

Abstract
Cellular checkpoints controlling entry into mitosis monitor the integrity of the DNA and delay mitosis onset until the alteration is fully repaired. However, this canonical response can weaken, leading to a spontaneous bypass of the checkpoint, a process referred to as checkpoint adaptation. Here, we have investigated the contribution of microcephalin 1 (MCPH1), mutated in primary microcephaly, to the decatenation checkpoint, a less-understood G2 pathway that delays entry into mitosis until chromosomes are properly disentangled. Our results demonstrate that, although MCPH1 function is dispensable for activation and maintenance of the decatenation checkpoint, it is required for the adaptive response that bypasses the topoisomerase II inhibition–mediated G2 arrest. MCPH1, however, does not confer adaptation to the G2 arrest triggered by the ataxia telangiectasia mutated– and ataxia telangiectasia and rad3 related–based DNA damage checkpoint. In addition to revealing a new role for MCPH1 in cell cycle control, our study provides new insights into the genetic requirements that allow cellular adaptation to G2 checkpoints, a process that remains poorly understood.—Arroyo, M., Kuriyama, R., Guerrero, I., Keifenheim, D., Cañuelo, A., Calahorra, J., Sánchez, A., Clarke, D. J., Marchal, J. A. MCPH1 is essential for cellular adaptation to the G2-phase decatenation checkpoint.
Keywords: cell cycle control, checkpoint adaptation, chromosome condensation, topoisomerase II, MCPH1
Checkpoints are specialized surveillance mechanisms that ensure coupling and completion of critical processes, such as DNA replication or chromosome segregation, within the cell cycle. Checkpoints controlling entry into mitosis critically monitor the integrity of the DNA. Accordingly, the pathways regulating normal mitosis onset are rewired under damaging conditions, triggering a reversible cell cycle arrest to allow efficient DNA repair before entry into mitosis (1). However, cells retain a capacity to spontaneously bypass the checkpoint arrest following a transient G2 delay despite the persistence of the damage that triggered it (2, 3). Unraveling the molecular requirements of this overlooked mechanism, named checkpoint adaptation, is of importance to better understand the checkpoint control system itself. Checkpoint adaptation mechanisms are also of particular relevance to cancer etiology.
Microcephalin 1 (MCPH1), mutated in primary microcephaly, regulates cell progression into mitosis in multiple contexts. During unperturbed cell division, it is required for coupling chromosome condensation and centrosome maturation in mitosis (4–7). The underlying pathways controlling the G2/M transition are also misregulated under undamaging conditions in MCPH1-deficient cells. Unscheduled activation of cyclin-dependent kinase 1 (Cdk1) and cell division cycle 25A (Cdc25A) and loss of centrosomal checkpoint kinase 1 (Chk1) during G2 are all consequences of MCPH1 down-regulation (6, 8, 9). How MCPH1 modulates Cdk1 and Cdc25A activation during unperturbed cell division remains to be clarified. Remarkably, those pathways retain sufficient capacity to allow scheduled G2 progression and mitosis onset in cells depleted of MCPH1 (7, 10).
A different scenario in which MCPH1 function has been explored is cell cycle control in the presence of DNA damage. MCPH1 patient cells, similar to ataxia telangiectasia and rad3 related (ATR)-mutated cells, display defective G2 arrest after treatments that activate the ATR signaling pathway [e.g., after UV irradiation induced DNA damage (8)]. MCPH1 is proposed to function downstream of Chk1 and upstream of Cdc25A in the ATR-related pathway (8). Several lines of evidences suggest that MCPH1 is however dispensable for the ataxia telangiectasia mutated (ATM)-related DNA damage checkpoint. A collection of studies using patient cell lines (11), DT40 (chicken bursal lymphoma cell line) knockout cells (12), or MCPH1−/− mouse cells (6, 13) demonstrated a proficient ATM checkpoint response in each case. Although previous analyses based on RNA interference (RNAi)-mediated MCPH1 depletion are discordant with these findings (14, 15), these discrepancies are likely explained by the incomplete loss of MCPH1 function or the up-regulation of redundant pathways within the cellular models investigated (16). In addition to contributing to G2 checkpoint responses, MCPH1 is a component of the DNA repair machinery. It localizes to sites of double strand breaks (DSBs) (17–19), and its interaction with SWI/SNF (switch or sucrose nonfermentable) nucleosome remodeling complex chromatin remodeling complexes is required for efficient repair of DSBs (20). This function could explain the compromised cell viability after exposure to DSB-inducing agents in cells lacking MCPH1 (13, 20, 21).
Although the contribution of MCPH1 to the DNA damage checkpoint has been extensively investigated, less is known about its potential importance for other G2 checkpoints. Of particular interest is the decatenation checkpoint, a less-understood pathway that delays entry into mitosis until chromosomes are properly disentangled (decatenated) (22). This checkpoint is triggered after inhibition of DNA topoisomerase II (topo II) with catalytic inhibitors such as 4-[2-(3,5-dioxo-1-piperazinyl)-1-methylpropyl]piperazine-2,6-dione (ICRF-193) and related bisdioxopiperazines, which block topo II activity without inducing DSBs (23–25). Interestingly, mitotic cells lacking MCPH1 function show impaired sister chromatid resolution and chromosome segregation problems (7, 26), which could be indicative of a defect in the decatenation checkpoint. That is, if MCPH1-deficient cells have an attenuated decatenation checkpoint, the expected phenotype would be entry into mitosis in the presence of catenated chromatids that cannot be resolved or segregated efficiently. Moreover, ATR signaling, which is essential for the decatenation checkpoint pathway (24), requires MCPH1 function to effectively stop the cell cycle in response to UV irradiation (8).
Considering this evidence, we investigated the proficiency of the decatenation checkpoint in cells that lack MCPH1 function. Our results have demonstrated that MCPH1 function is dispensable for the temporary G2 arrest that is triggered by the decatenation checkpoint. Unexpectedly, however, we find that MCPH1 is required for the adaptive response that triggers entry into mitosis despite topo II catalytic inhibition.
MATERIALS AND METHODS
Cell cultures and treatments
We have used standard and modified (H2B-Red1–tagged) HeLa cell lines. Also, we have employed lymphoblast cell lines (nontransformed EBV immortalized) from 1 patient with MCPH1 (S25X mutation) and 1 healthy control subject (4, 5). HeLa cell lines were grown following standard conditions using DMEM supplemented with 10% fetal bovine serum. Lymphoblast cell lines were grown under usual conditions in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 15% fetal bovine serum.
