

Abstract
Objective
To evaluate ixekizumab safety in adults with psoriatic arthritis (PsA).
Methods
Safety data from 2 integrated data sets are presented: 1) 24‐week, double‐blind, placebo‐controlled period of SPIRIT‐P1 and SPIRIT‐P2; and 2) all ixekizumab‐treated patients of SPIRIT‐P1 and SPIRIT‐P2 plus SPIRIT‐P3 open‐label period. We report adverse event (AE) frequency and exposure‐adjusted incidence rates per 100 patient‐years at 12‐week intervals to week 96.
Results
The placebo‐controlled period had 678 patients (safety population): 224 placebo, 229 ixekizumab every 4 weeks, and 225 ixekizumab every 2 weeks. Overall, 1,118 patients received ixekizumab (total exposure 1,373.4 patient‐years). In the placebo‐controlled period, the frequencies of ixekizumab‐treated patients experiencing ≥1 treatment‐emergent AE (TEAE) and those experiencing serious AEs were 68.1% (56.7% placebo) and 4.4% (2.7% placebo), respectively. Injection site reactions (ISRs) were very common (21.4% ixekizumab [4.5% placebo]), with ISR discontinuation rates of 1.1% (ixekizumab) and 0.4% (placebo). Through week 96, the incidence rates of ISRs decreased with increasing ixekizumab exposure. The frequencies of AEs of special interest were 32.8% (ixekizumab) and 27.7% (placebo); for serious infections, the frequencies were 1.3% and 0%, respectively; Candida infections, 2.6% and 0.4%; confirmed major adverse cardiac events, 0% and 0%; malignancy, 0.4% and 0%; hypersensitivities, 5.3% and 1.8%; and depression‐related, 1.8% and 1.3%. The frequency of Crohn's disease and ulcerative colitis (investigator‐reported) was 0% in both groups, and the frequencies of sponsor‐determined inflammatory bowel disease were 0.2% in the ixekizumab group and 0% in the placebo group. Overall, no active tuberculosis, invasive Candida infections, anaphylaxis, or suicide/self‐injury behaviors were reported.
Conclusion
The PsA ixekizumab safety integrated data set reached 1,373.4 patient‐years total exposure. Ixekizumab‐treated patients had higher rates of overall TEAEs, serious infections, mucocutaneous Candida, hypersensitivities (non‐anaphylactic), and ISRs than placebo‐treated patients. No unexpected safety outcomes were reported.
Introduction
Interleukin‐17A (IL‐17A) is a therapeutic target for psoriasis and psoriatic arthritis (PsA) given its role in pathological immune responses associated with those conditions 1. Secukinumab was the first IL‐17A inhibitor approved for the treatment of PsA 2, 3. Ixekizumab is a high‐affinity monoclonal antibody that selectively targets IL‐17A 4. In the SPIRIT‐P1 trial, ixekizumab was shown to be superior to placebo for reducing disease activity and radiographic disease progression and for improving physical function and patient‐reported quality of life in biologic‐naive patients with active PsA 5. In SPIRIT‐P2, in which patients with active PsA who previously had an inadequate response to a tumor necrosis factor inhibitor (TNFi) were enrolled, ixekizumab was also effective for improving the signs and symptoms of PsA 6, with a safety profile consistent with those seen in studies involving ixekizumab treatment in patients with PsA and psoriasis 5, 7, 8, 9.
Significance & Innovations.
Results from 1,373.4 patient‐years of ixekizumab exposure in patients with psoriatic arthritis (PsA) are reported.
A major strength of this study is that it combines 2 data sets, each of which has its own advantages. The placebo‐controlled period data set allows comparisons with placebo in a data set combining all data available to date. The all ixekizumab‐treated data set is larger, with increased numbers of patients and longer durations of exposure, allowing detection of adverse events that occur less frequently than in the placebo‐controlled period data set, and enabling assessment of how the incidence rates of adverse events of special interest evolve over time.
Overall, treatment‐emergent adverse events, injection site reactions, serious infections, mucocutaneous (but not systemic) Candida infection, and hypersensitivities (non‐anaphylactic) were observed more frequently in the ixekizumab group than in the placebo group.
The safety profile of ixekizumab for the treatment of PsA was consistent with the known safety profile of ixekizumab for the treatment of patients with moderate‐to‐severe plaque psoriasis, and no unexpected safety signals were observed.
The benefit/risk profile is an important consideration for any drug. Given the role of IL‐17A in host immunity, safety considerations for IL‐17A inhibitors include an increased risk of certain types of infections, including mucocutaneous Candida and upper respiratory tract infections 10, 11, 12, 13. Inflammatory bowel disease (IBD) is also a potential concern with regard to IL‐17 inhibitors, based on unexpected findings in studies in which an IL‐17 inhibitor was used 14, 15. General concerns more broadly for immunomodulatory agents, such as a TNFi, include serious infections (active tuberculosis [TB]), malignancies, and major adverse cardiovascular events (MACE) 16, 17, 18. Monoclonal antibody treatment may cause hypersensitivity, including anaphylaxis 16. Short‐ and long‐term safety analyses using integrated data sets from clinical trials were reported for ixekizumab and secukinumab in patients with plaque psoriasis 9, 19. In the current study, we report an integrated safety analysis of ixekizumab in patients with active PsA, using data pooled from phase III trials.
