

Abstract
Wingless/integrated (Wnt) signaling has emerged as a major mechanism for promoting bone formation and a target pathway for developing bone anabolic agents against osteoporosis. However, the downstream events mediating the potential therapeutic effect of Wnt proteins are not fully understood. Previous studies have indicated that increased glycolysis is associated with osteoblast differentiation in response to Wnt signaling, but direct genetic evidence for the importance of glucose metabolism in Wnt-induced bone formation is lacking. Here, we have generated compound transgenic mice to overexpress Wnt family member 7B (Wnt7b) transiently in the osteoblast lineage of postnatal mice, with or without concurrent deletion of the glucose transporter 1 (Glut1), also known as solute carrier family 2, facilitated glucose transporter member 1. Overexpression of Wnt7b in 1-mo-old mice for 1 wk markedly stimulated bone formation, but the effect was essentially abolished without Glut1, even though transient deletion of Glut1 itself did not affect normal bone accrual. Consistent with the in vivo results, Wnt7b increased Glut1 expression and glucose consumption in the primary culture of osteoblast lineage cells, and deletion of Glut1 diminished osteoblast differentiation in vitro. Thus, Wnt7b promotes bone formation in part through stimulating glucose metabolism in osteoblast lineage cells.—Chen, H., Ji, X., Lee, W.-C., Shi, Y., Li, B., Abel, E. D., Jiang, D., Huang, W., Long, F. Increased glycolysis mediates Wnt7b-induced bone formation.
Keywords: Glut1, Slc2a1, glucose, osteoblast, mouse
Wingless/integrated (Wnt) proteins are a family of secreted glycoproteins that are critical for bone development and remodeling (1). Human genetic studies first indicated that inactivating mutations in the Wnt coreceptor Low-density lipoprotein receptor-related protein 5 (LRP5) result in osteoporosis-pseudoglioma syndrome, whereas the gain-of-function mutations of LRP5 cause osteosclerosis (2–4). Subsequently, loss of expression or function of sclerostin, a secreted antagonist of Wnt proteins encoded by the Sclerostin (SOST) gene, was shown to underlie severe osteosclerosis syndromes (5, 6). More recently, deficiency of Wnt family member 1 (WNT1) was linked to early-onset osteoporosis or osteogenesis imperfecta in patients (7–10), and WNT16 was implicated in genome-wide association studies to associate with bone mineral density, cortical bone thickness, and nonvertebral fractures (11, 12). Similarly, in the mouse, deletion of Lrp5, Wnt1, or Wnt16 led to osteopenia, whereas expression of a hyperactive Lrp5 allele or deletion of SOST caused excessive bone formation (13–17). In addition, Wnt7b is normally expressed by the osteogenic perichondrium during murine long bone development, and its deletion causes a temporary delay in ossification in the mouse embryo (18, 19). Conversely, overexpression of Wnt7b in osteogenic cells markedly stimulates bone formation in the mouse (20). Thus, compelling genetic evidence from both humans and mice supports a critical role for Wnt signaling in controlling bone mass.
The intracellular mechanisms for Wnt signaling are complex and highly context dependent (21). Multiple mouse genetic studies have demonstrated the importance of β-catenin signaling in osteoblast differentiation both in the embryo and in postnatal life (22–27). Wnt7b, on the other hand, despite its potent osteogenic activity, does not activate β-catenin signaling in osteoblast lineage cells, but instead signals through protein kinase C (PKC)δ and mammalian/mechanistic target of rapamycin complex (mTORC)1 (18, 20, 28). In addition, Wnt signaling via Lrp5 has been shown to activate mTORC2, whereas removal of mTORC2 activity diminishes the bone anabolic function of an antisclerostin neutralizing antibody (29, 30). Thus, Wnt proteins signal through multiple intracellular mechanisms to promote bone formation.
Recent studies have uncovered a link between glycolysis and osteoblast differentiation (31). Both parathyroid hormone and the osteogenic Wnt proteins, such as Wnt3a and Wnt10b, stimulate aerobic glycolysis in osteoblast lineage cells (29, 32). Moreover, chemical suppression of glycolysis blunts the bone anabolic activity of intermittent parathyroid hormone in the mouse (32). Conversely, physiologic Notch signaling, which is known to suppress osteoblast differentiation, has recently been shown to reduce glycolysis in osteoblast lineage cells (33). In addition, runt-related transcription factor 2 (Runx2), a critical transcription factor for osteoblast differentiation, has been shown to induce the expression of glucose transporter 1 (Glut1), a main glucose transporter in osteoblasts, whereas glucose uptake suppresses Runx2 degradation to promote osteoblast differentiation (34). The studies so far, therefore, support increased glucose metabolism as an important mechanism underlying osteoblast differentiation and function.
Here, we have investigated the contribution of increased glycolysis to bone anabolism induced by Wnt7b in vivo. By genetically deleting Glut1 in the osteogenic cells we show that the bone anabolic function of Wnt7b critically depends on Glut1.
MATERIALS AND METHODS
Mice
The mouse strains of Osx-CreERT2, Rosa26-Wnt7b, Glut1f/f, and Ai9 are as previously described (20, 35–37). One-month-old mice were fed a tamoxifen (TAM) diet (TD.130859; Envigo, Somerset, NJ, USA) for 3 consecutive days and then harvested after 4 d. The Animal Studies Committee at Washington University has approved all mouse procedures in this study.
Studies of bone morphology
Radiographic images of the hindlimbs were acquired using a Faxitron X-ray System (Faxitron X-Ray, Tucson, AZ, USA) at 45 kv for 6.68 s. The right tibiae were scanned by a micro–computed tomography (μCT) system (μCT 40; Scanco Medical, Wangen-Brüttisellen, Switzerland). For quantifying trabecular bone parameters, 100 μCT slices (1-mm total) immediately below the growth plate of the right tibiae were analyzed. For quantifying cortical bone parameters, 50-μCT slices (0.5-mm total) starting from 3 mm above the inferior tibio-fibular junction of the right tibiae were analyzed.