For RNAi treatments, cells were transfected with 120 nM siRNA duplexes using Lipofectamine (Thermo Fisher Scientific, Waltham, MA, USA) at 50% confluency. Opti-MEM (Thermo Fisher Scientific) was used for cell transfection. The sequences of the MCPH1 and siRNA duplexes used to deplete specifically both major isoforms of MCPH1 and mRNA were based on a previous study (19). These validated siRNA oligos were purchased from Qiagen (Germantown, MD, USA) and knocked down the MCPH1 protein levels efficiently (7, 19). As a control in our experiments, we used negative scrambled siRNA duplexes (Thermo Fisher Scientific). Furthermore, as an additional control, we used cells treated under the same conditions but without adding siRNAs to the transfection mixture. Both controls showed similar cell cycle profiles under the same treatment conditions, which demonstrates that the cell cycle dynamics observed in our experiments were not induced by off-target effects or related to the transfection procedure. Synchronization of cells at G1/S was achieved by a double thymidine protocol. The inhibitors employed were nocodazole (final concentration 1.5 µM), ICRF-193 (final concentration 7 µM), caffeine (final concentration 2 mM), SB202190 (final concentration 18 µM), and etoposide (final concentration 25 µM). Untreated control cells were incubated in all cases with a similar volume of solvent.
Live-cell microscopy
The procedure was similar to one previously described (7). Cells were plated onto 35-mm tissue culture dishes fitted with glass coverslips (Cultureware; MatTek, Ashland, MA, USA). The siRNA transfection and cell synchrony were performed as described in the Results and Discussion section, except that upon release from the second thymidine the standard medium containing the thymidine was exchanged for DMEM without phenol red supplemented with 10% fetal bovine serum, penicillin or streptomycin, and 200 mM Trolox (Calbiochem, San Diego, CA, USA). The dishes were transferred to a microscope humidified stage incubator containing 5% CO2 at 37°C. Cells were filmed with 3–5 z sections using a Nikon Biostation IM microscope (Nikon, Tokyo, Japan) fitted with ×20 and 40/0.8 numerical aperture objectives and coupled with Biostation IM software. Images were stacked and processed using ImageJ (National Institutes of Health, Bethesda, MD, USA) software. Timing data were obtained after visual inspection of a minimum of 50 cells. Statistical comparisons were done using Statgraphics software (Statgraphics Technologies, The Plains, VA, USA).
Flow cytometry
Flow cytometry analyses were done using lymphoblast cell cultures in log-phase. One million cells approximately were recovered, washed in PBS, and fixed in ice-cold Ethanol 70 overnight. Phosphorylated histone H3–positive cells were detected with a rabbit anti-histone H3PS10 antibody (Abcam, Cambridge, MA, USA) at a dilution of 1:250 and a donkey anti-mouse IgG FITC-conjugated secondary antibody (Santa Cruz Biotechnology, Dallas, TX, USA). Propidium iodide was used as a counterstain for DNA content. Fluorescence detection was performed using an analytical flow cytometer (LSR Fortessa; BD Biosciences, San Jose, CA, USA) equipped with BD FACSDiva software (BD Biosciences) for data acquisition. Quantitative cell cycle analysis was done with Flowing Software v.2.5.1 as previously described (7).
Cytogenetic analyses
Cytogenetic preparations following standard protocols were obtained in parallel from the same log-phase cell cultures analyzed by flow cytometry. Chromosome preparations were fixed using Carnoy’s solution (methanol or glacial acetic acid, 3:1), stained with Giemsa (10%), and finally visualized by bright-field microscopy. The fraction of prophase-like cells (PLCs) and metaphases was determined after counting 1000 nuclei from coded slides. Microscopy images were captured with a charge-coupled device camera (DP70; Olympus, Tokyo, Japan) coupled to a microscope (BX51; Olympus) and finally managed with ImageJ software.
Immunofluorescence
Control and MCPH1 lymphoblast cells were treated with the corresponding inhibitors for 3 h and attached to glass coverslips pretreated with poly-l-lysine. HeLa cells growing directly in glass coverslips were previously synchronized at the G1/S border by double thymidine block and transfected with siRNAs during the release from the first thymidine block. The corresponding inhibitors were added 6 h after release from the second thymidine block, and cells were processed 3 h after. Cells were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 15 min at room temperature and permeabilized with ice-cold methanol for 30 min on ice. Cells were incubated with PBS containing 20% fetal bovine serum as a blocking agent for 30 min and then with mouse anti–γ-H2AX (MilliporeSigma, Burlington, MA, USA) overnight at 4°C. After being washed 3 times with PBS, cells were incubated with donkey anti-mouse IgG FITC-conjugated secondary antibody (Santa Cruz Biotechnology). After counterstaining with DAPI, coverslips were mounted with Vectashield and examined with a Zeiss Axioskop microscope (Carl Zeiss, Oberkochen, Germany) equipped with a cooled charge-coupled device camera. Grayscale images were pseudocolored and merged using ImageJ.
Western blot
Approximately 1 × 105 cells were suspended in 100 μl of lysis buffer, sonicated, and boiled for 2 min. Proteins were resolved by SDS-PAGE and transferred to Hybond-P PVDF membranes (Amersham, Little Chalfont, United Kingdom). The membrane was blocked with 2.5% (w/v) dry milk in Tris-buffered saline with Tween 20 [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.05% Tween 20]. Incubation with primary antibodies was performed in Tris-buffered saline with Tween 20 containing 1% bovine serum albumin and 0.05% sodium azide overnight at 4°C. Blots were developed by an ECL detection system (Amersham). Primary antibodies used were anti-total CDK1 (ab131450; Abcam), anti–phosphorylated Y15 CDK1 (ab47594; Abcam), and anti–α-tubulin (MilliporeSigma) as loading control.
RESULTS AND DISCUSSION
MCPH1 function is dispensable for activation of the G2 decatenation checkpoint but required to allow cellular adaptation to it
We first analyzed whether cells lacking MCPH1 function display a functional decatenation checkpoint. In order to do so, we made use of log-phase cultures of control and MCPH1 lymphoblasts, and we assayed the dynamics of mitotic entry after prolonged incubation with the topo II inhibitor ICRF-193 (Fig. 1A–D). Nocodazole was also added alone or combined with ICRF-193 to trap those cells entering into mitosis, which were detected by flow cytometry using histone H3PS10 as a mitotic marker. Our data revealed almost no accumulation of mitotic cells during the first 6 h of incubation in both control and patient cells (Fig. 1B) in comparison with the progressive accumulation observed when only nocodazole was added (Fig. 1A). Therefore, in response to topo II inhibition, an efficient G2 arrest is triggered in both control and patient cells (Fig. 1B, E–H). These results are in accordance with previous studies that demonstrated a proficient decatenation checkpoint in human lymphoblastoid cell lines (24, 27) and suggest that MCPH1 function is not required for decatenation checkpoint activation. Interestingly, this checkpoint is notoriously leaky, and G2 cells treated with ICRF-193 gradually enter mitosis with catenated, unresolved chromatids after delaying in G2 only transiently (23). This leakiness could be observed in control cells as the mitotic index increased notoriously over time following 6 h of incubation with ICRF-193. However, MCPH1 patient cells did not leak into mitosis at a rate similar to control cells. Instead, the mitotic index remained fairly constant during the time course of the experiment (Fig. 1B). These results suggest that MCPH1 cells are defective in the adaptive response that spontaneously bypasses the decatenation checkpoint G2 arrest.