Patients and Methods
Patients and study design
Data were derived from SPIRIT‐P1 (ClinicalTrials.gov identifier: NCT01695239) 5, SPIRIT‐P2 (ClinicalTrials.gov identifier: NCT02349295) 6, and SPIRIT‐P3 (ClinicalTrials.gov identifier: NCT02584855) (Table 1). Supplementary Listing 1 (available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract) shows key enrollment criteria. SPIRIT‐P1 and SPIRIT‐P2 are randomized, double‐blind, placebo‐controlled, phase III trials involving patients with active PsA 5, 6 (for details, see Supplementary Text 1, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract). SPIRIT‐P3 is a phase III study with a 36‐week to 64‐week open‐label treatment period during which the effects of treatment with ixekizumab administered every 2 weeks were examined, followed by a randomized withdrawal period in patients with active PsA who have an inadequate response to a conventional disease‐modifying antirheumatic drug (cDMARD) and also are biologic DMARD (bDMARD)–naive. SPIRIT‐P3 is ongoing; therefore, only data from the open‐label period are included. SPIRIT‐P2 is also ongoing.
Table 1.
Overview of the clinical trialsa
| Study | Study description | Population | Treatments according to study periodb | Statusc |
|---|---|---|---|---|
|
SPIRIT‐P1 (ClinicalTrials.gov identifier: NCT01695239), pivotal, phase III |
Multicenter, randomized, double‐blind, active and placebo‐controlled, parallel‐group study followed by long‐term extension; primary end point at week 24 | Adult bDMARD‐naive patients with active PsA (meet CASPAR criteria and ≥3 of 68 tender and ≥3 of 66 swollen joints, ≥1 disease‐related hand or foot joint erosion, or a CRP level >6 mg/liter) | Double‐blind placebo‐controlled treatment period (week 0 to week 24)IXE 80 mg Q4WIXE 80 mg Q2WADA 40 mg Q2WPlaceboExtended and long‐term treatment period (week 24 to week 156)IXE 80 mg Q4WIXE 80 mg Q2W | 24‐ and 52‐week analyses completed; ongoing long‐term extension treatment period |
|
SPIRIT‐P2 (ClinicalTrials.gov identifier: NCT02349295), pivotal, phase III |
Multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study followed by long‐term extension; primary end point at week 24 | Adult patients who are both bDMARD‐and cDMARD‐experienced and have active PsA (meet CASPAR criteria, and ≥3 of 68 tender and ≥3 of 66 swollen joints) | Double‐blind placebo‐controlled treatment period (week 0 to week 24)IXE 80 mg Q4WIXE 80 mg Q2WPlaceboExtension treatment period (week 24 to week 156)IXE 80 mg Q4WIXE 80 mg Q2W | 24‐week analysis completed; ongoing extension treatment period (week 52 database lock and analysis completed) |
|
SPIRIT‐P3 (ClinicalTrials.gov identifier: NCT02584855), support, phase III§ |
Multicenter, randomized, double‐blind withdrawal study, preceded by 36‐week open‐label treatment period | Adult patients with active PsA (meet CASPAR criteria, ≥3 of 68 tender, and ≥3 of 66 swollen joints) who are cDMARD‐inadequate responders and also bDMARD‐naive |
Open‐label treatment period (week 0 to weeks 36–64)IXE 80 mg Q2WRandomized double‐blind withdrawal period (after week 36)Patients meeting MDA for 3 months and had received IXE 80 mg Q2W for ≥6 monthsdIXE 80 mg Q2WPlaceboPatients who no longer met MDA and had received IXE 80 mg Q2WdPatients who did not meet MDAdIXE 80 mg Q2W |
Ongoing |
bDMARD = biologic disease‐modifying antirheumatic drug; PsA = psoriatic arthritis; CASPAR = Classification Criteria for Psoriatic Arthritis; CRP = C‐reactive protein; cDMARD = conventional DMARD; MDA = minimal disease activity.
During week 16 in SPIRIT‐P1 and SPIRIT‐P2, inadequate responders received rescue therapy as defined in the protocols, which is a modification of allowed concomitant medications. At week 16, inadequate responders who were originally assigned to adalimumab (ADA) or placebo were re‐randomized 1:1 to receive either ixekizumab (IXE) 80 mg every 2 weeks (Q2W) or every 4 weeks (Q4W), and those who were originally assigned to IXE 80 mg Q2W or Q4W continued their originally assigned dosing regimen up to week 24.
Status at the time of the data cutoff date for the safety summary described in this report.
Safety data from the open‐label treatment period of SPIRIT‐P3 are included in the analyses presented in the safety summary described in this report.