For histologic analyses, the right tibiae were fixed in 10% buffered formalin overnight at room temperature, followed by decalcification in 14% EDTA for 2 wk. After decalcification, the tibiae were processed for paraffin embedding and then sectioned at 6-μm thicknesses. Hematoxylin and eosin (H&E) and tartrate-resistant acid phosphatase staining were performed on paraffin sections according to the standard protocols. For dynamic histomorphometry, calcein and alizarin (MilliporeSigma, Burlington, MA, USA) were injected intraperitoneally at 20 mg/kg at d 7 and 2, respectively, prior to euthanasia. The bones were then fixed in 70% ethanol and embedded in methyl-methacrylate for sectioning. Both static and dynamic bone histomorphometry were performed with the software Bioquant Osteo 2017 (Nashville, TN, USA).
Serum biochemical assays
For serum cross-linked C-terminal telopeptide of type I collagen (CTX-I) bone resorption marker and amino-terminal propeptide of type I procollagen bone formation marker (P1NP) assays, sera were collected from mice via retroorbital bleeding after 6 h of remaining unfed. CTX-I and P1NP assays were performed with the RatLaps ELISA Kit and the Rat/Mouse P1NP EIA Kit (both from Immunodiagnostic Systems, The Boldons, United Kingdom), respectively.
Frozen sections and immunofluorescence
Mice were perfused with 4% paraformaldehyde according to an approved protocol before the right femurs were isolated and then fixed in 4% paraformaldehyde at 4°C overnight. After 3 d of decalcification in 14% EDTA, the femurs were embedded in optimal cutting temperature embedding medium and then sectioned at 10 μm using a Leica cryostat equipped with Cryojane (Leica Microsystems, Buffalo Grove, IL, USA). Immunostaining was performed on frozen sections using rabbit polyclonal Glut1 antibody (ab652; Abcam, Cambridge, MA, USA) or rabbit polyclonal Wnt7b antibody (NBP2-15163; Novus Biologicals, Littleton, CO, USA). Rabbit IgG (ab172730; Abcam) was used as a negative control for staining. The secondary antibody was Alexa Fluor 488 goat anti-rabbit IgG (A-11034; Thermo Fisher Scientific, Waltham, MA, USA). Sections were mounted with Vectashield Mounting Medium containing DAPI (Vector Laboratories, Burlingame, CA, USA). Images were acquired with a Leica confocal microscope (Leica Microsystems).
Cell culture studies
Mouse osteoblastic cells were harvested from the long bones of 1-mo-old mice according to a published protocol (38). Briefly, long bones (bilateral tibiae and femurs) from the mice were harvested and cleaned of soft tissue. After the epiphyseal ends were removed and the bone marrow discarded through centrifugation, the bones were cut into little pieces (1–2 mm3) with sterile scissors and digested in medium containing 1 mg/ml collagenase II (C6885-5G; MilliporeSigma) for 2 h at 37°C. Then, the bone chips were cultured in α-MEM containing 10% fetal bovine serum (referred as growth medium) to allow for cells to migrate from the chips and spread onto the culture dish. The medium was changed every 4 d, and the adherent cells were dissociated and reseeded at 4 × 104/cm2 after 14 d. In certain experiments, the cells were infected at ∼80% confluency with a Cre-expressing adenovirus (University of Iowa) at 25 multiplicity of infection for 8 h before being switched to growth medium. For glucose and lactate measurements and for Western blot analyses, the cells were harvested after 7 d of culture in the growth medium. Glucose and lactate levels were measured with a Glucose (HK) Assay Kit (MilliporeSigma) and an l-Lactate Assay Kit (Eton Biosciences, San Diego, CA, USA), respectively. A plate reader was used to obtain the optical density of the reaction solution (SAMLFTA, Gen5 software; BioTek Instruments, Winooski, VT, USA). For osteoblast differentiation, the cells were switched to the mineralization medium (growth medium plus 50 μg/ml ascorbic acid and 10 mM β-glycerophosphate) at d 2 after the viral infection. The mineralization medium was changed daily. After 14 d of differentiation, the cells were extracted for mRNA or stained for alkaline phosphatase (Alpl) activity or with alizarin red.
RNA was isolated from cell cultures with the Qiagen RNeasy Kit and was reverse transcribed into cDNA using a iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Real-time PCR was performed with Fast-start SYBR Green (Bio-Rad) in an ABI StepOne Plus machine. β-Actin RNA was used for normalization. Sequence information for the primers is provided in the Supplemental Table S1.
Proteins were extracted from cell cultures after 7 d in mineralization medium with RIPA buffer containing a phosphatase inhibitor mix (78428; Thermo Fisher Scientific, Rockford, IL, USA). Thirty micrograms of protein extracts were subsequently used for Western blot according to a standard protocol. The antibody for Glut1 was purchased from Abcam (ab652). Antibodies for β-actin were purchased from Cell Signaling Technology (4970; Beverly, MA, USA).
Statistical analyses
All quantitative data are presented as means ± sd with a minimum of 3 independent samples. Statistical significance was determined by a Student’s t test or 2-way ANOVA followed by Bonferroni post hoc test.
RESULTS
Inducible deletion of Glut1 and activation of Wnt7b by Osx-CreERT2 in osteoblast lineage cells
To confirm the targeting specificity of Osx-CreERT2 in response to TAM, we generated Osx-CreERT2;Ai9 animals so that tdTomato expression could be activated by TAM in Osx-expressing cells and their progenies. Those mice were fed with either TAM-containing or regular diet at 1 mo of age for 3 consecutive days and then harvested after 4 more days on regular chow (Fig. 1A). As expected, bones from the mice fed with regular diet did not show any red fluorescence signal (Fig. 1B). In contrast, the femurs of the TAM-fed mice exhibited red fluorescence on all bone surfaces (BSs), with the highest intensity in the trabecular bone of the primary ossification center (Fig. 1C). Examination at a higher magnification confirmed tdTomato expression in the trabecular bone of both primary and secondary ossification centers, as well as in the cortical bone, but not in the growth plate or the bone marrow (Fig. 1C1–C3). Within the bone, both osteoblasts on the surface and osteocytes within the bone matrix expressed tdTomato (Fig. 1C). These results therefore validate the utility of Osx-CreERT2 in achieving genetic modification in osteoblast and osteocytes in response to short-term feeding of a TM diet.
Figure 1.