Figure 1.

A–D) Frequency of histone H3PS10–positive cells in control and MCPH1 cells determined by flow cytometry after incubation with nocodazole alone (A) or combined with either ICRF-193 (B), caffeine (C), or both (D) for the indicated time points. Mean and range (bars) data from 2 independent experiments are presented. Pooled data from control and patient cells were compared by χ2 test of independence. E–H) Fold increase of G2 cell fraction, related to untreated samples, in control and MCPH1 cells incubated as described in A–D during 3 h (E), 6 h (F), 10 h (G), and 13 h (H). Mean and range (bars) data from 2 independent experiments are presented. For each time point, pooled data for each treatment were compared independently in control and patient cells by χ2 test of independence to nocodazole-treated cells. Furthermore, for each treatment and time point, control and patient data were pairwise compared by χ2 test of independence (underlined). CAFF, caffeine; NOC, nocodazole; ns, nonsignificant. *P < 0.01, **P < 0.001.
We next investigated the dynamics of mitotic entry after forced bypass of the decatenation checkpoint arrest. In order to do that, we made use of caffeine, a well-known inhibitor of ATM and ATR kinases, which override the ICRF-193–imposed G2 arrest (24). As shown in Fig. 1C, single incubation with caffeine did not alter the rate of mitotic entry in both control and patient cells. When caffeine was simultaneously combined with ICRF-193, however, we observed a striking inability of MCPH1 cells to enter mitosis compared with control cells. Thus, control cells bypassed the checkpoint G2 arrest and immediately entered into mitosis at a rate similar to untreated samples or caffeine-treated samples (Fig. 1D–H). However, patient cells had a prolonged arrest in G2, and only after 10 h of incubation did mitotic cells start to accumulate (Fig. 1D–H). When we analyzed the chromosome structure of control cells treated with ICRF-193 and caffeine, we observed a high frequency of mitotic cells with tangled, unresolved, and uncondensed chromosomes. Those chromosome alterations, which are a consequence of cells entering mitosis without active topo II (23, 27, 28), were barely observed in MCPH1 cells. Instead, an elevated frequency of PLCs was observed.
The prominent adaptive response of the decatenation checkpoint is not well understood. No cell lines have been examined in which cells permanently arrest in G2 in the presence of topo II inhibitors. Because MCPH1 cells lack this adaptive response, MCPH1 may be a crucial factor required for decatenation checkpoint bypass. We therefore sought to test directly whether MCPH1 is a component of this adaptation response. We employed HeLa cells depleted of MCPH1 function by siRNA because this strategy has been previously characterized (7). HeLa H2B-Red1 cells synchronized at G1/S using excess thymidine were transfected with siRNAs against MCPH1 and then monitored by fluorescent live-cell microscopy upon release into the cell cycle (experimental outline in Fig. 2A). We previously used this approach to demonstrate that the timing of mitotic entry is similar in control and MCPH1-depleted cells following release from the G1/S arrest (7). Here, ICRF-193 was added 7 h after release from the thymidine block at the time when cells are already in mid-G2 phase (7). Caffeine was added to parallel samples 1 h after ICRF-193. As depicted in Fig. 2B, control cells efficiently activated the G2 decatenation checkpoint in the presence of ICRF-193 with typical kinetics. That is, topo II inhibition immediately blocked entry of cells into mitosis and then, following 3 h, some cells began to spontaneously adapt to the arrest. As expected from the lack of functional topo II, these cells exhibited chromosome segregation problems (Fig. 2C). Therefore, robust decatenation checkpoint activation followed by checkpoint adaptation was readily observed. This was important to establish using the approach because decatenation checkpoint kinetics vary significantly between different human cell lines (22). Caffeine efficiently bypassed the G2 decatenation checkpoint as most control cells treated with ICRF-193 entered into mitosis without delay (Fig. 2B, D). Like in control cells, HeLa H2B-Red1 cells depleted of MCPH1 by RNAi efficiently activated the G2 decatenation checkpoint (Fig. 2B, C). However, unlike the controls but consistent with the MCPH1 cells, the MCPH1-depleted cells did not adapt to the decatenation checkpoint because no spontaneous override was observed. In the presence of caffeine, there was no immediate entry into mitosis as occurred in control cells. Strikingly, in this case, nearly 50% of cells remained arrested in G2 for many hours (Fig. 2B, D). Thus, unlike control cells, spontaneous decatenation checkpoint adaptation was severely impaired, and the ability of caffeine to induce adaptation in the presence of ICRF-193 was strongly perturbed in MCPH1-depleted cells. Single incubation with caffeine does not alter cell cycle progression and only produced a slight increase in the rate of mitotic entry in MCPH1-depleted cells (Fig. 2E).
Figure 2.