According to a report by Coates et al 27, “A patient is classified as achieving minimal disease activity (MDA) when meeting 5 of the 7 following criteria: tender joint count ≤1; swollen joint count ≤1; Psoriasis Activity and Severity Index ≤1 or body surface area ≤3; patient pain visual analog score (VAS) score ≤15; patient global disease activity VAS ≤20; Health Assessment Questionnaire ≤0.5; tender entheseal points ≤1.”
Integrated data sets
The placebo‐controlled period data set contains data from the placebo‐controlled periods (week 0 through week 24) of SPIRIT‐P1 and SPIRIT‐P2 but excludes data observed after week 16 for week‐16 inadequate responders. Adalimumab was used during the placebo‐controlled period of SPIRIT‐P1 only. Results for adalimumab are not presented herein, because the adalimumab safety profile in SPIRIT‐P1 was previously reported 5. The all ixekizumab‐treated data set consists of available ixekizumab safety data for ixekizumab‐exposed patients during all treatment periods of SPIRIT‐P1 and SPIRIT‐P2 and the open‐label period of SPIRIT‐P3.
The interim database locks used in this study for the placebo‐controlled period data set were September 15, 2016 (SPIRIT‐P1) and September 30, 2016 (SPIRIT‐P2). For the all ixekizumab‐treated data set, the database locks were February 22, 2017 (SPIRIT‐P1 and SPIRIT‐P3) and April 26, 2017 (SPIRIT‐P2).
Safety evaluations
For the safety analyses, we used data from the PsA Safety Population, defined as all randomized patients who received ≥1 dose of study drug (placebo‐controlled period data set) and all patients who received ≥1 dose of ixekizumab (all ixekizumab‐treated data set). Adverse events (AEs) are presented based on Medical Dictionary for Regulatory Activities (MedDRA) versions 19.0 and 19.1. A treatment‐emergent AE (TEAE) was defined as an event that first occurred or worsened in severity after the start of study treatment (baseline) and on or before the date of the last visit within the treatment period. The maximum severity for each TEAE during the baseline period was used as baseline severity. TEAEs were assigned to the treatment period during which they were considered treatment‐emergent.
A serious AE (SAE) was defined as an event causing death, initial or prolonged inpatient hospitalization, a life‐threatening experience, persistent or significant disability/incapacity, congenital anomaly/birth defect, or any other event considered by the investigator to be significant. AEs of special interest (AESIs) were prespecified, and selected AESIs are described in the current study. Injection site reactions (ISRs), in the plural form, refers to a grouping of injection site‐related TEAEs based on a prespecified list of terms, whereas ISR, in its singular form, refers to the individual preferred term (PT). The grouping of ISRs included all PTs defined by ISR‐related high‐level terms in the MedDRA dictionary and excluded 10 joint‐related PTs. The number of the PTs in ISR grouping depends on the MedDRA version. In MedDRA version 19.1, the ISRs high‐level term includes 78 PTs; therefore, the ISRs grouping included 68 PTs.
For neutrophils, the lower limit of normal (2.0 × 109/liter) was defined conservatively as an increment above Common Terminology Criteria for Adverse Events grade 1, equal to increments defining grades 1 through 4. Shift tables were produced for neutrophils: normal ≥2.0 × 109/liter; grade 1 <2.0 × 109/liter to ≥1.5 × 109/liter; grade 2 <1.5 × 109/liter to ≥1.0 × 109/liter; grade 3 <1.0 × 109/liter to ≥0.5 × 109/liter; and grade 4 <0.5 × 109/liter.
Infections temporally associated with neutropenia were defined as infections with an onset date ≤14 days before or after the date of laboratory‐identified neutropenia. Cerebrocardiovascular events, including MACE, were externally adjudicated by the Clinical Events Committee (CEC) at the Cleveland Clinic.
TB testing was conducted at baseline and then at yearly intervals in patients with no history of TB infection. In SPIRIT‐P1, patients who had a post‐baseline positive TB test during yearly testing were required to discontinue ixekizumab treatment. In SPIRIT‐P2 and SPIRIT‐P3, patients were allowed to continue if active TB was excluded and if they received a full course of latent TB treatment with no evidence of hepatotoxicity.
Statistical analysis
Overall exposure was summarized in total patient‐years, calculated as follows: patient‐year = sum of duration of ixekizumab exposure (days) for all patients in the treatment group/365.25. TEAEs were summarized by frequencies and incidence rates (IRs). For AESIs that maintained a constant hazard rate over the treatment period, the exposure‐adjusted IRs of the entire treatment period were also analyzed. The slope of the hazard rate plot for each AESI was visually assessed. If the line was approximately flat, the hazard rate was considered constant. IRs were calculated by dividing the total number of patients experiencing the TEAE for each PT by the sum of all patients' time (in 100 years) of exposure during the treatment period. The entire time on study during the treatment period was used for the calculations. IRs are expressed as the IR (95% confidence interval [95% CI])/100 patient‐years.
