = 5 each; Fig 1A–C). Our results demonstrated an early increase of tPA (2‐fold) and uPA (2.6‐fold) expression within 3 hours after insult compared with naive animals (p < 0.0001 each). PAI‐1 mRNA expression increased about 22‐fold at 3 hours after injury, reached a 305‐fold maximum 12 hours after CCI (vs naive, p < 0.0001; see Fig 1C), and remained elevated until 24 hours postinsult (202.5‐fold, p = 0.01). To determine brain endothelial PAI‐1 mRNA expression, EC were purified from pericontusional tissue and were compared with TB lysates of the same animals (6, 24, and 72 hours post‐CCI; n = 2 naive and n = 4 pooled samples per group, pooled from 2 animals each). PAI‐1 levels increased 2‐fold in the TB tissue fraction from 6 hours to 24 hours postinjury. Seventy‐two hours after injury, PAI‐1 expression dropped to the expression levels observed at 6 hours after TBI. In brain EC, PAI‐1 mRNA expression levels were higher compared with those in TB lysates (see Fig 1D). At 6 hours after trauma, endothelial PAI‐1 mRNA expression was 3‐fold higher compared with TB lysates (p = 0.02 vs TB). At 24 hours after TBI, PAI‐1 mRNA expression levels in brain EC were similar to those in TB. However, 72 hours after TBI, PAI‐1 mRNA expression in endothelial brain cells increased again compared with TB samples (2.5‐fold, p = 0.02 vs TB). In summary, our data demonstrated an upregulation of PAI‐1 mRNA expression in response to TBI following the early post‐traumatic peak expression of the plasminogen activators at 3 hours after TBI. PAI‐1 expression peaked at 12 hours in pericontusional tissue and remained elevated up to 72 hours after TBI in EC, indicating a long‐lasting shift to an antifibrinolytic state on the vascular level.
Figure 1.
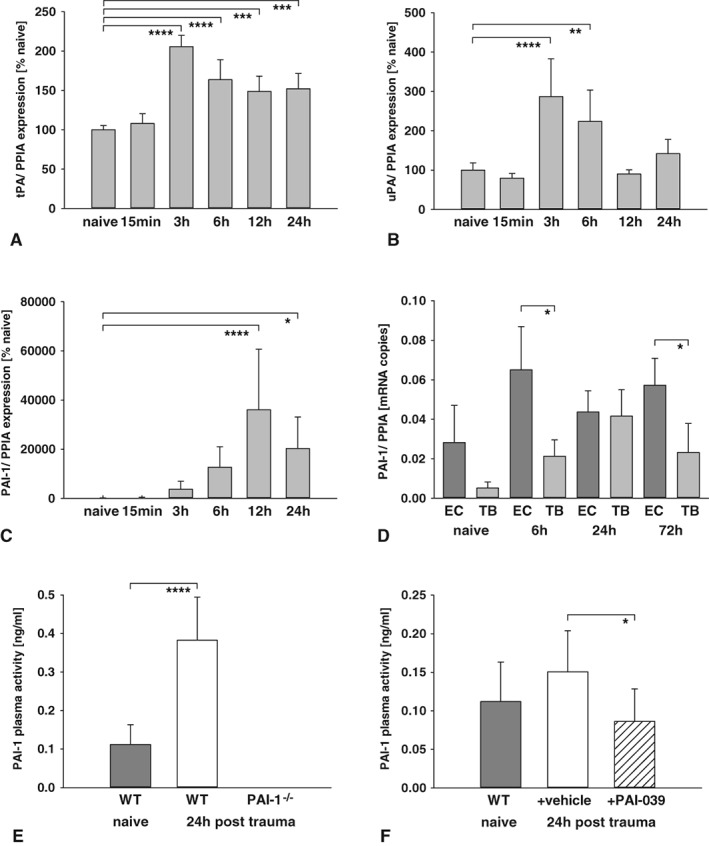
(A–C) Expressions of tissue‐type plasminogen activator (tPA), urokinase plasminogen activator (uPA), and plasminogen activator inhibitor‐1 (PAI‐1) were determined in naive and contused brain tissue samples (naive, 15 minutes, 3 hours, 6 hours, n = 6; contused, 12 hours, 24 hours, n = 5) and showed a moderate increase of tPA (2‐fold; A) and uPA (2.6‐fold; B) with a maximum at 3 hours postinjury. PAI‐1 was strongly upregulation with a 305‐fold peak at 12 hours postinsult (C). One‐way analysis of variance (ANOVA) was used with post hoc Dunnett multiple comparisons test. (D) Cerebrovascular PAI‐1 expression was determined in endothelial cells (EC) and total brain lysates (TB) from naive animals (n = 2 samples, each pooled from 2 animals) and 6, 24, and 72 hours postinjury (n = 4 samples per group, each pooled from 2 animals). PAI‐1 expression was 3‐fold at 6 hours and 2.5‐fold afterward in EC compared to TB. Welch t test was used. (E) Post‐traumatic PAI‐1 plasma activity increased in WT at 24 hours after injury, but was not detectible in PAI‐1–deficient mice (n = 9 each; naive, n = 7). One‐way ANOVA was used with post hoc Holm–Šidák comparisons test. (F) PAI‐039 reduced serum PAI‐1 activity compared to vehicle treatment (n = 9 each; naive, n = 7). Welch t test was used. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. PAI‐1−/− = PAI‐1 knockout animals; PPIA = cyclophilin A; WT = wild‐type animals. Data are presented as mean ± standard deviation.
Next, post‐traumatic changes in the vascular system were determined and showed increased plasma PAI‐1 protein activity levels 24 hours after brain injury (p < 0.0001, n = 9; see Fig 1E) compared with naive animals (n = 7). As expected, PAI‐1 activity was undetectable in PAI‐1–deficient mice (n = 9). PAI‐039 administration also inhibited the post‐traumatic activation of PAI‐1 (p = 0.01, n = 9 each; see Fig 1F).
mRNA expression analysis showed increased PAI‐1 levels in pericontusional tissue samples and in brain EC, along with elevated PAI‐1 plasma activity. These results suggest a shift in the endothelial–vascular milieu toward a procoagulatory state.