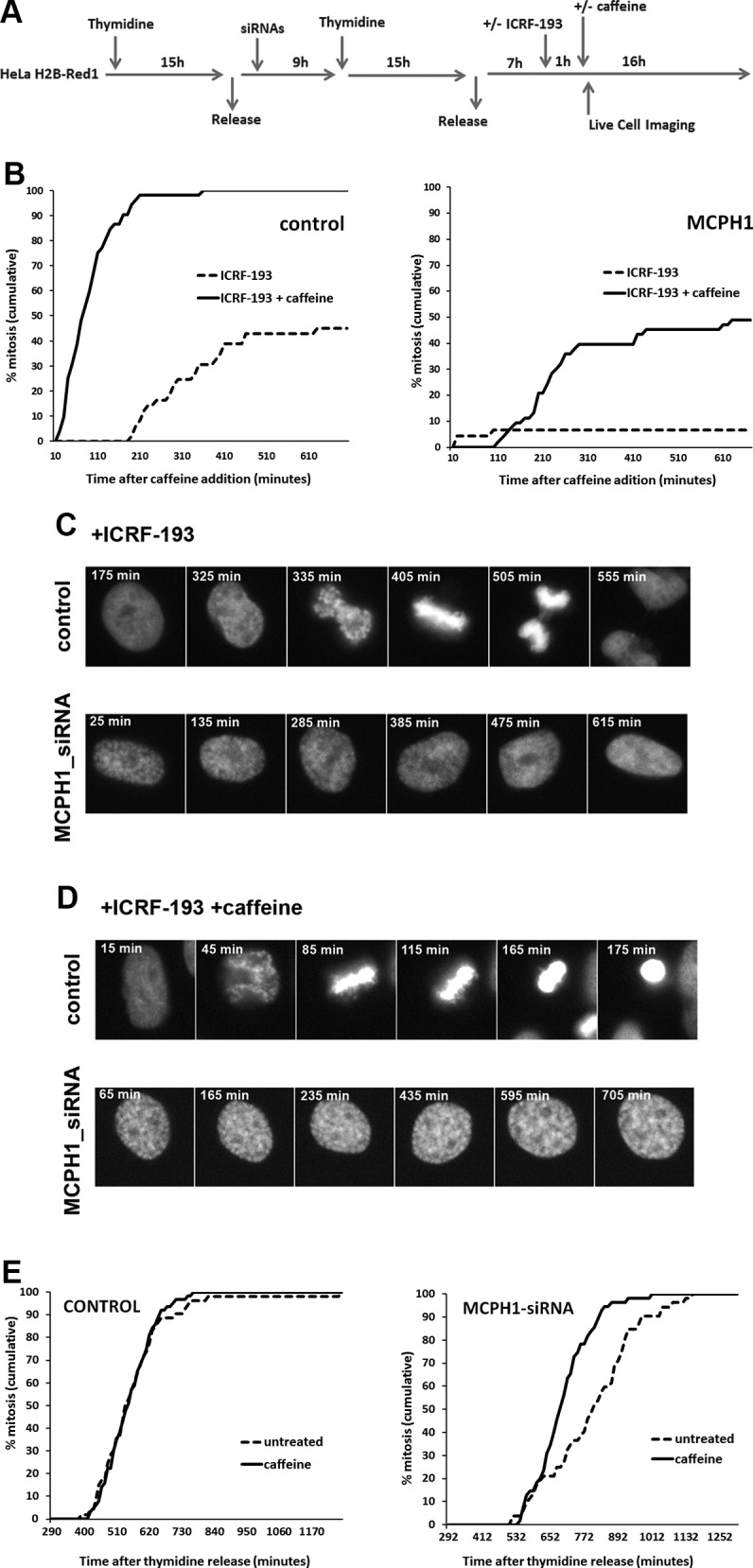
A) Description of the experimental procedure employed. HeLa cells stably expressing fluorescent histone H2B fused to Red1 were synchronized at the G1/S border by double thymidine block. Transfection with siRNAs was performed during the release from the first thymidine block. ICRF-193 was added 7 h after release from the second thymidine block to coincide with the occurrence of PLCs during G2 in the siRNA-treated cells. Caffeine (or an equal volume of medium) was added 1 h after ICRF-193 treatment. Time-lapse images were collected using a Nikon Biostation IM Cell Incubator immediately after caffeine (or medium) adding. B) Cumulative frequency chart showing the timing (min) of mitosis onset after the indicated treatments in cells transfected with either control scrambled or MCPH1 siRNAs and monitored in A. Time after caffeine (or solvent) addition is shown. Timing data were obtained after visual inspection of mitosis onset, revealed by nuclear envelope breakdown, of a minimum of 50 cells. Cells treated under the same conditions but without adding siRNAs to the transfection mixture showed cell cycle dynamics similar to the scrambled siRNA–transfected cells (unpublished results). C, D) Selected frames showing the mitotic progression of representative control and MCPH1-depleted HeLa cells treated with ICRF-193 alone (C) or combined with caffeine (D) as indicated in A. Time form caffeine (or medium) addition is indicated in minutes. E) Cumulative frequency chart showing the timing (min) of mitosis onset after incubation with caffeine or solvent (untreated). Time after release from the second thymidine block is shown. Caffeine was added 200 min after the release. At least 50 cells were analyzed in each case. Control cells were treated under the same conditions as MCPH1-siRNA–treated cells but without adding siRNAs to the transfection mixture.
Overall, our results demonstrate that MCPH1 function is dispensable for decatenation checkpoint activation but required for decatenation checkpoint adaptation. Our finding that MCPH1 is dispensable for activation and maintenance of the G2 decatenation checkpoint was a surprising outcome. The decatenation checkpoint depends on the ATR signaling pathway (24), and the ATR pathway requires MCPH1 function to efficiently stop the cell cycle in response to UV irradiation (8). Taken together, these data support the idea that although the pathways responding to either UV irradiation or catalytic inhibition of topo II both depend on ATR, they are nevertheless distinct in their mechanisms of activation and some of their signaling components.
The G2 decatenation checkpoint induces decondensation in PLCs dependent on the ATM and ATR pathway in MCPH1-deficient cells
Chromosome condensation is markedly uncoupled from mitosis in MCPH1-deficient cells, covering part of G2 and G1 phases of the cell cycle (7, 8). This altered timing explained the increased number of nonmitotic cells with visible chromosome condensation, referred to as PLCs (4) (representative example depicted in Fig. 3B). Therefore, we next analyzed the condensation dynamics of G2 cells lacking MCPH1 function during decatenation checkpoint activation. Our analyses showed that PLCs do not increase in parallel with the G2 fraction during prolonged incubation with ICRF-193. However, if caffeine was added simultaneously, PLCs clearly increased over time in parallel with the accumulation of cells in G2 phase (Fig. 3A). In control cells, PLCs were rarely observed, as expected. We next recapitulated these experiments in synchronized HeLa H2B-Red1 cells depleted of MCPH1 by siRNAs. During the incubation with ICRF-193, condensation was progressively abolished, and PLCs lacked any sign of condensation after 386 ± 126 min (Fig. 3C, D). However, when caffeine was added simultaneously with ICRF-193, condensation persisted in the PLCs for much longer (734 ± 235 min), and many cells even remained with condensed chromatin at the end of the movie (>1000 min) (Fig. 3C, D). Taken together, our results indicate that upon prolonged G2 arrest induced by topo II inhibition, cells lacking MCPH1 go through chromosome decondensation progressively unless ATR or ATM is inhibited by caffeine.
Figure 3.

A) Percentage of PLCs and G2 cells in control and MCPH1 lymphoblastoid cells observed after either 3 h (left) or 6 h (right) with the indicated treatments. PLCs were determined by microscopic analysis of cytogenetic preparations prepared simultaneously from the same samples shown in Fig. 1. More than 500 cells were scored per sample. G2 fraction was obtained by flow cytometry. Mean and range (bars) data from 2 independent experiments are presented. For each time point, pooled data for each treatment were compared independently in control and patient cells by c2 test of independence to nocodazole-treated cells. Furthermore, for each treatment and time point, control and patient data were pairwise compared by c2 test of independence (underlined). *P < 0.01, **P < 0.001. B) Representative images from cytogenetic preparations of control and MCPH1 cells treated simultaneously with ICRF-193 and caffeine. Arrows point to PLCs, a cellular phenotype characteristic of MCPH1 lack of function that is defined by visible chromosome condensation and intact nuclear envelope. Arrow heads point to cells with tangled, unresolved, and uncondensed chromosomes, characteristic of cells entering mitosis without topo II function. Scale bars, 5 μM. C) Box-plots showing the time (in minutes) that PLCs from MCPH1-siRNA–treated HeLa cells required to completely decondense their chromosomes after adding ICRF-193 alone or combined with caffeine. The red line indicates the mean value. Thirty PLCs were monitored. Statistical comparisons for the mean and median data were done by Student’s t and Wilcoxon tests, respectively. **P < 0.01. D) Selected frames showing the condensation dynamics of MCPH1-siRNA–treated HeLa cells while incubated with ICRF-193 alone or combined with caffeine as explained in Fig. 2A. Note that the PLC phenotype of the cells, visible from the first frames, is progressively reduced in ICRF-193–treated but not in ICRF-193– and caffeine-treated cells. Time from caffeine addition is indicated in minutes. I, ICRF-193; C, caffeine; ns, nonsignificant; untr., untreated. E) Immunoblot analyses of CDK1 levels in control and MCPH1 cells after incubation with ICRF-193 or combined with caffeine.