For the placebo‐controlled period data set, treatment comparisons for categorical data (frequency) were analyzed using the Cochran‐Mantel‐Haenszel test, stratified by study. For the all ixekizumab‐treated data set, the number and percentage of patients and the exposure‐adjusted IRs are presented by 12‐week intervals from week 0 to week 96 for patients with ≥1 TEAE, patients with ≥1 TEAE in each system organ class group, and for individual AESIs.
Results
Baseline characteristics and exposure
Overall, 454 ixekizumab‐treated patients (193.8 patient‐years) and 1,118 ixekizumab‐treated patients (1,373.4 patient‐years) were included in the placebo‐controlled period data set (Table 2) and the all ixekizumab‐treated safety data set, respectively (see Supplementary Table 1, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract); between‐group demographics were similar in the placebo‐controlled period data set. The median numbers of ixekizumab injections were 7 (range 2–14) during the placebo‐controlled period and 19 (range 1–79) among all ixekizumab‐treated patients. Supplementary Table 2 (available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract) shows study drug exposure.
Table 2.
Demographic and baseline characteristics of patients included in the placebo‐controlled period data set (SPIRIT‐P1 and SPIRIT‐P2), according to treatment groupa
| Characteristic | Placebo (N = 224) | IXEQ4W (N = 229) | IXEQ2W (N = 225) | Total IXE (N = 454) |
|---|---|---|---|---|
| Age, mean ± SD years | 51.1 ± 11.33 | 50.9 ± 12.16 | 50.9 ± 12.22 | 50.9 ± 12.18 |
| ≥65 years, no. (%) | 25 (11.2) | 34 (14.8) | 35 (15.6) | 69 (15.2) |
| Sex, no. (%) | ||||
| Male | 104 (46.4) | 108 (47.2) | 97 (43.1) | 205 (45.2) |
| Female | 120 (53.6) | 121 (52.8) | 128 (56.9) | 249 (54.8) |
| White race, no. (%)b | 207 (92.4) | 213 (93.0) | 208 (92.9) | 421 (92.9) |
| Weight, mean ± SD kg | 88.0 ± 21.58 | 87.8 ± 22.54 | 83.5 ± 19.25 | 85.7 ± 21.06 |
| BMI, mean ± SD kg/m2 b | 30.6 ± 7.25 | 30.6 ± 7.73 | 29.4 ± 6.70 | 30.0 ± 7.26 |
| Tobacco use, no. (%)c | 72 (32.3) | 89 (38.9) | 86 (38.1) | 175 (38.5) |
| cDMARD‐experienced, no. (%)d | 121 (54.0) | 128 (55.9) | 136 (60.2) | 264 (58.0) |
| MTX at baseline, no. (%)d | 99 (44.2) | 105 (45.9) | 114 (50.4) | 219 (48.1) |
| Corticosteroids at baseline, no. (%) | 31 (13.8) | 29 (12.7) | 44 (19.6) | 73 (16.1) |
| Time since PsA onset, mean ± SD yearsd | 10.4 ± 8.55 | 11.7 ± 10.21 | 10.9 ± 9.04 | 11.3 ± 9.65 |
| Time since psoriasis onset, mean ± SD yearsd | 17.5 ± 12.71 | 18.3 ± 13.17 | 18.6 ± 13.68 | 18.5 ± 13.41 |
| Active psoriasis with BSA ≥3%, no. (%)e | 134 (65.0) | 141 (67.1) | 127 (63.8) | 268 (65.5) |
| No. of tender joints (68 assessed), mean ± SDc | 21.2 ± 14.88 | 21.3 ± 13.88 | 23.4 ± 15.97 | 22.4 ± 14.98 |
| No. of swollen joints (66 assessed), mean ± SDc | 10.4 ± 7.29 | 12.3 ± 9.90 | 12.9 ± 9.79 | 12.6 ± 9.84 |
| CRP >6 mg/liter, no. (%)c | 122 (55.0) | 129 (57.1) | 107 (47.3) | 236 (52.2) |
Unless indicated, data are from the safety population. BMI = body mass index; BSA = body surface area; MTX = methotrexate (see Table 1 for other definitions).
Data are missing for some patients; the actual denominator of a particular baseline measure is the number of patients with no missing data for baseline measures.
Baseline data are missing for some patients; the actual denominator of a particular baseline measure is the number of patients with no missing data for baseline measures. Data are from the intent‐to‐treat (ITT) population (226 for IXEQ2W and 455 for total IXE).
Data are from the ITT population (226 for IXEQ2W and 455 for total IXE).
Among patients with plaque psoriasis in the ITT population (206 for placebo, 210 for IXEQ4W, 199 for IXEQ2W, and 409 for total IXE).
Safety overview
Placebo‐controlled period. The overall incidence of TEAEs in both ixekizumab groups was higher than that in the placebo group (Table 3). The most common TEAEs in the total ixekizumab group were ISRs, upper respiratory tract infection, and erythema at the injection site (Table 3). The frequencies of patients experiencing ≥1 SAE tended to be higher in the ixekizumab groups compared with those in the placebo group (Table 3). No SAE term was reported for >1 patient. Similar numbers of patients in the total ixekizumab and placebo groups discontinued treatment because of AEs. There were no clinically meaningful (author assessment) differences between ixekizumab doses for overall TEAEs, SAEs, or treatment discontinuation (Table 3). There were no deaths.