PAI‐1 Inhibition Limits Secondary Lesion Expansion of Traumatized Brain Tissue
Our mRNA expression and plasma activity data suggest that TBI‐induced PAI‐1 may impair fibrinolysis, thereby aggravating secondary brain lesions. To test this hypothesis, PAI‐1 protein activity was inhibited with the selective small molecule tiplaxtinin (chemical designation PAI‐039; 1 mg/kg, n = 9) 30 minutes and 6 hours after trauma induction. Brain damage was determined by volumetry, and was compared with the primary lesion 15 minutes after trauma (n = 7, separate set of animals), which allowed for the estimation of secondary brain damage. Accordingly, brain lesion volume increased from 14.2 mm3 at 15 minutes to 30.2 mm3 at 24 hours in vehicle‐treated mice (p < 0.0001, n = 8). In contrast, the pharmacological inhibition of PAI‐1 with PAI‐039 reduced the secondary brain lesion to 23.4 mm3 (−42.5% of secondary brain damage) compared with vehicle (p = 0.004; Fig 2A). Then, histological slides were screened for intraparenchymal hemorrhage. Neither the vehicle nor treated animals showed any histological signs of intraparenchymal bleeding. To examine whether the observed effects were linked to PAI‐1, we subjected PAI‐1–deficient mice treated or mice treated not with PAI‐039 (n = 6 each) to TBI. In these experiments, PAI‐039 failed to reduce secondary brain damage and did not show any protective effects (see Fig 2B).
Figure 2.
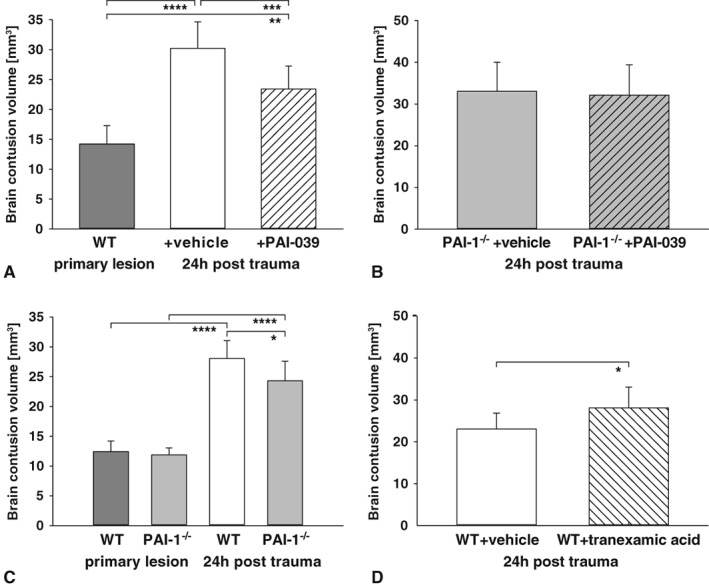
(A) To determine the role of plasminogen activator inhibitor‐1 (PAI‐1), the specific small molecule PAI‐039 (1 mg/kg) was applied to pharmacologically block PAI‐1–mediated impairment of fibrinolysis. Treatment reduced secondary lesion volume at 24 hours postinsult compared to vehicle (PAI‐039, n = 9; vehicle, n = 8; primary lesion, n = 7). One‐way analysis of variance (ANOVA) was used with post hoc Holm–Šidák comparisons test. (B) To determine the specificity, PAI‐039 was administered to PAI‐1–deficient animals. In these mice, no difference was present between PAI‐039 and vehicle (n = 6 each). Welch t test was used. (C) As prove of principle, wild‐type (WT) or PAI‐1 knockout (PAI‐1−/−) animals were randomized to controlled cortical impact. Secondary lesion volume at 24 hours was significantly less in PAI‐1−/− compared to control (PAI‐1−/− and WT, n = 9; primary lesion, n = 7 each). One‐way ANOVA was used with post hoc Holm–Šidák comparisons test. (D) Inhibition of fibrinolysis by the antifibrinolytic drug tranexamic acid enhanced brain damage after trauma. The lesion size increased about 25% compared with vehicle (n = 7 each). Welch t test was used. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are presented as mean ± standard deviation.
To confirm that PAI‐1 aggravated secondary brain lesion following TBI, PAI‐1–deficient and wild‐type mice were subjected to TBI (primary lesion groups, n = 7 each; 24‐hour groups, n = 9 each; see Fig 2C). In wild‐type animals, the brain lesion volume increased 2.3‐fold from 15 minutes (12.4 mm3) to 24 hours (28.1 mm3, p < 0.0001). In contrast, the secondary brain lesion was significantly attenuated in PAI‐1–deficient animals (24.3 mm3, p = 0.02).
To discern whether the neuroprotective effects of genetic and pharmacological inactivation of PAI‐1 is related to the reversal of antifibrinolytic activity, the effect of antifibrinolytic tranexamic acid (which prevents plasmin from binding to and degrading fibrin) was investigated. To this end, 0.5 g/kg tranexamic acid or vehicle solution was administered 30 minutes after experimental TBI (n = 7 each; see Fig 2D). Brain lesion volume was determined at 24 hours after TBI, resulting in a 25% increase of lesion volume in tranexamic acid–treated animals compared with vehicle (p = 0.03). Our data show that antifibrinolytic treatment aggravates secondary brain damage after TBI, suggesting that the inhibition of PAI‐1 facilitates fibrinolysis.
PAI‐1 Inhibition Limits Microvascular Thrombosis in Pericontusional Brain Tissue
To study the formation of platelet aggregates following TBI, brain tissue sections were double‐immunolabeled with platelet integrin alpha‐IIb (CD41) and claudin‐5 as endothelial‐specific markers of the blood–brain barrier. At 24 hours after injury, CD41‐labeled platelet aggregates were found in pericontusional brain tissue (Fig 3). However, the great majority of aggregates were found to be perivascular, whereas intravascular aggregation was rarely observed, likely due to the post‐traumatic disintegration of the vessel structure.41, 42 In support of this notion, CD41‐labeled platelets were not detectable in the noninjured hemisphere contralateral to the lesion site. Although the reduction of CD41‐labeled platelet aggregates was suggestive in PAI‐1–deficient mice (see Fig 3B) and PAI‐039–treated animals (Fig 4B) compared with wild‐type (see Fig 3A) and vehicle‐treated (see Fig 4A) mice, we experienced technical constraints using perfused and fixed brain tissue, which prevented the reliable quantification of intravascular platelet aggregation.
Figure 3.
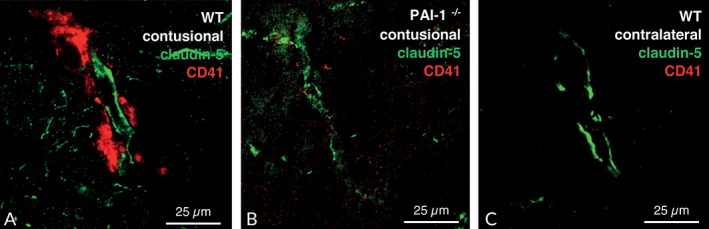
Fluorescence double staining of claudin‐5 (green) as an endothelial cell marker, and CD41 (red) as a platelet marker was performed to visualize accumulation of platelets in cerebral tissue after controlled cortical impact. At 24 hours after injury, the CD41 signal was stronger in the pericontusional brain tissue and colocalized with claudin‐5 (A). Less signal was present in plasminogen activator inhibitor‐1 knockout (PAI‐1−/−) mice (B), and CD41 fluorescence signal was barely detectible in the healthy contralateral hemisphere (C). WT = wild‐type animals.