PLCs have prematurely condensed chromatin in the G2 phase because MCPH1 deficiency results in inappropriate CDK1 activation in G2 (7, 8). Therefore, chromosome decondensation induced by ICRF-193 treatment would be an expected consequence if prolonged activation of the decatenation checkpoint ultimately abolishes CDK1 activation. This hypothetical scenario would also explain why decondensation is prevented when this pathway is inhibited by caffeine. To verify this hypothesis, we compared the protein levels of total and phosphorylated Y15 (inactive) CDK1 in control and patient samples (Fig. 3E). After 6 h of ICRF-193 treatment, the levels of inactive CDK1 increased in both control and patient cells, whereas simultaneous addition of caffeine reduced those levels enormously, as expected. Therefore, in patient cells, the duration of chromosome condensation correlates inversely with the levels of inactive CDK1. By contrast, the levels of total CDK1 remained quite similar in all cases. Our data thus confirm that PLCs progressively reverse their condensation state while arrested in G2 by decatenation checkpoint activation. However, decondensation is prevented by caffeine because the ATR and ATM signaling pathways initiated by the decatenation checkpoint are required for sustaining CDK1 inactivation, and thus decondensation, in PLCs. Of interest, the levels of inactive phosphorylated Y15 CDK1 were increased after 3 h of ICRF-193 incubation in patient but not in control cells (Fig. 3E). Because a robust G2 arrest was equally observed during the first hours of ICRF-193 incubation in both (see Fig. 1A, D), this result suggests that the ICRF-193–induced checkpoint arrest occurs despite substantial CDK1 activity, in line with previous reports (29).
ICRF-193–induced G2 arrest is not dependent on the p38 MAPK pathway in MCPH1-deficient cells
MCPH1-deficient cells condense their chromosomes in G2 because of premature Cdk1 activation to a level that is enough to start condensation but is insufficiently high to induce entry into mitosis. Indeed, MCPH1-deficient cells perform nuclear envelope breakdown and enter prometaphase with the same timing as controls cells (7). The lack of decatenation checkpoint bypass in the presence of ICRF-193 and caffeine suggest that Cdk1 activity does not reach the threshold needed for mitosis, and this indicates that MCPH1 might act between ATM or ATR and Cdk1. Another explanation is the possibility that MCPH1 deficiency and topo II inhibition synergize in a more complex manner, perhaps leading to the activation of another G2 checkpoint pathway. One such pathway is the antephase checkpoint. We considered this in part because PLCs decondense their chromosomes while arrested in G2 in the presence of ICRF-193, and this phenotype resembles the activity of the G2/M antephase checkpoint. This checkpoint acts at the end of G2 in response to a variety of cellular stresses, including hypotonic shock, UV irradiation, and chromatin topological perturbations, and triggers chromatin decondensation in early prophase cells (30, 31). It is distinct from DNA damage response checkpoints because it does not require ATM or ATR; however, it is critically dependent on p38 MAPK signaling (31, 32). These facts, together with the caffeine-insensitive nature of the G2 arrest triggered by ICRF-193 in MCPH1-deficient cells, prompted us to examine whether the persistent G2 arrest could be explained by activation of p38 MAPK signaling. In order to test this, we inhibited p38 MAPK with the well-characterized inhibitor SB202190 (33) in control and MCPH1 patient lymphoblastoid cells and determined whether checkpoint adaptation is restored. Flow cytometry analyses revealed that SB202190 did not abolish the G2 arrest imposed by ICRF-193 in either control or MCPH1-deficient cells (Fig. 4A). Therefore, the G2 arrest is independent of p38 kinase signaling. To corroborate the results in patient cells, experiments were next performed in HeLa H2B-Red1 cells depleted of MCPH1 using siRNAs. Cells synchronized at the G1/S transition were released, and 7 h later, ICRF-193 was added, followed by addition of SB202190 2 h later (Fig. 4B). Our results revealed that MCPH1 siRNA–treated cells did not perform nuclear envelope breakdown and remained arrested in G2 at the end of the movie, whereas in control cells a small fraction was able to override the ICRF-193–induced G2 arrest and enter mitosis during the incubation period (Fig. 4C, D). Therefore, like in the MCPH1 cells, the HeLa cells depleted of MCPH1 did not arrest in G2 dependent on the p38 kinase antephase checkpoint. Importantly, SB202190 alone did not affect the progression through G2 and the rate of mitosis entrance in the lymphobast and HeLa cell models investigated (Fig. 4A, E), in accordance with previous reports (33, 34). Taken together, our results show that p38 MAPK is unlikely to trigger G2 arrest upon ICRF-193 treatment either in MCPH1-deficient cells or in control cells, in agreement with some previous studies (22).
Figure 4.

A) Frequency of histone H3PS10–positive cells in control and MCPH1 cells determined by flow cytometry after incubation with nocodazole alone or combined with either SB202190 or SB202190 and ICRF-193 for the indicated time points. Mean and range (bars) data from 2 independent experiments are presented. For each time point, pooled data for each treatment were compared independently in control and patient cells by χ2 test of independence to untreated cells. Furthermore, for each treatment and time point, control and patient data were pairwise compared by χ2 test of independence (underlined). B) Experimental procedure employed in HeLa H2B-Red1 cells to monitor cell cycle progression by live-cell microscopy after incubation with ICRF-193 and SB202190. C) Selected frames showing the cell cycle dynamic of MCPH1 siRNA–treated HeLa cells while incubated with ICRF-193 and SB202190. Time from SB202190 addition is indicated in minutes. D) Cumulative frequency chart showing the timing (in minutes) of mitosis onset for cells transfected with either control scrambled or MCPH1 siRNAs and treated as explained in B. Time after SB202190 addition is shown. At least 50 cells were analyzed in each case. Cells treated under the same conditions but without adding siRNAs to the transfection mixture showed similar cell cycle dynamics to the scrambled siRNA–transfected cells (unpublished results). E) Cumulative frequency chart showing the timing (min) of mitosis onset after single incubation with SB202190. Time after release from the second thymidine block is shown. SB202190 was added 300 min after the release. Control cells were treated under the same conditions as MCPH1-siRNA–treated cells but without adding siRNAs to the transfection mixture. At least 50 cells were analyzed in each case. Note that all cells were later arrested in mitosis as a consequence of p38 inhibition. NOC, nocodazole; ns, nonsignificant. **P < 0.001.