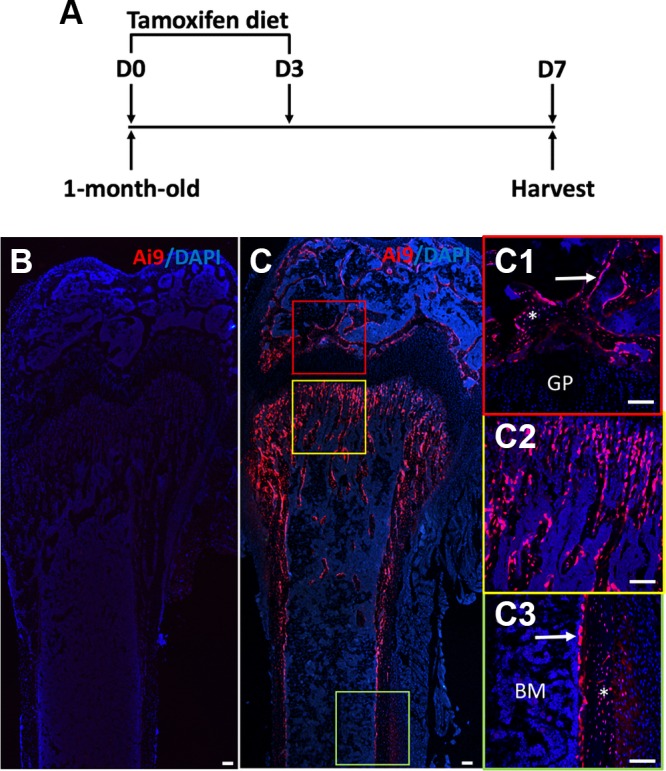
Osx-CreERT2 targets osteoblasts and osteocytes. A) A schematic of treatment strategy. One-month-old mice were fed with TAM diet from d 1 (D0) through d 3 (D3), and harvested at d 7 (D7). B–C3) Confocal fluorescence images of longitudinal femoral sections from mice with the genotype of Osx-CreERT2;Ai9 fed with regular chow (B) or TAM diet (C). The sections were counterstained for nuclei with DAPI (blue) before direct imaging of red fluorescence expressed from Ai9 allele. C1–C3) Boxed regions in C are shown at a higher magnification for secondary ossification center (C1), trabecular bone (C2), and cortical bone (C3). Arrows and asterisks denote osteoblasts and osteocytes, respectively. BM, bone marrow; GP, growth plate. Scale bars, 100 μm.
We next used Osx-CreERT2 to activate Wnt7b expression with or without simultaneous deletion of Glut1. Specifically, we crossed mice with the genotype of Osx-CreERT2;R26-Wnt7b;Glut1f/f with those of Glut1f/f to generate littermate animals with the genotypes of Osx-CreERT2;Glut1f/+, Osx-CreERT2;Glut1f/f, Osx-CreERT2;R26-Wnt7b; Glut1f/+, or Osx-CreERT2;R26-Wnt7b;Glut1f/f, together with the others that did not carry Osx-CreERT2. In this scheme, Osx-CreERT2 in response to TAM was expected to delete the Glutf allele and also to activate Wnt7b expression from the R26-Wnt7b allele in osteoblast lineage cells. The mice were raised until 1 mo of age and then fed TAM-chow for 3 d before they were harvested 4 d later. Immunofluorescence staining with a Glut1 antibody detected Glut1 protein in the growth plate chondrocytes as well as the trabecular bone osteoblasts and the bone marrow cells on sections of the femur from the Osx-CreERT2;R26-Wnt7b;Glut1f/+ mouse (Fig. 2A). However, the Glut1 signal was notably reduced in the trabecular bone in the Osx-CreERT2;R26-Wnt7b;Glut1f/f mouse, despite normal levels in the chondrocytes and the marrow, indicating that deletion of Glut1 in osteoblasts was effective (Fig. 2B). In addition, immunofluorescence staining of Wnt7b detected a relatively spotty expression pattern on the trabecular BS in the Osx-CreERT2;Glut1f/+ mouse, but the Wnt7b-positive osteoblasts were more numerous in the trabecular bone of the Osx-CreERT2;R26-Wnt7b;Glut1f/+ mouse (Fig. 2C, C1, D, D1). Staining with a control IgG did not detect any signal in cartilage, bone, or marrow, indicating the specificity of the Glut1 and Wnt7b antibodies (Fig. 2E, E1). Quantification of the immunostaining results confirmed that the Osx-CreERT2;R26-Wnt7b;Glut1f/f sections contained significantly fewer Glut1+ osteoblasts than the Osx-CreERT2;R26-Wnt7b;Glut1f/+ samples (Fig. 2F, left). Similarly, more Wnt7b+ osteoblasts were present in Osx-CreERT2;R26-Wnt7b;Glut1f/+ than in Osx-CreERT2;Glut1f/+ sections (Fig. 2F, right). Thus, Osx-CreERT2 was effective in deleting Glut1 and activating Wnt7b expression in osteoblast lineage cells in response to TAM.
Figure 2.

Osx-CreERT2 induces Glut1 deletion and Wnt7b overexpression in osteoblast lineage cells. A–E1) Representative images of immunofluorescence staining on longitudinal sections of the femur with antibodies against Glut1 (A, B), Wnt7b (C, D), or control IgG (E). Boxed areas shown at a higher magnification to the right for growth plate (A1, B1, E1) or trabecular bone region (A2, B2, C1, D1). Immunostaining signal in green, DAPI nuclear staining in blue. F) Quantification of immunostaining data. Number (N.) of Glut1+ or Wnt7b+ cells normalized to trabecular BS in primary ossification center of proximal end of tibia. Red arrow, osteoblast; yellow arrow, chondrocyte; white arrow, blood cells. Scale bars, 100 μm. ** P < 0.01, Student’s t test (n = 3 mice, 1 section/mouse).