Figure 4.
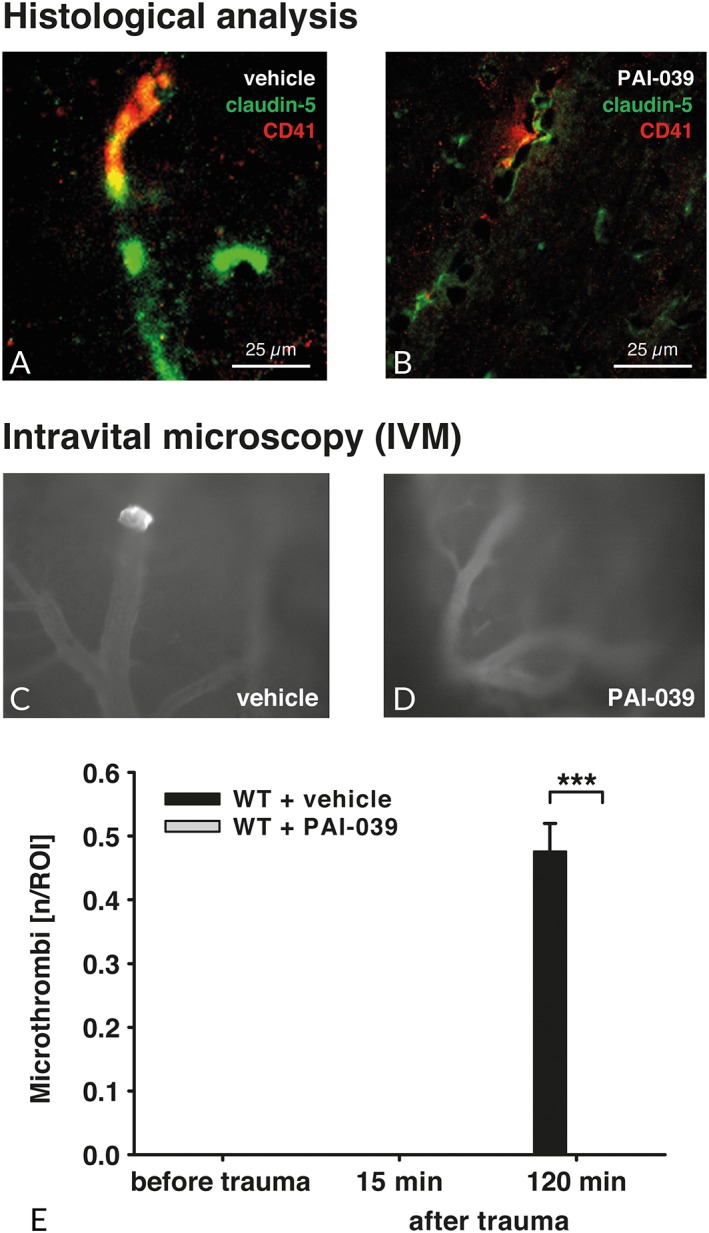
(A, B) To localize the accumulation of platelets in cerebral tissue after controlled cortical impact, fluorescence double staining with claudin‐5 (green) as an endothelial‐specific marker of the blood–brain barrier and CD41 (red) as a platelet marker was performed. At 24 hours after injury, the CD41 signal was strong in the pericontusional brain tissue and colocalized with claudin‐5 (A) in vehicle‐treated animals. Less signal was present in PAI‐039–treated mice (B). (C–E) In a separate set of experiments using intravital microscopy (n = 5 per group), we were able to confirm this finding and showed an increased number of microthrombi or vessel wall–bound clots in brain cortical vessels at 120 minutes postinjury. In contrast with vehicle‐treated animals, PAI‐039 treatment ameliorated clot formation (D). Repeated measures 2‐way analysis of variance was used, followed by Šidák multiple comparisons test. ***p < 0.001. ROI = region of interest; WT = wild‐type animals. Data are presented as mean ± standard deviation.
Because of this issue, we next performed additional experiments, in which mice were subjected to intravital microscopy to directly investigate and quantify platelet aggregation following TBI (n = 5 each; see Fig 4C–E). Because PAI‐039 proved to be a specific PAI‐1 inhibitor of potential therapeutic value, our experiments were focused on PAI‐039–treated and vehicle‐treated animals. Mice were imaged at 15 minutes pre‐TBI, and 15 minutes and 120 minutes after TBI. Whereas stable, on the vessel wall fixed microthrombi occurred in the vehicle group 120 minutes post‐CCI, no clot formation was detectable in PAI‐1–inhibited mice (p < 0.0001; see Fig 4E). To determine whether cerebral perfusion was changed by PAI‐039 treatment, perfusion was investigated in normal perilesional cortical vasculature by using the cerebral perfusion categories (0–3). Cerebral perfusion was not different between the groups at 15 minutes before (vehicle, 3 ± 0; PAI‐039, 3 ± 0), 15 minutes after (vehicle, 2.9 ± 0.4; PAI‐039, 3 ± 0) and 120 minutes after (vehicle, 2.5 ± 1.1; PAI‐039, 2.6 ± 0.5) CCI.
Influence of PAI‐1 Inhibition and Deficiency on Inflammatory and Apoptotic Markers
Because intravital microscopy clearly showed that PAI‐1 inhibition via PAI‐039 prevented clot formation in the microvasculature after TBI, we assumed that restored fibrinolysis and the associated improved perfusion of brain tissue may modulate the inflammatory response and neuronal cell death.
To investigate whether PAI‐039 influences the expression of inflammatory marker genes, the mRNA expression of TNF‐α and IL‐1β was determined at 24 hours in pericontusional brain tissue (Fig 5A, B; PAI‐039, n = 9; vehicle, n = 8). TNF‐α expression increased within 15 minutes after insult. At 24 hours after insult expression, levels reached 20.3‐fold in vehicle‐treated animals. The expression levels in PAI‐039‐treated mice did not change (25.4‐fold increase). In contrast to TNF‐α, IL‐1β was not induced within 15 minutes, but showed significantly 2.4‐fold higher levels at 24 hours after insult in vehicle‐treated animals. In line with TNF‐α levels, IL‐1β expression was not influenced by PAI‐039 (3.3‐fold increase). These results are consistent with those in PAI‐1–deficient mice (see Fig 5C, D; n = 9 each). The expression of the inflammatory marker gene TNF‐α increased over time to 136.3‐fold (wild type) or 148.9‐fold (PAI‐1−/−) at 24 hours postinjury. Similarly, IL‐1β expression increased to 9.5‐fold (wild type) or 10‐fold (PAI‐1−/−) at 24 hours post‐CCI compared with naive animals. Likewise, the expression of inflammatory markers was not different between wild‐type and PAI‐1–deficient mice, indicating an inflammation‐independent modulation of lesion expansion.