G2 arrest after DNA damage induced by topo II poisons requires ATR and ATM in MCPH1-deficient cells
ICRF-193 is a catalytic inhibitor of topo II that blocks the strand passage reaction without causing DNA breakage. In contrast, there are chemicals known as topo II poisons that inhibit the strand passage reaction by trapping the covalent enzyme DNA complex in such a way as to generate massive DNA breakage in cells (22). In this way, topo II poisons arrest cells in G2 exclusively because of DNA damage checkpoint activation that requires ATM and ATR (25, 35, 36). Studies have revealed that the canonical ATM-dependent G2 checkpoint that responds to DNA damage induced by irradiation functions normally in cells lacking MCPH1 (6, 11, 13). Therefore, we sought to determine whether MCPH1-deficient cells respond like control cells when treated with topo II poisons—that is, whether, unlike the caffeine-insensitive G2 arrest triggered by ICRF-193, MCPH1-depleted cells would behave like control cells after incubation with topo II poisons. To test this, we examined cell cycle dynamics in control and MCPH1 cells incubated with etoposide (alone or combined with caffeine), an extensively studied topo II poison that is used widely as a cancer therapeutic, for 4 h. Nocodazole was included to arrest cells in mitosis. As shown in Fig. 5A, mitosis entrance is reduced in both control and patient cells in the presence of etoposide; however, the G2 arrest is efficiently bypassed by caffeine.
Figure 5.

A) Fraction of histone H3PS10–positive cells, determined by flow cytometry, in control and MCPH1 cells after 4 h of incubation with nocodazole alone or combined with either etoposide or etoposide and caffeine. Mean and range (bars) data from 2 independent experiments are presented. Pooled data for each treatment were compared independently in control and patient cells by χ2 test of independence to untreated cells. Furthermore, for each treatment control and patient, data were pairwise compared by χ2 test of independence (underlined). B) Experimental procedures employed in HeLa H2B-Red1 cells to monitor cell cycle progression by live-cell microscopy after incubation with etoposide alone or combined with caffeine. C) Cumulative frequency chart showing the timing (in minutes) of mitosis onset for cells transfected with either control scrambled or MCPH1- siRNAs and treated with etoposide and caffeine as explained in B. Time after caffeine addition is shown. At least 50 cells were analyzed in each case. Cells treated under the same conditions but without adding siRNAs to the transfection mixture showed similar cell cycle dynamics to scrambled siRNA–transfected cells (unpublished results). D) Cumulative frequency chart showing the timing (min) of mitosis onset after single incubation with etoposide. Time after solvent addition, which replaced caffeine, is shown. Control cells were treated under the same conditions as MCPH1-siRNA–treated cells but without adding siRNAs to the transfection mixture. At least 50 cells were analyzed in each case. E, F) Selected frames showing the mitotic progression of representative control and MCPH1-depleted HeLa cells treated with etoposide and caffeine (E) or etoposide alone (F) as indicated in B. Time from etoposide addition is indicated in minutes. Note that in control and MCPH1-depleted cells treated with etoposide and caffeine, cells enter mitosis immediately, and chromosome segregation is further altered probably as a consequence of the DNA damage induced by etoposide. NOC, nocodazole; ns, nonsignificant. *P < 0.01, **P < 0.001.
Similar results were obtained after live-cell analyses of HeLa H2B-Red1 cells depleted of MCPH1 function by siRNAs. Cells synchronized at the G1/S transition were released, and etoposide was added 7 h later. Caffeine, or an equal volume of medium, was added 1 h after the etoposide (Fig. 5B). In this case, caffeine induced rapid entrance into mitosis in control and MCPH1-depleted, etoposide-treated cells (Fig. 5C, E). Without caffeine, etoposide-treated cells remained arrested in G2 (Fig. 5D, F). Therefore, the DNA damage response induced by a topo II poison was efficiently activated in MCPH1-deficient cells, and, moreover, this G2 checkpoint arrest was entirely reliant on the ATM and ATR kinases that are inhibited by caffeine.
In order to verify that catalytic inhibition of topo II with ICRF-193, in contrast to etoposide, did not induce a DNA damage response in MCPH1 cells, we performed immunofluorescence analyses using a well-recognized DNA damage marker, γ-H2AX, on cells treated with either ICRF-193 or etoposide. As shown in Fig. 6, both control and MCPH1 cells exhibited a spectacular increase in γ-H2AX–positive cells after 3 h of etoposide treatment; however, 3 h of incubation with ICRF-193 induced no detectable γ-H2AX staining above background levels in control cells, whereas in patient cells a very minor increase was observed. Similar results were also observed in HeLa cells depleted of MCPH1 function by siRNAs (unpublished results). These results indicate that ICRF-193 does not induce massive DSB formation either in control cells, in agreement with previous reports (25, 36), or in cells lacking MCPH1 function.
Figure 6.

A, B) Representative images from immunofluorescence analyses using an antibody against γ-H2AX (MilliporeSigma) in proliferating lymphoblasts from control (A) and MCPH1 cells (B) treated with either DMSO (untreated), ICRF-193, or etoposide. Scale bars, 5 μM. C) Fraction of cells containing more than 5 γ-H2AX foci from the described analyses in A, B. At least 200 cells from 2 independent experiments were counted; mean and range (bars) data are presented. Pooled data for each treatment were compared independently in control and patient cells by χ2 test of independence to untreated cells. Furthermore, for each treatment, control and patient data were pairwise compared by χ2 test of independence (underlined). Ns, nonsignificant. *P < 0.01, **P < 0.001.
Together, our results demonstrate that the checkpoint response to DNA damage induced by etoposide is caffeine-sensitive in MCPH1-depleted cells. Thus, although MCPH1 function is essential for G2 checkpoint adaptation in the presence of ICRF-193, it is dispensable for bypass of the ATM-dependent G2 checkpoint that operates in response to etoposide-induced DNA damage. Our results are in accordance with previous irradiation analyses in patient cells (11) and with other studies that reported a canonical ATM-based checkpoint response in MCPH1-depleted cells (6, 12, 13). Importantly, the opposite results observed here with ICRF-193 and etoposide, both specific topo II inhibitors, provide support to the notion that the G2 DNA damage checkpoint and the decatenation checkpoint are distinct signaling pathways (13, 22, 36).
Because cells entering mitosis with altered DNA are a source of genomic instability, checkpoint adaptation is considered a potential mechanism contributing to tumorigenesis and cancer resistance during genotoxic therapy (37, 38). Nevertheless, the genetic requirements that allow cellular adaptation are poorly understood. Our current data highlight that G2 cells challenged by DNA catenation present different requirements for mitotic entry compared with cells damaged by UV or ionizing radiation, with MCPH1 function being essential only in the first scenario.