Inducible deletion of Glut1 eliminates bone accrual caused by Wnt7b overexpression
We next assessed the bone phenotypes by X-ray radiography and µCT. Comparison of the tibia by µCT indicated no difference in either trabecular or cortical bone of the tibia between the Glut1f/+ and the Osx-CreERT2;Glut1f/+ mice (Supplemental Fig. S1). We therefore used the Osx-CreERT2;Glut1f/+ mice as a normal control for the subsequent analyses. For brevity, we refer to Osx-CreERT2;Glut1f/+ as control, Osx-CreERT2;Glut1f/f as Glut1 conditional knockout (Glut1CKO), Osx-CreERT2;R26-Wnt7b; Glut1f/+ as Wnt7b over-expresser (Wnt7bOE), and Osx-CreERT2;R26-Wnt7b;Glut1f/f as Wnt7bOE;Glut1CKO. Contact X-ray radiography of the hindlimb detected an increase in the radiodensity of the trabecular bone in the proximal tibia, as well as the distal femur in Wnt7bOE over control or Glut1CKO (Fig. 3A–C compared with A1, A2, B1, B2). Importantly, Wnt7bOE; Glut1CKO showed a lower radiodensity than Wnt7bOE (Fig. 3D compared with C1, C2). On the other hand, the overall lengths of the femur or the tibia as measured from the X-ray images were not different across all 4 genotypes (Supplemental Fig. S2). The 3-dimensional reconstruction from the µCT scans of the proximal tibia confirmed that Wnt7bOE possessed the highest trabecular bone volume, whereas the diaphyseal cortical bone mass appeared to be similar across the genotypes (Fig. 3E–H). Quantification of the µCT data in both male and female mice indicated that the trabecular bone mass (bone volume/total volume) was normal in Glut1CKO but more than 40% higher in Wnt7bOE, and it was back to normal in Wnt7bOE; Glut1CKO (Fig. 3I and Supplemental Fig. S3). For simplicity, only data from males are presented in the subsequent figures. The increase in trabecular bone mass in Wnt7bOE was accounted for by the increase in both trabeculae number and thickness concurrent with a decrease in trabecular spacing (Fig. 3J–L). Interestingly, trabeculae number but not trabeculae thickness was normalized in Wnt7bOE;Glut1CKO (Fig. 3J, K). In contrast to the trabecular bone, the cortical bone parameters were normal in all groups (Fig. 3M, N). To assess whether Glut1 heterozygosity might impact bone accumulation caused by Wnt7b overexpression, we generated Osx-CreERT2;R26-Wnt7b mice and subjected them to the same TAM regimen as previously described. Those mice, according to μCT analyses, exhibited no difference in the bone parameters from the Osx-CreERT2;R26-Wnt7b;Glut1f/+ mice (Supplemental Fig. S4). Thus, inducible deletion of Glut1 for 1 wk at 1 mo of age did not affect normal bone growth but essentially eliminated the excessive bone accretion caused by Wnt7b overexpression.
Figure 3.

Inducible deletion of Glut1 diminishes Wnt7b-induced bone accrual. A–D2) Representative X-ray radiographs of the hind limbs. Boxed areas at a higher magnification (A1, A2, B1, B2, C1, C2, D1, D2). Red arrow, distal femur; white arrow, proximal tibia. E–H) μCT 3-dimensional reconstruction of trabecular bone (upper) from the proximal end or cortical bone (lower) from the midshaft of the tibia. Scale bars, 100 μm. I–N) Trabecular (I–L) and cortical (M, N) bone parameters from μCT analyses. BA, bone area; BV, bone volume; Ct. Th, cortical thickness; TA, total area; Tb. N, trabecular number; Tb. Sp, trabecular separation; Tb. Th, trabecular thickness; TV, tissue volume. **P < 0.01, ***P < 0.001, by 2-way ANOVA followed by Bonferroni’s post hoc test (n = 7).
Glut1 deletion diminishes Wnt7b-induced bone formation
We next investigated whether Glut1 deletion decreased bone mass in the Wnt7bOE; Glut1CKO mice by altering bone formation or resorption. We first measured the level of P1NP in the serum, which is the N-terminal propeptide of collagen type I and a marker for bone formation. The P1NP level was significantly higher in Wnt7bOB than control but was normal in Glut1CKO and Wnt7bOE;Glut1CKO (Fig. 4A). In contrast, CTX-1 levels in the serum, an indicator for the overall bone resorption activity, were not different across the different genotypes, indicating that bone resorption was not the driving force for the differences in bone mass (Fig. 4B). We next performed static and dynamic histomorphometry with the tibia. H&E staining of the tibial sections confirmed an increase in trabecular bone underneath the proximal growth plate in Wnt7bOE relative to the other groups (Fig. 4C–F). Quantification of trabecular osteoblast numbers within the primary ossification center indicated a significantly higher value in Wnt7bOE and no difference between Wnt7bOE and Wnt7bOE;Glut1CKO (Fig. 4G). In contrast, the growth plate height was the same across the different genotypes (Fig. 4H). Quantification of osteoclasts with tartrate-resistant acid phosphatase staining also did not reveal any difference across the genotypes (Supplemental Fig. S5). Dynamic histomorphometry showed a notable increase in the distance between the 2 labels in Wnt7bOE compared with the other mice (Fig. 5A). Quantification of the double-labeling data indicated that both mineral apposition rate and the percentage of mineralizing surface (mineralizing surface/BS) were increased in Wnt7bOE over control, resulting in a significant increase in bone formation rate (bone formation rate/BS) in Wnt7bOE (Fig. 5B–E). Mineral apposition rate was largely normalized in Wnt7bOE;Glut1CKO compared with Wnt7bOE even though mineralizing surface/BS remained high (Fig. 5B–D). As a result, bone formation rate/BS was significantly lower in Wnt7bOE;Glut1CKO than Wnt7bOE (Fig. 5E). These results indicate that Glut1 deletion diminishes Wnt7b-induced osteoblast activity without reducing the osteoblast number.
Figure 4.

Glut1 deletion impairs Wnt7b-induced bone formation. A, B) Serum levels of P1NP (A) or CTX-1 (B). C–F) Representative H&E tibial sections used for histomorphometric quantification. Boxed areas shown at a higher magnification to the right for growth plate (upper) or trabecular bone region (lower). Scale bars, 100 μm. G) Quantification of trabecular osteoblast numbers within the primary ossification center of the proximal end of the tibia. The region of interest was defined as a rectangular box of a fixed size at 100 μm below the growth plate with the long axis parallel to the growth plate. H) Measurements of the growth plate height. N. Ob, number of osteoblasts. *P < 0.05, **P < 0.01 by 2-way ANOVA followed by Bonferroni’s post hoc test (n = 5).
Figure 5.