Figure 5.
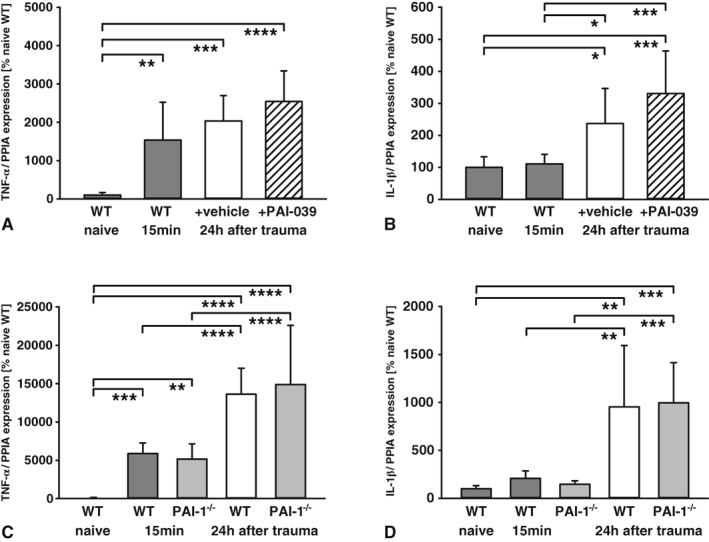
(A, B) The inflammatory marker genes tumor necrosis factor (TNF)‐α (A) and interleukin (IL)‐1β (B) increased over time with no difference between PAI‐039 and the vehicle group in pericontusional brain tissue (naive, n = 6; primary lesion [15 minutes], n = 7; vehicle, n = 8; PAI‐039, n = 9). (C, D) Similar regulation was observed in plasminogen activator inhibitor‐1 (PAI‐1)‐deficient animals for TNF‐α (C) and IL‐1β (D; naive, n = 6; primary lesion [15 minutes], n = 7 per group; wild type [WT] and PAI‐1 knockout [PAI‐1−/−], each n = 9). One‐way analysis of variance was used with post hoc Holm–Šidák comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. PPIA, cyclophilin A. Data are presented as mean ± standard deviation.
To examine neuronal cell death after TBI, the dentate gyrus of the traumatized hemisphere was investigated by TUNEL staining (Fig 6A). Granule cells in this circumscribed anatomical region have been shown to degenerate in response to post‐traumatic hypoxia.43 The counting of TUNEL‐stained cells revealed a substantial decrease of apoptotic cells in PAI‐039–treated mice compared with the vehicle group by 29.8% (PAI‐039, 14 ± 3 cells/ROI; vehicle, 47 ± 8 cells/ROI; p < 0.0001, n = 7 each). The detection of less DNA fragmentation indicates less apoptotic cell death signaling in PAI‐039–treated animals. To determine whether calpain‐dependent cell death is of key importance, we next performed immunoblotting of αII‐spectrin fragments in contused brain tissue (see Fig 6B; PAI‐039, n = 9; vehicle, n = 8). The 150 kDa αII‐spectrin breakdown product (SBDP150) was used as a marker for calpain‐dependent cell death.44 In this analysis, only a trend toward decreased SBDP150 levels was present in PAI‐039–treated animals. These results may suggest that other cell death mechanisms aside from calpain are attenuated by PAI‐1 inhibition.
Figure 6.
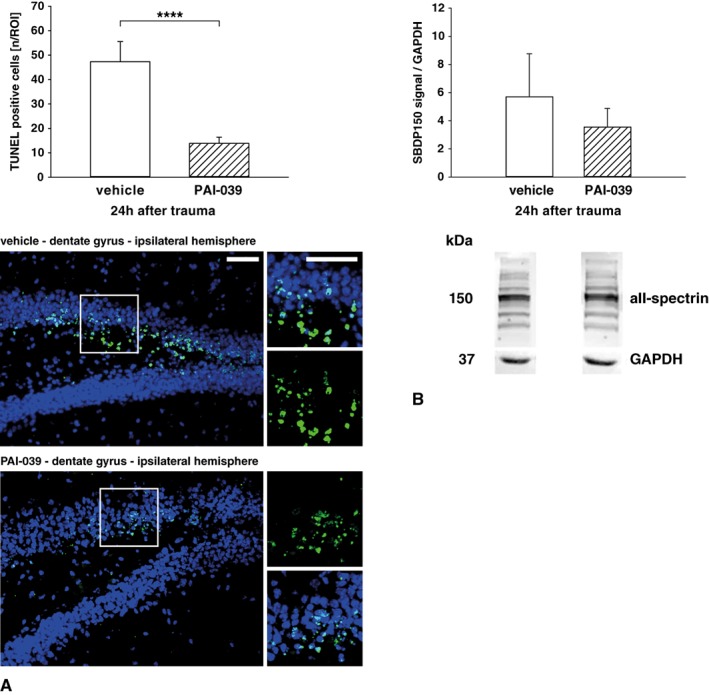
(A) The number of apoptotic cells (green) were visualized in TdT‐mediated dUTP‐biotin nick end labeling (TUNEL)‐stained slices of the ipsilateral dentate gyrus in vehicle or PAI‐039–treated animals. Fewer apoptotic cells were counted in TUNEL‐stained sections of PAI‐039‐treated animals (−29.8% vs vehicle, n = 7 per group scale bar = 75 μm). (B) To determine the role of calpain‐dependent cell death, 150kD αII‐spectrin fragments were quantified in contused brain tissue. The 150 kDa spectrin levels were not different between groups (PAI‐039, n = 9; vehicle, n = 8). Welch t test was used. ****p < 0.0001. GAPDH = glyceraldehyde‐3‐phosphate dehydrogenase; SBDP150 = 150 kDa αII‐spectrin breakdown product; ROI = region of interest. Data are presented as mean ± standard deviation.
PAI‐1 Inhibition Reduces Functional Impairment and Delayed Brain Damage
To determine whether the observed reduction of brain damage at 24 hours postinsult translated into a persistent histological and neurofunctional effect at a delayed time point, we monitored the effects of pharmacological PAI‐1 inhibition over 5 days. At 5 days after trauma, brains were dissected and processed for Nissl staining and lesion volumetry. The brain lesion volume in PAI‐039–treated mice (n = 13) was significantly smaller (−43%, p < 0.0001) compared with vehicle (n = 12; Fig 7). Neurocognitive deficits were tested with a neurofunctional score (0–15 points), and demonstrated a highly significant reduced impairment of PAI‐039 animals on testing days 1, 3, and 5 after injury compared with vehicle treatment. In line with our results indicating early inflammation‐independent effects of PAI‐1 inhibition, we did not observe any pharmacological effects on the mRNA expression of the proinflammatory markers IL‐1β and TNF‐α at 5 days after TBI. Accordingly, the number of perilesional Iba‐1–positive microglia/macrophages remained unchanged between PAI‐039–treated and vehicle‐treated mice. In summary, treatment with PAI‐039 significantly improved functional and histological outcomes after experimental TBI.