Table 3.
Frequency of adverse events in patients included in the placebo‐controlled data set (SPIRIT‐P1 and SPIRIT‐P2), according to treatment groupa
| Event type or category | Placebo (n = 224) | IXEQ4W (n = 229) | IXEQ2W (n = 225) | Total IXE (n = 454) |
|---|---|---|---|---|
| Patients with ≥1 TEAE | 127 (56.7) | 153 (66.8)b | 156 (69.3)b | 309 (68.1)b |
| Mildc | 60 (26.8) | 91 (39.7) | 81 (36.0) | 172 (37.9) |
| Moderatec | 63 (28.1) | 54 (23.6) | 61 (27.1) | 115 (25.3) |
| Severe | 4 (1.8) | 8 (3.5) | 14 (6.2)b | 22 (4.8) |
| Patients discontinuing study drug because of an AE | 8 (3.6) | 7 (3.1) | 12 (5.3) | 19 (4.2) |
| Patients with ≥1 SAE | 6 (2.7) | 9 (3.9) | 11 (4.9) | 20 (4.4) |
| Deaths | 0 | 0 | 0 | 0 |
| Patients with ≥1 most frequent TEAE (preferred term)d | ||||
| Injection site reaction | 1 (0.4) | 22 (9.6)b | 32 (14.2)b | 54 (11.9)b |
| Upper respiratory tract infection | 16 (7.1) | 16 (7.0) | 15 (6.7) | 31 (6.8) |
| Injection site erythema | 0 | 9 (3.9)b | 17 (7.6)b | 26 (5.7)b |
| Nasopharyngitis | 9 (4.0) | 15 (6.6) | 7 (3.1) | 22 (4.8) |
| Diarrhea | 6 (2.7) | 7 (3.1) | 10 (4.4) | 17 (3.7) |
| Headache | 4 (1.8) | 10 (4.4) | 6 (2.7) | 16 (3.5) |
| Sinusitis | 5 (2.2) | 9 (3.9) | 6 (2.7) | 15 (3.3) |
| Patients with ≥1 AE of special interest | ||||
| Hepatice | 10 (4.5) | 7 (3.1) | 11 (4.9) | 18 (4.0) |
| Infections | 62 (27.7) | 77 (33.6) | 72 (32.0) | 149 (32.8) |
| Serious infection | 0 | 1 (0.4) | 5 (2.2)b | 6 (1.3) |
| Candida infectionf | 1 (0.4) | 4 (1.7) | 8 (3.6)b | 12 (2.6) |
| Esophageal candidiasis | 0 | 0 | 1 (0.4) | 1 (0.2) |
| Active tuberculosis | 0 | 0 | 0 | 0 |
| Latent tuberculosisg | 0 | 0 | 0 | 0 |
| Injection site reactionsh | 10 (4.5) | 40 (17.5)b | 57 (25.3)i | 97 (21.4)b |
| Allergic reaction/hypersensitivity | 4 (1.8) | 10 (4.4) | 14 (6.2)b | 24 (5.3)b |
| Confirmed cerebrocardiovascular event | 2 (0.9) | 0 | 0 | 0b |
| Confirmed MACE event | 0 | 0 | 0 | 0 |
| Malignancy | 0 | 2 (0.9) | 0 | 2 (0.4) |
| Depression‐related | 3 (1.3) | 4 (1.7) | 4 (1.8) | 8 (1.8) |
| Inflammatory bowel disease (narrow and broad terms) j | 0 | 0 | 1 (0.4) | 1 (0.2) |
| Crohn's disease | 0 | 0 | 0 | 0 |
| Ulcerative colitis | 0 | 0 | 0 | 0 |
Values are the number (%). TEAE = treatment‐emergent adverse event (AE); SAE = serious AE; MACE = major adverse cardiovascular event (see Table 1 for other abbreviations).
P ≤ 0.05 vs. placebo, by Cochran‐Mantel‐Haenszel test.
Comparisons between treatment and placebo were not performed.
AEs are listed according to the preferred terms (PTs) in Medical Dictionary for Regulatory Activities (MedDRA). Shown are AEs occurring in ≥3% of patients in the combined (total) ixekizumab group.
Potentially drug‐related hepatic disorders using the MedDRA PTs contained any of the following MedDRA Queries: broad and narrow terms in the liver‐related investigations, signs and symptoms; broad and narrow terms in the cholestasis and jaundice of hepatic origin; broad and narrow terms in the hepatitis, non‐infectious; broad and narrow terms in the hepatic failure, fibrosis, cirrhosis, and other liver damage; narrow terms in the liver‐related coagulation and bleeding disturbances.