Finally, from our results it could be concluded that the deficiencies during chromosome resolution and segregation previously reported in cells lacking MCPH1 function (7, 26) are not a direct consequence of a weakened G2 decatenation checkpoint as originally thought. This was important to establish because decatenation failure is proposed to be the pathogenic mechanism that accounts for microcephaly in patients with condensin II–mutated forms (39), and a similar mechanism could hypothetically contribute to the pathogenesis of MCPH1 syndrome. This assumption relies on some observations, as do the interaction of MCPH1 with condensin II and the alterations on chromosome structure and segregation as previously described (7, 26, 40). Despite MCPH1-deficient cells displaying a robust G2 decatenation checkpoint, further catenation problems arising at mitosis would not be corrected by this mechanism. In this scenario, a mitotic signaling pathway depending of PKC-ε is responsible for monitoring and resolving chromosome catenations (41). Given this context, further investigations are required to elucidate whether the observed mitotic deficiencies in MCPH1 syndrome are related to an excess of catenation that arises exclusively during mitosis or are the result of a different deficiency.
ACKNOWLEDGMENTS
The authors express gratitude to H. Neitzel (Charite Virchow-Klinikum Hospital, Berlin, Germany) for providing the lymphoblast cell lines used in this study, G. Marques (University of Minnesota–Minneapolis, Minneapolis, MN, USA) for technical assistance, and J. F. Gimenez-Abián [Centre for Biological Research (CIB), Madrid, Spain] and V. Rodriguez-Bravo (Sidney Kimmel Cancer Center, Baltimore, MD, USA) for helpful discussions. Technical and human support provided by Centro de Instrumentación Científico-Técnica [CICT; Universidad de Jaén, Ministry of Economy and Competitiveness (MINECO), Junta de Andalucía, Federación Española de Enfermedades Raras (FEDER)] is gratefully acknowledged. This work was supported by Junta de Andalucía (funding program Ayudas a Grupos de Investigación, BIO 220). M.A. was provided with travelling grants to perform short-term stays at the University of Minnesota by EMBO and Escuela de Doctorado (UJA), respectively. Research at the laboratory of R.K. was financially supported by the National Science Foundation (MCB1140033). Studies performed in the laboratory of D.J.C. were funded by U.S. National Institutes of Health (NIH), National Institute of General Medical Sciences Grants R01GM112793 and R01GM130858. The authors declare no conflicts of interest.
Glossary
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia and rad3 related
- Cdc25A
cell division cycle 25A
- CDK1
cyclin-dependent kinase 1
- CHK1
checkpoint kinase 1
- DSB
double strand break
- ICRF-193
4-[2-(3,5-dioxo-1-piperazinyl)-1-methylpropyl]piperazine-2,6-dione
- MCPH1
microcephalin 1
- PLC
prophase-like cell
- RNAi
RNA interference
- topo II
topoisomerase II
AUTHOR CONTRIBUTIONS
D. J. Clarke and J. A. Marchal designed the experiments; M. Arroyo, I. Guerrero, D. Keifenheim, J. Calahorra, and J. A Marchal performed the experiments; M. Arroyo, R. Kuriyama, A. Cañuelo, A. Sánchez, and J. A. Marchal analyzed the results; D. J. Clarke and J. A. Marchal wrote the main manuscript text; and all authors reviewed the manuscript.
REFERENCES
- 1.Van Vugt M. A. T. M., Brás A., Medema R. H. (2004) Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 15, 799–811 [DOI] [PubMed] [Google Scholar]
- 2.Paulovich A. G., Toczyski D. P., Hartwell L. H. (1997) When checkpoints fail. Cell 88, 315–321 [DOI] [PubMed] [Google Scholar]
- 3.Serrano D., D’Amours D. (2016) Checkpoint adaptation: keeping Cdc5 in the T-loop. Cell Cycle 15, 3339–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neitzel H., Neumann L. M., Schindler D., Wirges A., Tönnies H., Trimborn M., Krebsova A., Richter R., Sperling K. (2002) Premature chromosome condensation in humans associated with microcephaly and mental retardation: a novel autosomal recessive condition. Am. J. Hum. Genet. 70, 1015–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trimborn M., Bell S. M., Felix C., Rashid Y., Jafri H., Griffiths P. D., Neumann L. M., Krebs A., Reis A., Sperling K., Neitzel H., Jackson A. P. (2004) Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am. J. Hum. Genet. 75, 261–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gruber R., Zhou Z., Sukchev M., Joerss T., Frappart P. O., Wang Z. Q. (2011) MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat. Cell Biol. 13, 1325–1334 [DOI] [PubMed] [Google Scholar]
- 7.Arroyo M., Kuriyama R., Trimborn M., Keifenheim D., Cañuelo A., Sánchez A., Clarke D. J., Marchal J. A. (2017) MCPH1, mutated in primary microcephaly, is required for efficient chromosome alignment during mitosis. Sci. Rep. 7, 13019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alderton G. K., Galbiati L., Griffith E., Surinya K. H., Neitzel H., Jackson A. P., Jeggo P. A., O’Driscoll M. (2006) Regulation of mitotic entry by microcephalin and its overlap with ATR signalling. Nat. Cell Biol. 8, 725–733 [DOI] [PubMed] [Google Scholar]
- 9.Tibelius A., Marhold J., Zentgraf H., Heilig C. E., Neitzel H., Ducommun B., Rauch A., Ho A. D., Bartek J., Krämer A. (2009) Microcephalin and pericentrin regulate mitotic entry via centrosome-associated Chk1. J. Cell Biol. 185, 1149–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaplet M., Rai R., Jackson-Bernitsas D., Li K., Lin S. Y. (2006) BRIT1/MCPH1: a guardian of genome and an enemy of tumors. Cell Cycle 5, 2579–2583 [DOI] [PubMed] [Google Scholar]
- 11.Gavvovidis I., Pöhlmann C., Marchal J. A., Stumm M., Yamashita D., Hirano T., Schindler D., Neitzel H., Trimborn M. (2010) MCPH1 patient cells exhibit delayed release from DNA damage-induced G2/M checkpoint arrest. Cell Cycle 9, 4893–4899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown J. A., Bourke E., Liptrot C., Dockery P., Morrison C. G. (2010) MCPH1/BRIT1 limits ionizing radiation-induced centrosome amplification. Oncogene 29, 5537–5544 [DOI] [PubMed] [Google Scholar]
- 13.Zhou Z. W., Tapias A., Bruhn C., Gruber R., Sukchev M., Wang Z. Q. (2013) DNA damage response in microcephaly development of MCPH1 mouse model. DNA Repair (Amst.) 12, 645–655 [DOI] [PubMed] [Google Scholar]