Figure 7.
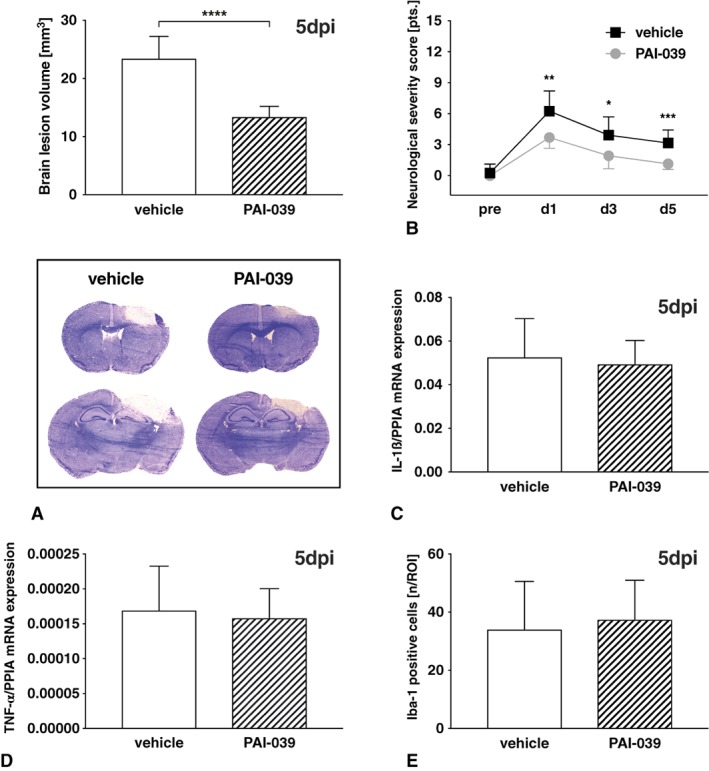
(A) Long‐term effects were determined 5 days after brain injury (5 dpi) in male C57Bl6 mice treated with PAI‐039 (n = 13) or vehicle (n = 12). Lesion volumes were determined in Nissl‐stained sections and were significantly lower in PAI‐039–treated animals. Welch t test was used. (B) At postoperative days 1, 3, and 5, neurofunctional impairment was determined using a neurological severity score and demonstrated a significantly lower degree of impairment in PAI‐039–treated animals. Repeated measures 2‐way analysis of variance was used followed by Šidák multiple comparisons test. (C, D) Markers of cerebral inflammation were determined in these animals by quantitative polymerase chain reaction in lesioned brain tissue and failed to demonstrate an influence on interleukin (IL)‐1β (C) and tumor necrosis factor (TNF)‐α (D) mRNA levels. (E) The number of perilesional ionized calcium‐binding adapter molecule (Iba)‐1–positive microglia/macrophages was also not different between PAI‐039–treated and vehicle‐treated mice. Welch t test was used. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. PPIA = cyclophilin A; ROI = region of interest. Data are presented as mean ± standard deviation. [Color figure can be viewed at www.annalsofneurology.org]
Discussion
The findings of this study demonstrate that impaired fibrinolysis is a critical process in brain injury, with secondary brain damage and functional impairment attenuated by pharmacologic or genetic inhibition of PAI‐1 activity. The reduction of PAI‐1 plasma activity was shown to limit pericontusional cortical microclot formation as demonstrated by intravital microscopy. Our data further support the hypothesis that the inhibition of PAI‐1 reactivates the fibrinolytic pathway, preventing the shift to a procoagulatory state in the early phase after brain trauma and reducing the risk of microthrombosis in the cerebral microvasculature.
Following the mechanical destruction of the brain parenchyma, the rupture of the cerebral vasculature induces a series of pathophysiological events contributing to the progression of the primary lesion into surrounding healthy tissue. This secondary lesion expansion is enhanced further by platelet and leukocyte aggregation, which impairs cerebral blood flow and causes cerebral ischemia. Clot formation has been reported in injured humans as well as in experimental models.6, 45 In addition, platelet aggregation occurs in the early post‐traumatic period in hemorrhagic brain tissue,5 along with the pericontusional region, which leads to impaired cerebral perfusion.6 To limit the overshooting of clot formation and prevent vascular occlusion, tPA is released from the injured endothelium.46 Interestingly, an early peak of tPA/uPA expression was observed; however, this expression is counteracted by a 305‐fold upregulation of the endogenous inhibitor PAI‐1. To confirm the endothelial expression of PAI‐1, brain EC were isolated and analyzed for PAI‐1 expression. These cells were shown to have a high baseline expression of PAI‐1, with a 2‐fold increase from 6 to 72 hours postinjury. These data suggest that EC are a relevant source of PAI‐1 in our TBI model. However, TB lysates also showed a post‐traumatic induction of PAI‐1 expression, albeit on a lower scale. Therefore, brain parenchyma can be considered a putative source of PAI‐1, with astrocytes an important cell type showing increased PAI‐1 levels upon ischemia.47 Regarding the vascular route, both EC and platelets have been identified as key sources of PAI‐1 in pathological conditions and after stimulation with thrombin.14, 46 After acute myocardial infarction, elevated PAI‐1 plasma levels (particularly those observed in platelet‐rich clots) were associated with failing thrombolytic therapy.48 In the present study, PAI‐1 plasma activity was increased 24 hours after injury, supporting the hypothesis that endogenous PAI‐dependent fibrinolysis inhibition may counteract the increased tPA and uPA levels. In support of this notion, PAI‐1–overexpressing mice show increased infarct size in a model of thrombotic occlusion of the middle cerebral artery.49
To clarify the in vivo role of PAI‐1 after TBI, PAI‐1 was inhibited with the selective small molecule PAI‐039 (tiplaxtinin).31 PAI‐039 inhibits both nonglycosylated and glycosylated PAI‐1 isoforms and interacts with free but not vitronectin‐bound PAI‐1.13 The application of PAI‐039 resulted in a significant reduction in active PAI‐1 plasma levels. Moreover, PAI‐039 application reduced brain lesion volume by 43% at 5 days after insult. This protective effect of PAI‐039 was not observed in PAI‐1–deficient animals. These findings provide evidence that the effects of PAI‐039 on brain lesions are mediated via PAI‐1.