Patients with ≥1 TEAE of Candida infections (high‐level plus clinical terms). The esophageal candidiasis case was an SAE.
Two ixekizumab‐treated patients had positive results of an interferon‐γ (IFNγ) release assay. At screening, these patients had negative results using a purified protein derivative test, which later appeared to be poorly documented; this prompted the study site to perform an IFNγ release assay, which appeared to be positive (in the absence of any baseline IFNγ release assay).
Composite of several injection site reaction–related terms.
P ≤ 0.05, IXE vs. placebo and IXEQ2W vs. IXEQ4W, by Cochran‐Mantel‐Haenszel test.
Composite of inflammatory bowel disease (IBD; narrow terms) and events that can occur with IBD (broad terms).
All ixekizumab‐treated
When AEs were analyzed in 12‐week intervals (week 0 to week 96), the IR of overall TEAEs decreased with longer ixekizumab exposure (Figure 1A). The most common TEAEs were ISRs, upper respiratory tract infection, and nasopharyngitis. When overall SAEs were analyzed at 12‐week intervals, there was some variation in IRs, but there was no clinically meaningful increase in the exposure‐adjusted IR with longer ixekizumab exposures (Figure 1B). SAEs that occurred in >1 patient were cholelithiasis (IR 0.3 [95% CI 0.1–0.8]/100 patient‐years); pneumonia (IR 0.2 [95% CI 0.1–0.7]/100 patient‐years); and lower respiratory tract infection, acute cholecystitis, latent TB, esophageal candidiasis, carotid artery stenosis, cerebrovascular accident, transient ischemic attack, fall, meniscus injury, acute myocardial infarction, unstable angina, coronary artery disease, lumbar spine stenosis, and osteoarthritis (IR 0.1 [95% CI 0.0–0.6]/100 patient‐years each). Four deaths (IR 0.3 [95% CI 0.1–0.8]/100 patient‐years) were reported (pneumonia [pathogen not identified], cerebrovascular accident, cardiorespiratory arrest, and drowning). TEAEs causing ixekizumab discontinuation included positive results of an interferon‐γ release assay (IR 0.7 [95% CI 0.4–1.4]/100 patient‐years); latent TB (IR 0.4 [95% CI 0.2–1.0]/100 patient‐years); ISRs (IR 0.3 [95% CI 0.1–0.8]/100 patient‐years); positive tuberculin test (IR 0.2 [95% CI 0.1–0.7]/100 patient‐years); and pneumonia, myalgia, and cerebrovascular accident (IR 0.1 [95% CI 0.0–0.6]/100 patient‐years each).
Figure 1.
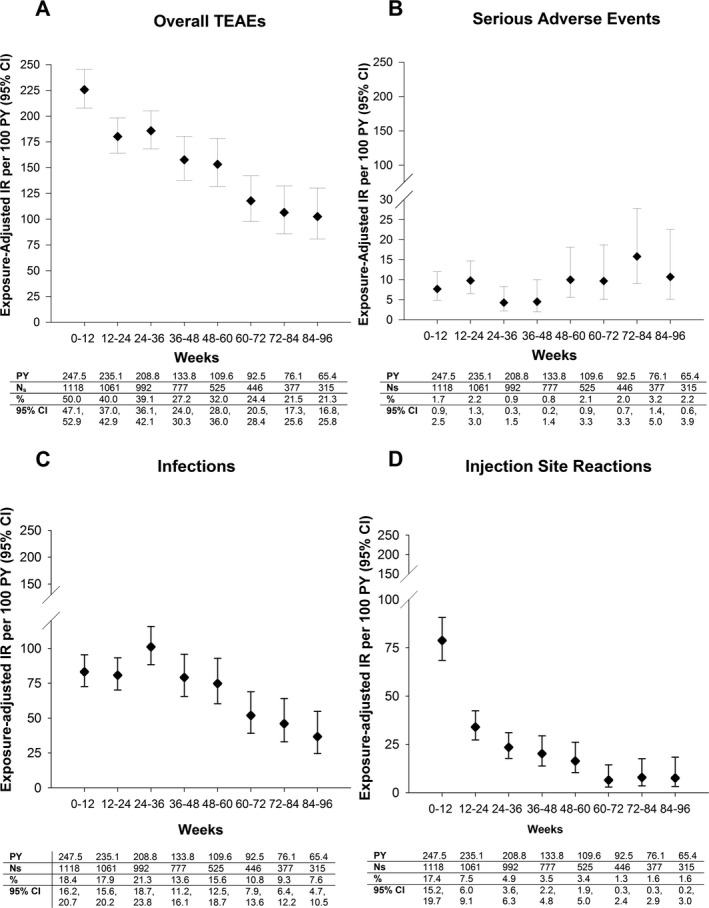
Exposure‐adjusted incidence rates (IRs) of treatment‐emergent adverse events (TEAEs) at 12‐week intervals (week 0 to week 96) in the all ixekizumab‐treated data set (SPIRIT‐P1, SPIRIT‐P2, and SPIRIT‐P3). A, Overall TEAEs. B, Serious AEs. C, Infections (System Organ Class). D, Injection site reactions (this is a composite of several injection site reaction–related preferred terms). The 95% confidence intervals (95% CIs) for the IRs are from likelihood ratio test of treatment effect from the Poisson regression model. AEs were coded using Medical Dictionary for Regulatory Activities version 19.1. Values are from a binomial model. PY = patient‐year; Ns = number of patients entered in each time interval.