Glut1 deletion diminishes osteoblast activity induced by Wnt7b. A) Representative micrographs of double labeling at endosteal BS of the tibia. B) Representative micrographs of double labeling in trabecular bone region of primary ossification center at proximal end of the tibia. Boxed areas shown at a higher magnification to the right. Scale bars, 100 μm. C–E) Quantification of bone formation parameters at proximal tibial trabecular region. BFR, bone formation rate; MAR, mineral apposition rate; MS, mineralizing surface. *P < 0.05, **P < 0.01, ***P < 0.001 by 2-way ANOVA followed by Bonferroni’s post hoc test (n = 5).
Glut1 contributes to osteoblast differentiation in vitro
To explore further the role of Glut1 and Wnt7b in osteoblast differentiation, we established an osteogenic protocol with osteoblast lineage cells isolated from the long bones (hereafter termed “bone-chip cells”) of adult mice. After 14 d of culture in mineralization medium, the bone-chip cells from wild-type mice exhibited more alizarin red staining, a readout for osteoblast activity, than those in the growth medium (Supplemental Fig. S6A). Quantification of mRNA by quantitative PCR (qPCR) showed that the common osteoblast markers Alpl, Bglap (also known as osteocalcin), Col1a2, and Dmp1, but not Runx2 or Sp7, were induced by the mineralization media (Supplemental Fig. S6B). Having validated the differentiation protocol, we next isolated bone-chip cells from mice with the genotype of Glut1f/+, Glut1f/f, R26-Wnt7b;Glut1f/+, or R26-Wnt7b;Glut1f/f and infected the cells with adenovirus expressing Cre (Ad-Cre) before culturing them in mineralization media for 14 d. Alpl or alizarin red staining indicated that osteoblast differentiation was reduced in Glut1f/f cells but increased in R26-Wnt7b;Glut1f/+ cells over Glut1f/+ cells (Fig. 6A). The staining also showed that differentiation was diminished in R26-Wnt7b;Glut1f/f cells compared with R26-Wnt7b;Glut1f/+ cells (Fig. 6A). qPCR revealed a marked increase of Wnt7b mRNA in R26-Wnt7b;Glut1f/+ or R26-Wnt7b;Glut1f/f cells but a significant decrease of Glut1 in the Glut1f/f or R26-Wnt7b;Glut1f/f cells, thus confirming the efficacy of Ad-Cre in mediating DNA recombination in vitro (Fig. 6B, C). Importantly, Wnt7b overexpression in the R26-Wnt7b;Glut1f/+ cells markedly increased the mRNA levels of all common osteoblast markers, including Alpl, Runx2, Sp7, Bglap, Col1a2, and Dmp1, but they were all decreased by concurrent deletion of Glut1 in R26-Wnt7b;Glut1f/f cells (Fig. 6D–I). As we have previously shown that genes participating in protein anabolism are induced during osteoblast differentiation, we examined the expression of several representative genes involved in the endoplasmic reticulum unfolded protein response (Atf4, Ddit3, Chac1), amino acid synthase (Asns), or tRNA aminoacylation (Tars, Lars) (39). All of those genes were induced in R26-Wnt7b;Glut1f/+ over Glut1f/+ cells, but the induction was blunted in R26-Wnt7b;Glut1f/f cells (Fig. 6J–O). In addition, most of the osteoblast markers and protein anabolism genes examined were reduced in Glut1f/f cells compared with Glut1f/+ cells (Fig. 6D–O). Thus, Glut1 is necessary for optimal osteoblast differentiation both under basal conditions and in response to Wnt7b overexpression in vitro.
Figure 6.

Glut1 deletion impedes osteoblast differentiation in vitro. Bone-chip cells of various genotypes were infected with Ad-Cre before culture in mineralization media for 14 d. A) Alpl activity or alizarin red staining. B–O) Quantification of mRNA by qPCR. Data normalized to β-actin and expressed as relative values to the level in Glut1f/+ cell designated 1. *P < 0.05, **P < 0.01, ***P < 0.001 by 2-way ANOVA followed by Bonferroni’s post hoc test (n = 5).
We next sought to gain further insight into the relationship between Wnt7b and glucose metabolism. Western blots showed that Glut1 protein was reduced by ∼60% in Glut1f/f or R26-Wnt7b;Glut1f/f cells relative to Glut1f/+ cells; the decrease was similar to that of the mRNA (Figs. 6C and 7A). In contrast, Glut1 protein was increased by 400% in R26-Wnt7b;Glut1f/+ cells relative to Glut1f/+ cells, which exceeded the ∼ 30% increase in Glut1 mRNA, indicating posttranscriptional stimulation of Glut1 protein levels by Wnt7b (Figs. 6C and 7A). Glut2, 3, or 4 was not affected by either deletion of Glut1 or overexpression of Wnt7b in the bone-chip cells (Fig. 7B–D). To determine the functional relevance of Glut1 deletion or Wnt7b overexpression to glucose metabolism, we assayed for glucose consumption and lactate production in the cells after 7 d of culture in the growth medium. Compared with Glut1f/+ cells, Glut1f/f cells consumed ∼50% less glucose and R26-Wnt7b;Glut1f/+ cells ∼30% more, whereas R26-Wnt7b;Glut1f/f cells used a normal amount of glucose (Fig. 7E). Lactate levels in the medium were normal with Glut1f/f cells, but increased with either R26-Wnt7b;Glut1f/+ or R26-Wnt7b;Glut1f/f cells (Fig. 7F). Finally, Glut1 deletion decreased the number of Glut1f/f and R26-Wnt7b;Glut1f/f cells relative to Glut1f/+ and R26-Wnt7b;Glut1f/+ cells, respectively, whereas overexpression of Wnt7b increased the number of R26-Wnt7b;Glut1f/+ cells compared with Glut1f/+ cells (Fig. 7G). Thus, Wnt7b overexpression increases Glut1 levels and glucose consumption in osteoblast lineage cells in vitro.
Figure 7.

Wnt7b stimulates glucose metabolism in osteoblast lineage cells. A) Western blot analyses of total cell lysates from bone-chip cells infected with Ad-Cre and then cultured in growth medium for 7 d. Glut1 protein abundance normalized to β-actin and expressed in relative values (means + sd) to level in Glut1f/+ cells designated 1. B–D) Expression analysis of mRNA by qPCR in bone-chip cells after Ad-Cre infection and 14 d of culture in mineralization medium. Data normalized to β-actin and then expressed as values relative to the level in Glut1f/+ cells that was designated 1. E, F) Glucose consumption and lactate production by bone-chip cells infected with Ad-Cre and then cultured in growth medium for 7 d. G) Cell number per well in 12-well plates after bone-chip cells were infected with Ad-Cre and then cultured in growth medium for 7 d. *P < 0.05, **P < 0.01, ***P < 0.001 by 2-way ANOVA followed by Bonferroni’s post hoc test [n = 3 (A); n = 5 (B–G)].