The present results support the hypothesis that impaired fibrinolysis aggravates brain damage, and are in line with findings demonstrating accelerated lysis and the improved perfusion of microvessels through the inactivation of PAI‐1 in a model of platelet‐rich clot formation in rat mesenteric arterioles50 and in PAI‐1 deficient mice.51 In addition, several studies of diseases such as stroke or myocardial infarction have demonstrated that an increase in tPA:PAI‐1 complex formation is associated with impaired fibrinolysis.52, 53 In stroke, increased PAI‐1 activity appears to cause a hypercoagulable state and recurrent ischemic stroke.54 Immunohistological staining in contused brain tissue suggested pronounced clot formation in wild‐type and vehicle‐treated mice. Platelets were present in proximity to vessels, but not at all times were they intravascular. We ascribe this finding to the post‐traumatic impairment of blood–brain barrier integrity, which was also observed in rat CCI. In this model, thrombosis was shown to have a peak at 1 to 4 days after injury, and to be intravascular in the lesion boundary zone, as well extravascular in the lesion core zone.6 To provide evidence for the PAI‐1–dependent impairment of fibrinolysis after TBI, we performed intravital microscopy, which showed a strong reduction of microthrombi formation in the pericontusional brain tissue of mice treated with a PAI‐1 inhibitor.
The beneficial effects we observed on brain lesion volume were supported by less neuronal cell death as evaluated by TUNEL staining in the dentate gyrus, a region highly sensitive to hypoxia.43 These results support an effect mediated by improved cerebral perfusion as an underlying mechanism to limit secondary brain in the early phase after brain injury. Recently, Xia et al showed that the lack of tPA in knockout mice impedes long‐term recovery of white matter and neurological function after TBI, which was restored by recombinant tPA through intranasal application without increasing intracerebral hemorrhage volumes.8 These findings suggest that genetic PAI‐1 deficiency or pharmacologic PAI‐1 blockade may also have a direct effect on the structural and functional integrity of white matter, with postinjury compensatory sprouting in corticofugal projections occurring through the reestablished tPA activity.
Interestingly, current trauma guidelines recommend the administration of tranexamic acid in patients with severe multiple injuries to enhance clot formation and strength, and to prevent overshooting fibrinolysis. This recommendation is based primarily on the CRASH‐2 trial (Clinical Randomization of an Anti‐fibrinolytic in trauma patients with Significant Hemorrhage), which showed a reduction in the risk of death from major bleeding by tranexamic acid in multiple trauma patients.55 In our experimental setting, we focused on isolated brain injury, which was not complicated by additional injures or intracerebral hemorrhage. Therefore, our data and results should not be compared to a setting of major bleeding with acquired coagulopathy. Approximately 46% of TBI patients develop intracranial bleeding, mostly within 24 to 48 hours after injury.56 Therefore, in humans, it is not clear whether patients with isolated head injury benefit from the routine administration of tranexamic acid (see Nishida et al for review57). To investigate this question, the CRASH‐3 trail was initiated with a focus on tranexamic acid in TBI patients.58 Presently, the trial is recruiting, but hopefully it will be completed by 2020.
In our experiments, we screened for the macroscopic signs of major intracerebral bleeding and for signs of intraparenchymal bleeding in histological slides. Neither PAI‐1–deficient mice nor animals treated with PAI‐1 inhibitors presented with any signs of bleeding at 24 hours or 5 days after insult. We cannot rule out the presence of microhemorrhage. However, we observed in some animals extravasal platelet signals (CD41) in immunohistochemistry. This may present a histological correlate of microhemorrhage or a histologic artifact from the perfusion fixation step.
Furthermore, the cerebrovascular expression of PAI‐1 was not characterized by Western blotting or immunohistochemistry using negative controls such as tissue from PAI‐1–deficient animals. Therefore, we tested different commercial antibodies but failed to identify a specific PAI‐1 antibody. All the tested antibodies demonstrated strong signals in PAI‐1 knockout mice (data not shown); thus, we are not able to provide valid protein data. A further limitation of the present study is the use of mice to investigate fibrinolysis. The analysis of PAI‐1 blood activity suggested lower post‐traumatic PAI‐1 levels in vehicle‐treated animals. Although post‐traumatic PAI‐1 levels were significantly reduced by PAI‐039 compared with vehicle, we cannot rule out that 0.5% DMSO as a vehicle interfered with the blood levels or measurements, which may have introduced a systematic bias. In addition, species differences between humans and rodents regarding the coagulation system warrant future investigation to determine the role of fibrinolysis in human TBI.
Conclusions
In summary, the present study provides the first evidence for post‐traumatic inhibition of tPA/uPA‐mediated fibrinolysis via the PAI‐1 pathway. In addition, this study provides data for a PAI‐1–dependent procoagulatory state in the early phase after brain injury, contributing in concert with other pathophysiological mechanisms to secondary brain damage. Therefore, it is crucial to maintain a tight balance between coagulation and fibrinolysis to prevent deleterious vascular occlusion and cerebral ischemia, as well as the progression of intracerebral bleeding. In cases without systemic coagulopathy or major bleeding, the pharmacological restoration of fibrinolysis via PAI‐039 may be a promising approach to balancing fibrinolysis and coagulation and to limiting secondary brain damage after TBI.
Data Availability
Datasets generated or analyzed during this study are included in this published article or are available from the corresponding author upon reasonable request.
Author Contributions
S.C.T. and E.‐V.G. contributed to the conception and design of the study. All authors contributed to the acquisition and analysis of data, drafting the text, and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Supplementary Table 1 Experimental groups.
Supplementary Table 2 Overview on applied statistical tests.
Acknowledgment
This work was supported by grants from the Focus Program Translational Neurosciences of Johannes Gutenberg University (S.C.T.) and Advancement Program of the University Medical Center of the Johannes Gutenberg University Mainz (E.‐V.G.).
We thank F. Kornes and T. Hirnet for their excellent technical assistance and Dr D. Wollschläger for statistical advice.
Some of the data in this article are from the doctoral thesis of I.P. and the professorial dissertation (Habilitation) of E.‐V.G., both presented to Johannes Gutenberg University Mainz.