AEs of special interest
Infections. Placebo‐controlled period. The frequency of patients with ≥1 infection‐related TEAE was slightly higher in the total ixekizumab group than in the placebo group and was similar in the 2 ixekizumab dosage groups (Table 3). The most common infections occurring in ixekizumab‐treated patients included upper respiratory tract infection, nasopharyngitis, and sinusitis. Six ixekizumab‐treated patients (5 in the group that received ixekizumab every 2 weeks and 1 in the group that received ixekizumab once every 4 weeks) experienced ≥1 serious infection (none occurred in the placebo group); no serious infection type was experienced by >1 patient. Three patients discontinued ixekizumab because of a nonserious infection (folliculitis, subcutaneous abscess, and urinary tract infection, respectively).
The frequency of mucocutaneous Candida infections was higher in the ixekizumab treatment groups compared with that in the placebo group (Table 3). No Candida infection led to treatment discontinuation. There were no deep organ or systemic fungal infections. Predefined opportunistic infections (see Supplementary Listing 2, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract) in the ixekizumab groups were oral candidiasis (0.4% in the group receiving ixekizumab every 4 weeks and 1.8% in the group receiving ixekizumab every 2 weeks), esophageal candidiasis (0.4% in the group receiving ixekizumab every 2 weeks), and unidermatomal herpes zoster (0.4% in the group receiving ixekizumab every 2 weeks).
A 65‐year old woman (in the group receiving ixekizumab every 2 weeks) who had been taking oral prednisone for >2 years was hospitalized on day 7 of ixekizumab treatment because of esophageal candidiasis. She was successfully treated with a 16‐day course of oral fluconazole. Endoscopy was not performed. Ixekizumab dosing in this patient was interrupted and resumed after resolution of the infection.
All ixekizumab‐treated. There was no increase in the IRs of infection‐related TEAEs with increasing durations of ixekizumab exposure (Figure 1C). Ten TEAEs (IR 0.7 [95% CI 0.4–1.4]/100 patient‐years) of latent TB were reported. The incidence of serious infections was low (IR 1.2 [95% CI 0.7–1.9]/100 patient‐years). Serious infections occurring in >1 patient were pneumonia (IR 0.2 [95% CI 0.1–0.7]/100 patient‐years) and latent TB (considered serious due to testing), lower respiratory tract infection, and esophageal candidiasis (IR 0.1 [95% CI 0.0–0.6]/100 patient‐years each).
Two patients reported SAEs of esophageal candidiasis (one reported during the placebo‐controlled period and described above). In the other patient (a 48‐year‐old man), esophageal candidiasis led to hospitalization. Endoscopy showed esophageal mucosa covered in its majority by whitish exudate. The patient was treated with fluconazole. At a follow‐up visit after hospital discharge, the investigator maintained treatment with ixekizumab and methotrexate without lowering the dosage; the outcome of the infection had not been reported at the time of database lock. The 2 esophageal candidiasis cases were the only serious Candida infections in the all ixekizumab‐treated data sets. No treatment‐emergent Candida infection resulted in ixekizumab discontinuation. There were no reports of deep organ or bloodstream Candida infections. Among predefined opportunistic infections (see Supplementary Listing 2, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract), only mucocutaneous Candida infections and localized herpes zoster infection were reported. There were no disseminated or central nervous system herpes simplex infections, endemic mycoses, invasive aspergillosis, or other deep fungal infections.
Eighteen patients (IR 1.3 [95% CI 0.8–2.1]/100 patient‐years) discontinued ixekizumab because of infections. Of these, 6 patients (IR 0.4 [95% CI 0.2–1.0]/100 patient‐years) and 2 patients (IR 0.1 [95% CI 0.0–0.6]/100 patient‐years) discontinued because of latent TB and pneumonia (1 case of pneumonia was fatal), respectively. For all patients who discontinued due to latent TB, the screening test was negative and patients tested positive during protocol‐required yearly testing. There were no cases of active TB. One patient with a history of hepatitis B virus (HBV) infection discontinued the study drug, as required by protocol, when a protocol‐required test for HBV DNA was positive according to the local laboratory despite a result below the level of quantification. Three weeks later, testing was negative for HBV DNA as well as on 3 subsequent samples. The remaining treatment discontinuations were due to bacterial arthritis, bronchitis, nasopharyngitis, staphylococcal infection, tooth abscess, and tonsillitis, in addition to the folliculitis, subcutaneous abscess, and urinary tract infection described above.