DISCUSSION
We have investigated the role of Glut1 in mediating Wnt7b-induced osteoblast differentiation and function in vivo and in vitro. We show that deletion of Glut1 in osteoblast lineage cells largely blocks the bone anabolic function of Wnt7b in vivo, and also impairs osteoblast differentiation and mineralization in vitro. The results therefore provide genetic evidence that glucose metabolism is necessary to support Wnt7b-induced bone formation.
Both glucose and fatty acid metabolism have been implicated in bone anabolism in response to Wnt signaling (40). Similar to Wnt7b presented here, Wnt3a and Wnt10b, both also known to stimulate osteoblast differentiation, have previously been shown to enhance aerobic glycolysis (metabolism of glucose to lactate under aerobic conditions) in osteoblast progenitors (29). Moreover, the extent of glycolysis correlates positively with the state of Lrp5-mediated Wnt signaling and bone formation in vivo (29). Similarly, fatty acid metabolism has been reported to decrease in osteoblasts upon deletion of Lrp5 but increased by expression of a high-bone-mass variant of Lrp5 (41). Here, we have provided genetic evidence supporting the importance of glucose metabolism, but future studies are necessary to determine whether fatty acid metabolism is also necessary for the bone anabolic function of Wnt signaling in vivo.
It is worth noting that deletion of Glut1 for up to 7 d in 1-mo-old mice did not affect the basal level of bone formation. This may be explained by the short duration of Glut1 deficiency combined with a relatively low basal rate of bone formation, or due to potential compensatory mechanisms. Previously, deletion of Glut1 with Prx1-Cre had little impact on osteoblast differentiation in the mouse embryo even though the mineralizing activity appeared to be partially impaired (42). In vitro experiments in the current study showed that Glut1 deletion markedly reduced glucose metabolism by osteoblast lineage cells, thus arguing against functional compensation by the other glucose transporters. The dominant role of Glut1 was consistent with our qRT-PCR results (data not shown) that the mRNA levels of Glut2, 3, or 4 were normally at least one order of magnitude lower than those of Glut1 in bone-chip cells, and were not induced upon Glut1 deletion in vitro. Assuming glucose metabolism is similarly disrupted by Glut1 deletion in vivo, the defect may be compensated by increased utilization of other energy substrates such as fatty acids and glutamine, both of which may contribute to osteoblast bioenergetics (41, 43). Future experiments measuring substrate utilization in vivo are warranted to discern those possibilities.
Several in vitro observations warrant further investigation. Contrary to the lack of an effect on basal-level bone formation in vivo, Glut1 deletion in vitro diminished both the number and differentiation of osteoblast lineage cells regardless of Wnt7b overexpression. Considering that the current cell culture medium contained little fatty acids, it would be interesting to test in the future whether supplementation with fatty acids rescues those defects. In addition, the in vitro studies showed that Wnt7b increased Glut1 protein more than mRNA, but the mechanism is yet unknown. As Wnt7b is known to activate mTORC1, future experiments are necessary to determine whether Wnt7b stimulates Glut1 translation via mTORC1 (20).
The mechanism underlying the importance of Glut1 in Wnt-induced bone formation remains to be investigated. Loss of Glut1 may significantly reduce intracellular ATP levels and intermediate metabolites necessary for meeting the requirements for energy and building blocks during bone formation. Glut1 may also be necessary for stabilizing Runx2 protein to promote osteoblast differentiation as previously indicated (34). Besides glucose metabolism as reported here, increased glutamine metabolism has been previously shown to underlie bone anabolism induced by hyperactive Lrp5 signaling (43). Thus, Wnt signaling likely exerts bone anabolic function by stimulating metabolism of multiple fuel substrates.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health (NIH) National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant AR060456 (to F.L.), NIH National Institute of Diabetes and Digestive and Kidney Diseases Grant DK111212 (to F.L.), and the Washington University Musculoskeletal Research Center (NIH P30 AR057235). H.C. is a visiting scholar supported by the China Scholarship Council (201608500093). The authors declare no conflicts of interest.
Glossary
- μCT
micro–computed tomography
- Ad-Cre
adenovirus expressing Cre
- Alpl
alkaline phosphatase
- BS
bone surface
- CKO
conditional knockout
- CTX-I
cross-linked C-terminal telopeptide of type I collagen
- Glut1
glucose transporter 1
- H&E
hematoxylin and eosin
- mTORC
mammalian/mechanistic targe of rapamycin complex
- OE
overexpresser
- P1NP
amino-terminal propeptide of type I procollagen, bone formation marker
- qPCR
quantitative PCR
- Runx2
runt-related transcription factor 2
- TAM
tamoxifen
- Wnt7b
wingless/integrated family member 7B
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
H. Chen, X. Ji, W.-C. Lee, Y. Shi, and B. Li conducted experiments; E. D. Abel contributed reagent; H. Chen, D. Jiang, W. Huang, and F. Long analyzed data; H. Chen and F. Long wrote the manuscript; and F. Long designed research.