References
- 1. Nguyen R, Fiest KM, McChesney J, et al. The international incidence of traumatic brain injury: a systematic review and meta‐analysis. Can J Neurol Sci 2016;43:774–785. [DOI] [PubMed] [Google Scholar]
- 2. Gennarelli TA. Mechanisms of brain injury. J Emerg Med 1993;11:5–11. [PubMed] [Google Scholar]
- 3. Bullock R, Sakas D, Patterson J, et al. Early post‐traumatic cerebral blood flow mapping: correlation with structural damage after focal injury. Acta Neurochir Suppl (Wien) 1992;55:14–17. [DOI] [PubMed] [Google Scholar]
- 4. Huber A, Dorn A, Witzmann A, Cervos‐Navarro J. Microthrombi formation after severe head trauma. Int J Legal Med 1993;106:152–155. [DOI] [PubMed] [Google Scholar]
- 5. Dietrich WD, Alonso O, Halley M. Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J Neurotrauma 1994;11:289–301. [DOI] [PubMed] [Google Scholar]
- 6. Schwarzmaier SM, Kim SW, Trabold R, Plesnila N. Temporal profile of thrombogenesis in the cerebral microcirculation after traumatic brain injury in mice. J Neurotrauma 2010;27:121–130. [DOI] [PubMed] [Google Scholar]
- 7. Morales D, McIntosh T, Conte V, et al. Impaired fibrinolysis and traumatic brain injury in mice. J Neurotrauma 2006;23:976–984. [DOI] [PubMed] [Google Scholar]
- 8. Xia Y, Pu H, Leak RK, et al. Tissue plasminogen activator promotes white matter integrity and functional recovery in a murine model of traumatic brain injury. Proc Natl Acad Sci U S A 2018;115:E9230–E9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hijazi N, Abu Fanne R, Abramovitch R, et al. Endogenous plasminogen activators mediate progressive intracerebral hemorrhage after traumatic brain injury in mice. Blood 2015;125:2558–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Giesen PL, Nemerson Y. Tissue factor on the loose. Semin Thromb Hemost 2000;26:379–384. [DOI] [PubMed] [Google Scholar]
- 11. Emeis JJ. Regulation of the acute release of tissue‐type plasminogen activator from the endothelium by coagulation activation products. Ann N Y Acad Sci 1992;667:249–258. [DOI] [PubMed] [Google Scholar]
- 12. Lawrence DA, Olson ST, Muhammad S, et al. Partitioning of serpin‐proteinase reactions between stable inhibition and substrate cleavage is regulated by the rate of serpin reactive center loop insertion into beta‐sheet A. J Biol Chem 2000;275:5839–5844. [DOI] [PubMed] [Google Scholar]
- 13. Gorlatova NV, Cale JM, Elokdah H, et al. Mechanism of inactivation of plasminogen activator inhibitor‐1 by a small molecule inhibitor. J Biol Chem 2007;282:9288–9296. [DOI] [PubMed] [Google Scholar]
- 14. Brogren H, Karlsson L, Andersson M, et al. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood 2004;104:3943–3948. [DOI] [PubMed] [Google Scholar]
- 15. Simpson AJ, Booth NA, Moore NR, Bennett B. Distribution of plasminogen activator inhibitor (PAI‐1) in tissues. J Clin Pathol 1991;44:139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hultman K, Blomstrand F, Nilsson M, et al. Expression of plasminogen activator inhibitor‐1 and protease nexin‐1 in human astrocytes: response to injury‐related factors. J Neurosci Res 2010;88:2441–2449. [DOI] [PubMed] [Google Scholar]
- 17. Huang J, Sabater‐Lleal M, Asselbergs FW, et al. Genome‐wide association study for circulating levels of PAI‐1 provides novel insights into its regulation. Blood 2012;120:4873–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emeis JJ, Kooistra T. Interleukin 1 and lipopolysaccharide induce an inhibitor of tissue‐type plasminogen activator in vivo and in cultured endothelial cells. J Exp Med 1986;163:1260–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Hinsbergh VW, Kooistra T, van den Berg EA, et al. Tumor necrosis factor increases the production of plasminogen activator inhibitor in human endothelial cells in vitro and in rats in vivo. Blood 1988;72:1467–1473. [PubMed] [Google Scholar]
- 20. van der Poll T, Levi M, Buller HR, et al. Fibrinolytic response to tumor necrosis factor in healthy subjects. J Exp Med 1991;174:729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Griemert EV, Recarte Pelz K, Engelhard K, et al. PAI‐1 but not PAI‐2 gene deficiency attenuates ischemic brain injury after experimental stroke. Transl Stroke Res 2018. doi: 10.1007/s12975-018-0644-9. PMID: 29978354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tuttolomondo A, Pinto A, Corrao S, et al. Immuno‐inflammatory and thrombotic/fibrinolytic variables associated with acute ischemic stroke diagnosis. Atherosclerosis 2009;203:503–508. [DOI] [PubMed] [Google Scholar]
- 23. Lip GY, Blann AD, Farooqi IS, et al. Sequential alterations in haemorheology, endothelial dysfunction, platelet activation and thrombogenesis in relation to prognosis following acute stroke: the West Birmingham Stroke Project. Blood Coagul Fibrinolysis 2002;13:339–347. [DOI] [PubMed] [Google Scholar]
- 24. Dietzmann K, von Bossanyi P, Krause D, et al. Expression of the plasminogen activator system and the inhibitors PAI‐1 and PAI‐2 in posttraumatic lesions of the CNS and brain injuries following dramatic circulatory arrests: an immunohistochemical study. Pathol Res Pract 2000;196:15–21. [DOI] [PubMed] [Google Scholar]
- 25. Beuth W, Kotschy M, Kasprzak HA, et al. Tissue plasminogen activator (T‐PA) and tissue plasminogen activator inhibitor (PAI‐1) in patients after head injury [in Polish]. Neurol Neurochir Pol 1996;30:427–434. [PubMed] [Google Scholar]
- 26. Sashindranath M, Sales E, Daglas M, et al. The tissue‐type plasminogen activator‐plasminogen activator inhibitor 1 complex promotes neurovascular injury in brain trauma: evidence from mice and humans. Brain 2012;135(pt 11):3251–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Soeda S, Koyanagi S, Kuramoto Y, et al. Anti‐apoptotic roles of plasminogen activator inhibitor‐1 as a neurotrophic factor in the central nervous system. Thromb Haemost 2008;100:1014–1020. [DOI] [PubMed] [Google Scholar]
- 28. Yang D, Nemkul N, Shereen A, et al. Therapeutic administration of plasminogen activator inhibitor‐1 prevents hypoxic‐ischemic brain injury in newborns. J Neurosci 2009;29:8669–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carmeliet P, Kieckens L, Schoonjans L, et al. Plasminogen activator inhibitor‐1 gene‐deficient mice. I. Generation by homologous recombination and characterization. J Clin Invest 1993;92:2746–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thal SC, Wyschkon S, Pieter D, et al. Selection of endogenous control genes for normalization of gene expression analysis after experimental brain trauma in mice. J Neurotrauma 2008;25:785–794. [DOI] [PubMed] [Google Scholar]
- 31. Elokdah H, Abou‐Gharbia M, Hennan JK, et al. Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor‐1: design, synthesis, and preclinical characterization. J Med Chem 2004;47:3491–3494. [DOI] [PubMed] [Google Scholar]