- 14.Xu X., Lee J., Stern D. F. (2004) Microcephalin is a DNA damage response protein involved in regulation of CHK1 and BRCA1. J. Biol. Chem. 279, 34091–34094 [DOI] [PubMed] [Google Scholar]
- 15.Rai R., Dai H., Multani A. S., Li K., Chin K., Gray J., Lahad J. P., Liang J., Mills G. B., Meric-Bernstam F., Lin S. Y. (2006) BRIT1 regulates early DNA damage response, chromosomal integrity, and cancer. Cancer Cell 10, 145–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mekel-Bobrov N., Gilbert S. L., Evans P. D., Vallender E. J., Anderson J. R., Hudson R. R., Tishkoff S. A., Lahn B. T. (2005) Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science 309, 1720–1722 [DOI] [PubMed] [Google Scholar]
- 17.Wood J. L., Singh N., Mer G., Chen J. (2007) MCPH1 functions in an H2AX-dependent but MDC1-independent pathway in response to DNA damage. J. Biol. Chem. 282, 35416–35423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeffers L. J., Coull B. J., Stack S. J., Morrison C. G. (2008) Distinct BRCT domains in Mcph1/Brit1 mediate ionizing radiation-induced focus formation and centrosomal localization. Oncogene 27, 139–144 [DOI] [PubMed] [Google Scholar]
- 19.Gavvovidis I., Rost I., Trimborn M., Kaiser F. J., Purps J., Wiek C., Hanenberg H., Neitzel H., Schindler D. (2012) A novel MCPH1 isoform complements the defective chromosome condensation of human MCPH1-deficient cells. PLoS One 7, e40387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng G., Lin S. Y. (2009) The linkage of chromatin remodeling to genome maintenance: contribution from a human disease gene BRIT1/MCPH1. Epigenetics 4, 457–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang Y., Gao H., Lin S. Y., Goss J. A., Du C., Li K. (2015) Mcph1/Brit1 deficiency promotes genomic instability and tumor formation in a mouse model. Oncogene 34, 4386–4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Damelin M., Bestor T. H. (2007) The decatenation checkpoint. Br. J. Cancer 96, 201–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Downes C. S., Clarke D. J., Mullinger A. M., Giménez-Abián J. F., Creighton A. M., Johson R. T. (1994) A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells/. Nature 372, 467–470; erratum: 710 [DOI] [PubMed] [Google Scholar]
- 24.Deming P. B., Cistulli C. A., Zhao H., Graves P. R., Piwnica-Worms H., Paules R. S., Downes C. S., Kaufmann W. K. (2001) Proc. Natl. Acad. Sci. USA The human decatenation checkpoint. 98, 12044–12049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo K., Yuan J., Chen J., Lou Z. (2009) Topoisomerase IIalpha controls the decatenation checkpoint. Nat. Cell Biol. 11, 204–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arroyo M., Trimborn M., Sánchez A., Hirano T., Neitzel H., Marchal J. A. (2015) Chromosome structure deficiencies in MCPH1 syndrome. Chromosoma 124, 491–501 [DOI] [PubMed] [Google Scholar]
- 27.Bower J. J., Zhou Y., Zhou T., Simpson D. A., Arlander S. J., Paules R. S., Cordeiro-Stone M., Kaufmann W. K. (2010) Revised genetic requirements for the decatenation G2 checkpoint: the role of ATM. Cell Cycle 9, 1617–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giménez-Abián J. F., Clarke D. J., Devlin J., Giménez-Abián M. I., De la Torre C., Johnson R. T., Mullinger A. M., Downes C. S. (2000) Premitotic chromosome individualization in mammalian cells depends on topoisomerase II activity. Chromosoma 109, 235–244 [DOI] [PubMed] [Google Scholar]
- 29.Deming P. B., Flores K. G., Downes C. S., Paules R. S., Kaufmann W. K. (2002) ATR enforces the topoisomerase II-dependent G2 checkpoint through inhibition of Plk1 kinase. J. Biol. Chem. 277, 36832–36838 [DOI] [PubMed] [Google Scholar]
- 30.Rieder C. L., Cole R. (2000) Microtubule disassembly delays the G2-M transition in vertebrates. Curr. Biol. 10, 1067–1070 [DOI] [PubMed] [Google Scholar]
- 31.Mikhailov A., Shinohara M., Rieder C. L. (2004) Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J. Cell Biol. 166, 517–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsusaka T., Pines J. (2004) Chfr acts with the p38 stress kinases to block entry to mitosis in mammalian cells. J. Cell Biol. 166, 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee K., Kenny A. E., Rieder C. L. (2010) P38 mitogen-activated protein kinase activity is required during mitosis for timely satisfaction of the mitotic checkpoint but not for the fidelity of chromosome segregation. Mol. Biol. Cell 21, 2150–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kukkonen-Macchi A., Sicora O., Kaczynska K., Oetken-Lindholm C., Pouwels J., Laine L., Kallio M. J. (2011) Loss of p38gamma MAPK induces pleiotropic mitotic defects and massive cell death. J. Cell Sci. 124, 216–227 [DOI] [PubMed] [Google Scholar]
- 35.Clarke D. J., Vas A. C., Andrews C. A., Díaz-Martínez L. A., Giménez-Abián J. F. (2006) Topoisomerase II checkpoints: universal mechanisms that regulate mitosis. Cell Cycle 5, 1925–1928 [DOI] [PubMed] [Google Scholar]
- 36.Bower J. J., Karaca G. F., Zhou Y., Simpson D. A., Cordeiro-Stone M., Kaufmann W. K. (2010) Topoisomerase IIalpha maintains genomic stability through decatenation G(2) checkpoint signaling. Oncogene 29, 4787–4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Syljuåsen R. G. (2007) Checkpoint adaptation in human cells. Oncogene 26, 5833–5839 [DOI] [PubMed] [Google Scholar]
- 38.Serrano D., D’Amours D. (2014) When genome integrity and cell cycle decisions collide: roles of polo kinases in cellular adaptation to DNA damage. Syst. Synth. Biol. 8, 195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin C. A., Murray J. E., Carroll P., Leitch A., Mackenzie K. J., Halachev M., Fetit A. E., Keith C., Bicknell L. S., Fluteau A., Gautier P., Hall E. A., Joss S., Soares G., Silva J., Bober M. B., Duker A., Wise C. A., Quigley A. J., Phadke S. R., Wood A. J., Vagnarelli P., Jackson A. P.; Deciphering Developmental Disorders Study (2016) Mutations in genes encoding condensin complex proteins cause microcephaly through decatenation failure at mitosis. Genes Dev. 30, 2158–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trimborn M., Schindler D., Neitzel H., Hirano T. (2006) Misregulated chromosome condensation in MCPH1 primary microcephaly is mediated by condensin II. Cell Cycle 5, 322–326 [DOI] [PubMed] [Google Scholar]
- 41.Brownlow N., Pike T., Zicha D., Collinson L., Parker P. J. (2014) Mitotic catenation is monitored and resolved by a PKCε-regulated pathway. Nat. Commun. 5, 5685 [DOI] [PMC free article] [PubMed] [Google Scholar]