REFERENCES
- 1.Zhong Z., Ethen N. J., Williams B. O. (2014) WNT signaling in bone development and homeostasis. Wiley Interdiscip. Rev. Dev. Biol. 3, 489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gong Y., Slee R. B., Fukai N., Rawadi G., Roman-Roman S., Reginato A. M., Wang H., Cundy T., Glorieux F. H., Lev D., Zacharin M., Oexle K., Marcelino J., Suwairi W., Heeger S., Sabatakos G., Apte S., Adkins W. N., Allgrove J., Arslan-Kirchner M., Batch J. A., Beighton P., Black G. C., Boles R. G., Boon L. M., Borrone C., Brunner H. G., Carle G. F., Dallapiccola B., De Paepe A., Floege B., Halfhide M. L., Hall B., Hennekam R. C., Hirose T., Jans A., Jüppner H., Kim C. A., Keppler-Noreuil K., Kohlschuetter A., LaCombe D., Lambert M., Lemyre E., Letteboer T., Peltonen L., Ramesar R. S., Romanengo M., Somer H., Steichen-Gersdorf E., Steinmann B., Sullivan B., Superti-Furga A., Swoboda W., van den Boogaard M. J., Van Hul W., Vikkula M., Votruba M., Zabel B., Garcia T., Baron R., Olsen B. R., Warman M. L.; Osteoporosis-Pseudoglioma Syndrome Collaborative Group (2001) LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107, 513–523 [DOI] [PubMed] [Google Scholar]
- 3.Boyden L. M., Mao J., Belsky J., Mitzner L., Farhi A., Mitnick M. A., Wu D., Insogna K., Lifton R. P. (2002) High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med. 346, 1513–1521 [DOI] [PubMed] [Google Scholar]
- 4.Little R. D., Carulli J. P., Del Mastro R. G., Dupuis J., Osborne M., Folz C., Manning S. P., Swain P. M., Zhao S. C., Eustace B., Lappe M. M., Spitzer L., Zweier S., Braunschweiger K., Benchekroun Y., Hu X., Adair R., Chee L., FitzGerald M. G., Tulig C., Caruso A., Tzellas N., Bawa A., Franklin B., McGuire S., Nogues X., Gong G., Allen K. M., Anisowicz A., Morales A. J., Lomedico P. T., Recker S. M., Van Eerdewegh P., Recker R. R., Johnson M. L. (2002) A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet. 70, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balemans W., Patel N., Ebeling M., Van Hul E., Wuyts W., Lacza C., Dioszegi M., Dikkers F. G., Hildering P., Willems P. J., Verheij J. B., Lindpaintner K., Vickery B., Foernzler D., Van Hul W. (2002) Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 39, 91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunkow M. E., Gardner J. C., Van Ness J., Paeper B. W., Kovacevich B. R., Proll S., Skonier J. E., Zhao L., Sabo P. J., Fu Y., Alisch R. S., Gillett L., Colbert T., Tacconi P., Galas D., Hamersma H., Beighton P., Mulligan J. (2001) Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 68, 577–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fahiminiya S., Majewski J., Mort J., Moffatt P., Glorieux F. H., Rauch F. (2013) Mutations in WNT1 are a cause of osteogenesis imperfecta. J. Med. Genet. 50, 345–348 [DOI] [PubMed] [Google Scholar]
- 8.Keupp K., Beleggia F., Kayserili H., Barnes A. M., Steiner M., Semler O., Fischer B., Yigit G., Janda C. Y., Becker J., Breer S., Altunoglu U., Grünhagen J., Krawitz P., Hecht J., Schinke T., Makareeva E., Lausch E., Cankaya T., Caparrós-Martín J. A., Lapunzina P., Temtamy S., Aglan M., Zabel B., Eysel P., Koerber F., Leikin S., Garcia K. C., Netzer C., Schönau E., Ruiz-Perez V. L., Mundlos S., Amling M., Kornak U., Marini J., Wollnik B. (2013) Mutations in WNT1 cause different forms of bone fragility. Am. J. Hum. Genet. 92, 565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laine C. M., Joeng K. S., Campeau P. M., Kiviranta R., Tarkkonen K., Grover M., Lu J. T., Pekkinen M., Wessman M., Heino T. J., Nieminen-Pihala V., Aronen M., Laine T., Kröger H., Cole W. G., Lehesjoki A. E., Nevarez L., Krakow D., Curry C. J., Cohn D. H., Gibbs R. A., Lee B. H., Mäkitie O. (2013) WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N. Engl. J. Med. 368, 1809–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pyott S. M., Tran T. T., Leistritz D. F., Pepin M. G., Mendelsohn N. J., Temme R. T., Fernandez B. A., Elsayed S. M., Elsobky E., Verma I., Nair S., Turner E. H., Smith J. D., Jarvik G. P., Byers P. H. (2013) WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am. J. Hum. Genet. 92, 590–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng H. F., Tobias J. H., Duncan E., Evans D. M., Eriksson J., Paternoster L., Yerges-Armstrong L. M., Lehtimäki T., Bergström U., Kähönen M., Leo P. J., Raitakari O., Laaksonen M., Nicholson G. C., Viikari J., Ladouceur M., Lyytikäinen L. P., Medina-Gomez C., Rivadeneira F., Prince R. L., Sievanen H., Leslie W. D., Mellström D., Eisman J. A., Movérare-Skrtic S., Goltzman D., Hanley D. A., Jones G., St Pourcain B., Xiao Y., Timpson N. J., Smith G. D., Reid I. R., Ring S. M., Sambrook P. N., Karlsson M., Dennison E. M., Kemp J. P., Danoy P., Sayers A., Wilson S. G., Nethander M., McCloskey E., Vandenput L., Eastell R., Liu J., Spector T., Mitchell B. D., Streeten E. A., Brommage R., Pettersson-Kymmer U., Brown M. A., Ohlsson C., Richards J. B., Lorentzon M. (2012) WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet. 8, e1002745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medina-Gomez C., Kemp J. P., Estrada K., Eriksson J., Liu J., Reppe S., Evans D. M., Heppe D. H., Vandenput L., Herrera L., Ring S. M., Kruithof C. J., Timpson N. J., Zillikens M. C., Olstad O. K., Zheng H. F., Richards J. B., St Pourcain B., Hofman A., Jaddoe V. W., Smith G. D., Lorentzon M., Gautvik K. M., Uitterlinden A. G., Brommage R., Ohlsson C., Tobias J. H., Rivadeneira F. (2012) Meta-analysis of genome-wide scans for total body BMD in children and adults reveals allelic heterogeneity and age-specific effects at the WNT16 locus. PLoS Genet. 8, e1002718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato M., Patel M. S., Levasseur R., Lobov I., Chang B. H., Glass D. A., II, Hartmann C., Li L., Hwang T. H., Brayton C. F., Lang R. A., Karsenty G., Chan L. (2002) Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 157, 303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui Y., Niziolek P. J., MacDonald B. T., Zylstra C. R., Alenina N., Robinson D. R., Zhong Z., Matthes S., Jacobsen C. M., Conlon R. A., Brommage R., Liu Q., Mseeh F., Powell D. R., Yang Q. M., Zambrowicz B., Gerrits H., Gossen J. A., He X., Bader M., Williams B. O., Warman M. L., Robling A. G. (2011) Lrp5 functions in bone to regulate bone mass. Nat. Med. 17, 684–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joeng K. S., Lee Y. C., Lim J., Chen Y., Jiang M. M., Munivez E., Ambrose C., Lee B. H. (2017) Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J. Clin. Invest. 