- 32. Schaible EV, Steinstrasser A, Jahn‐Eimermacher A, et al. Single administration of tripeptide alpha‐MSH(11‐13) attenuates brain damage by reduced inflammation and apoptosis after experimental traumatic brain injury in mice. PLoS One 2013;8:e71056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Timaru‐Kast R, Wyschkon S, Luh C, et al. Delayed inhibition of angiotensin II receptor type 1 reduces secondary brain damage and improves functional recovery after experimental brain trauma. Crit Care Med 2012;40:935–944. [DOI] [PubMed] [Google Scholar]
- 34. Staib‐Lasarzik I, Kriege O, Timaru‐Kast R, et al. Anesthesia for euthanasia influences mRNA expression in healthy mice and after traumatic brain injury. J Neurotrauma 2014;31:1664–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Timaru‐Kast R, Herbig EL, Luh C, et al. Influence of age on cerebral housekeeping gene expression for normalization of quantitative polymerase chain reaction after acute brain injury in mice. J Neurotrauma 2015;32:1777–1788. [DOI] [PubMed] [Google Scholar]
- 36. Neuhaus W, Schlundt M, Fehrholz M, et al. Multiple antenatal dexamethasone treatment alters brain vessel differentiation in newborn mouse pups. PLoS One 2015;10:e0136221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zehendner CM, Sebastiani A, Hugonnet A, et al. Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Sci Rep 2015;5:13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sebastiani A, Golz C, Werner C, et al. Proneurotrophin binding to P75 neurotrophin receptor (P75ntr) is essential for brain lesion formation and functional impairment after experimental traumatic brain injury. J Neurotrauma 2015;32:1599–1607. [DOI] [PubMed] [Google Scholar]
- 39. Faul F, Erdfelder E, Buchner A, Lang AG. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav Res Methods 2009;41:1149–1160. [DOI] [PubMed] [Google Scholar]
- 40. Schucany WR, Tony Ng HK. Preliminary goodness‐of‐fit tests for normality do not validate the one‐sample Student t. Commun Stat Theory Methods 2006;35:2275–2286. [Google Scholar]
- 41. Logsdon AF, Lucke‐Wold BP, Turner RC, et al. Role of microvascular disruption in brain damage from traumatic brain injury. Compr Physiol 2015;5:1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lu D, Mahmood A, Goussev A, et al. Delayed thrombosis after traumatic brain injury in rats. J Neurotrauma 2004;21:1756–1766. [DOI] [PubMed] [Google Scholar]
- 43. Feng JF, Zhao X, Gurkoff GG, et al. Post‐traumatic hypoxia exacerbates neuronal cell death in the hippocampus. J Neurotrauma 2012;29:1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Z, Larner SF, Liu MC, et al. Multiple alphaII‐spectrin breakdown products distinguish calpain and caspase dominated necrotic and apoptotic cell death pathways. Apoptosis 2009;14:1289–1298. [DOI] [PubMed] [Google Scholar]
- 45. Stein SC, Chen XH, Sinson GP, Smith DH. Intravascular coagulation: a major secondary insult in nonfatal traumatic brain injury. J Neurosurg 2002;97:1373–1377. [DOI] [PubMed] [Google Scholar]
- 46. Fujii S, Hopkins WE, Sobel BE. Mechanisms contributing to increased synthesis of plasminogen activator inhibitor type 1 in endothelial cells by constituents of platelets and their implications for thrombolysis. Circulation 1991;83:645–651. [DOI] [PubMed] [Google Scholar]
- 47. Xin H, Chopp M, Shen LH, et al. Multipotent mesenchymal stromal cells decrease transforming growth factor beta1 expression in microglia/macrophages and down‐regulate plasminogen activator inhibitor 1 expression in astrocytes after stroke. Neurosci Lett 2013;542:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huber K. Plasminogen activator inhibitor type‐1 (part two): role for failure of thrombolytic therapy. PAI‐1 resistance as a potential benefit for new fibrinolytic agents. J Thromb Thrombolysis 2001;11:195–202. [DOI] [PubMed] [Google Scholar]
- 49. Nagai N, Suzuki Y, Van Hoef B, et al. Effects of plasminogen activator inhibitor‐1 on ischemic brain injury in permanent and thrombotic middle cerebral artery occlusion models in mice. J Thromb Haemost 2005;3:1379–1384.15978095 [Google Scholar]
- 50. Rupin A, Martin F, Vallez MO, et al. Inactivation of plasminogen activator inhibitor‐1 accelerates thrombolysis of a platelet‐rich thrombus in rat mesenteric arterioles. Thromb Haemost 2001;86:1528–1531. [PubMed] [Google Scholar]
- 51. Smith LH, Dixon JD, Stringham JR, et al. Pivotal role of PAI‐1 in a murine model of hepatic vein thrombosis. Blood 2006;107:132–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Johansson L, Jansson JH, Boman K, et al. Tissue plasminogen activator, plasminogen activator inhibitor‐1, and tissue plasminogen activator/plasminogen activator inhibitor‐1 complex as risk factors for the development of a first stroke. Stroke 2000;31:26–32. [DOI] [PubMed] [Google Scholar]
- 53. Wiman B, Andersson T, Hallqvist J, et al. Plasma levels of tissue plasminogen activator/plasminogen activator inhibitor‐1 complex and von Willebrand factor are significant risk markers for recurrent myocardial infarction in the Stockholm Heart Epidemiology Program (SHEEP) study. Arterioscler Thromb Vasc Biol 2000;20:2019–2023. [DOI] [PubMed] [Google Scholar]
- 54. Zhuang P, Wo D, Xu ZG, et al. Dynamic changes in plasma tissue plasminogen activator, plasminogen activator inhibitor‐1 and beta‐thromboglobulin content in ischemic stroke. J Clin Neurosci 2015;22:1123–1127. [DOI] [PubMed] [Google Scholar]
- 55. CRASH‐2 Collaborators, Intracranial Bleeding Study . Effect of tranexamic acid in traumatic brain injury: a nested randomised, placebo controlled trial (CRASH‐2 Intracranial Bleeding Study). BMJ 2011;343:d3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luh C, Gierth K, Timaru‐Kast R, et al. Influence of a brief episode of anesthesia during the induction of experimental brain trauma on secondary brain damage and inflammation. PLoS One 2011;6:e19948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nishida T, Kinoshita T, Yamakawa K. Tranexamic acid and trauma‐induced coagulopathy. J Intensive Care 2017;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mahmood A, Roberts I, Shakur H. A nested mechanistic sub‐study into the effect of tranexamic acid versus placebo on intracranial haemorrhage and cerebral ischaemia in isolated traumatic brain injury: study protocol for a randomised controlled trial (CRASH‐3 Trial Intracranial Bleeding Mechanistic Sub‐Study [CRASH‐3 IBMS]). Trials 2017;18:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Experimental groups.
Supplementary Table 2 Overview on applied statistical tests.
Data Availability Statement
Datasets generated or analyzed during this study are included in this published article or are available from the corresponding author upon reasonable request.