127, 2678–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Movérare-Skrtic S., Henning P., Liu X., Nagano K., Saito H., Börjesson A. E., Sjögren K., Windahl S. H., Farman H., Kindlund B., Engdahl C., Koskela A., Zhang F. P., Eriksson E. E., Zaman F., Hammarstedt A., Isaksson H., Bally M., Kassem A., Lindholm C., Sandberg O., Aspenberg P., Sävendahl L., Feng J. Q., Tuckermann J., Tuukkanen J., Poutanen M., Baron R., Lerner U. H., Gori F., Ohlsson C. (2014) Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat. Med. 20, 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X., Ominsky M. S., Niu Q. T., Sun N., Daugherty B., D'Agostin D., Kurahara C., Gao Y., Cao J., Gong J., Asuncion F., Barrero M., Warmington K., Dwyer D., Stolina M., Morony S., Sarosi I., Kostenuik P. J., Lacey D. L., Simonet W. S., Ke H. Z., Paszty C. (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 23, 860–869 [DOI] [PubMed] [Google Scholar]
- 18.Tu X., Joeng K. S., Nakayama K. I., Nakayama K., Rajagopal J., Carroll T. J., McMahon A. P., Long F. (2007) Noncanonical Wnt signaling through G protein-linked PKCdelta activation promotes bone formation. Dev. Cell 12, 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joeng K. S., Long F. (2014) Wnt7b can replace Ihh to induce hypertrophic cartilage vascularization but not osteoblast differentiation during endochondral bone development. Bone Res. 2, 14004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J., Tu X., Esen E., Joeng K. S., Lin C., Arbeit J. M., Rüegg M. A., Hall M. N., Ma L., Long F. (2014) WNT7B promotes bone formation in part through mTORC1. PLoS Genet. 10, e1004145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Amerongen R., Nusse R. (2009) Towards an integrated view of Wnt signaling in development. Development 136, 3205–3214 [DOI] [PubMed] [Google Scholar]
- 22.Hu H., Hilton M. J., Tu X., Yu K., Ornitz D. M., Long F. (2005) Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development 132, 49–60 [DOI] [PubMed] [Google Scholar]
- 23.Day T. F., Guo X., Garrett-Beal L., Yang Y. (2005) Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750 [DOI] [PubMed] [Google Scholar]
- 24.Hill T. P., Später D., Taketo M. M., Birchmeier W., Hartmann C. (2005) Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727–738 [DOI] [PubMed] [Google Scholar]
- 25.Rodda S. J., McMahon A. P. (2006) Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133, 3231–3244 [DOI] [PubMed] [Google Scholar]
- 26.Chen J., Long F. (2013) β-catenin promotes bone formation and suppresses bone resorption in postnatal growing mice. J. Bone Miner. Res. 28, 1160–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song L., Liu M., Ono N., Bringhurst F. R., Kronenberg H. M., Guo J. (2012) Loss of wnt/β-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J. Bone Miner. Res. 27, 2344–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu X., Tu X., Joeng K. S., Hilton M. J., Williams D. A., Long F. (2008) Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell 133, 340–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esen E., Chen J., Karner C. M., Okunade A. L., Patterson B. W., Long F. (2013) WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 17, 745–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun W., Shi Y., Lee W. C., Lee S. Y., Long F. (2016) Rictor is required for optimal bone accrual in response to anti-sclerostin therapy in the mouse. Bone 85, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esen E., Long F. (2014) Aerobic glycolysis in osteoblasts. Curr. Osteoporos. Rep. 12, 433–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Esen E., Lee S. Y., Wice B. M., Long F. (2015) PTH promotes bone anabolism by stimulating aerobic glycolysis via IGF signaling. J. Bone Miner. Res. 30, 1959–1968; erratum: 2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee S. Y., Long F. (2018) Notch signaling suppresses glucose metabolism in mesenchymal progenitors to restrict osteoblast differentiation. J. Clin. Invest. 128, 5573–5586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei J., Shimazu J., Makinistoglu M. P., Maurizi A., Kajimura D., Zong H., Takarada T., Lezaki T., Pessin J. E., Hinoi E., Karsenty G. (2015) Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell 161, 1576–1591; erratum: 162, 1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maes C., Kobayashi T., Selig M. K., Torrekens S., Roth S. I., Mackem S., Carmeliet G., Kronenberg H. M. (2010) Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 19, 329–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Young C. D., Lewis A. S., Rudolph M. C., Ruehle M. D., Jackman M. R., Yun U. J., Ilkun O., Pereira R., Abel E. D., Anderson S. M. (2011) Modulation of glucose transporter 1 (GLUT1) expression levels alters mouse mammary tumor cell growth in vitro and in vivo. PLoS One 6, e23205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R., Lein E. S., Zeng H. (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bakker A. D., Klein-Nulend J. (2012) Osteoblast isolation from murine calvaria and long bones. Methods Mol. Biol. 816, 19–29 [DOI] [PubMed] [Google Scholar]
- 39.Karner C. M., Lee S. Y., Long F. (2017) Bmp induces osteoblast differentiation through both Smad4 and mTORC1 signaling. Mol. Cell. Biol. 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karner C. M., Long F. (2017) Wnt signaling and cellular metabolism in osteoblasts. Cell. Mol. Life Sci. 74, 1649–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frey J. L., Li Z., Ellis J. M., Zhang Q., Farber C. R., Aja S., Wolfgang M. J., Clemens T. L., Riddle R. C. (2015) Wnt-Lrp5 signaling regulates fatty acid metabolism in the osteoblast. Mol. Cell. Biol. 35, 1979–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee S. Y., Abel E. D., Long F. (2018) Glucose metabolism induced by Bmp signaling is essential for murine skeletal development. Nat. Commun. 9, 4831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karner C. M., Esen E., Okunade A. L., Patterson B. W., Long F. (2015) Increased glutamine catabolism mediates bone anabolism in response to WNT signaling. J. Clin. Invest. 125, 551–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.