Neutropenia. Placebo‐controlled period. The percentage of ixekizumab‐treated patients with a treatment‐emergent low absolute neutrophil count (<2.0 × 109/liter) was 9.7% (43 of 444) versus 2.7% (6 of 219) of placebo‐treated patients (P = 0.001). The frequencies of grade 1 or worse and grade 2 or worse neutropenia were higher in ixekizumab‐treated patients than those in placebo‐treated patients. (see Supplementary Table 3, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract). No grade 3 or grade 4 neutropenia was reported. Approximately 50% of ixekizumab‐treated patients (and 83% of placebo‐treated patients) who had a treatment‐emergent low absolute neutrophil count at an earlier visit had values within the normal range at all subsequent visits.
All ixekizumab‐treated. The percentage of patients with ≥1 treatment‐emergent low neutrophil count (<2.0 × 109/liter) was 14.5% (158 of 1,092). Fifty percent of ixekizumab‐treated patients with treatment‐emergent low absolute neutrophil counts at an earlier visit had values within the normal range at all subsequent visits. Four patients had grade 3 neutropenia that improved to a normal or baseline grade at subsequent visits during which patients were receiving treatment, without reoccurrence of grade 3 neutropenia during the observation period. No patient developed grade 4 neutropenia. There was no tendency for an increase or a decrease in the IR of treatment‐emergent neutropenia with increased ixekizumab exposure (data not shown).
The frequency of infection‐related TEAEs was comparable in patients with neutrophil counts <2.0 × 109/liter (54.4%) versus patients whose counts were above this cutoff (49.0%) and in patients with neutrophil counts <1.5 × 109/liter (42.0%) versus patients whose counts were above the latter (<1.5 × 109/liter) cutoff (50.1%). Seventeen patients (IR 1.2 [95% CI 0.8–2.0]/100 patient‐years) had ≥1 infection‐related TEAE that was temporally associated with grade 1 neutropenia; the infection‐related TEAEs occurring in >1 patient were nasopharyngitis (IR 0.3 [95% CI 0.1–0.8]/100 patient‐years), oral herpes (IR 0.1 [95% CI 0.0–0.6]/100 patient‐years), and upper respiratory tract infection (IR 0.1 [95% CI 0.0–0.6]/100 patient‐years). No patients had infections that were temporally associated with grade 2, 3, or 4 neutropenia.
ISRs. Placebo‐controlled period. ISRs were common in ixekizumab‐treated patients and occurred less frequently in the group receiving ixekizumab every 4 weeks than in the group receiving ixekizumab every 2 weeks (Table 3). The most common types of reported ISRs were injection site reaction, injection site erythema, and injection site hypersensitivity. Most ISRs were mild or moderate in severity. No serious ISRs were reported. Five ixekizumab‐treated patients (1.1% versus 0.4% of placebo‐treated patients) discontinued treatment because of ISRs (4 in the group receiving ixekizumab every 2 weeks and 1 in the group receiving ixekizumab every 4 weeks).
All ixekizumab‐treated. When the incidence of ISRs was analyzed at 12‐week intervals (week 0 to week 96), the incidence was shown to decrease substantially over time (Figure 1D). The most common types of reported ISRs were injection site reaction, injection site erythema, and injection site pain. There were 4.0 ISRs per 100 active injections. Injection site pain, injection site swelling, and injection site discoloration were the first types of ISRs to appear, with median onset times of 1, 2, and 2 days, respectively. The median duration of ISRs was 3 days. Injection site pain and injection site discoloration had the shortest median duration (1 day), whereas injection site hematoma (7 days) and injection site warmth (13 days) had the longest median duration.
There were no serious ISRs; 6 patients (IR 0.4 [95% CI 0.2–1.0]/100 patient‐years) discontinued treatment because of ISRs. There was no clear association between the development of treatment‐emergent antidrug antibodies and ISRs (see Supplementary Table 4 and Supplementary Text 2, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract).
Hypersensitivity events. Placebo‐controlled period. The frequencies of hypersensitivity events in the ixekizumab and placebo treatment groups were 5.3% and 1.8%, respectively (see Supplementary Listing 3, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract). No SAEs of hypersensitivity or anaphylaxis were reported. Two patients treated with ixekizumab every 4 weeks discontinued treatment because of hypersensitivity (hypersensitivity and pruritic rash).
All ixekizumab‐treated. When hypersensitivity was analyzed at 12‐week intervals (week 0 to week 96, the IR of hypersensitivity events did not increase with increasing duration of ixekizumab exposure (Figure 2A). No anaphylaxis was reported, but 1 patient (IR 0.1 [95% CI 0.0–0.5]/100 patient‐years) experienced an SAE of angioedema. Six patients (IR 0.4 [95% CI 0.2–1.0]/100 patient‐years) (including the patient with angioedema) discontinued treatment due to hypersensitivity (drug eruption, rash, pruritic rash, solar urticaria, hypersensitivity). There was no clear association between the development of treatment‐emergent antidrug antibodies and hypersensitivity events (see Supplementary Table 5 and Supplementary Listing 3, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract).
Figure 2.
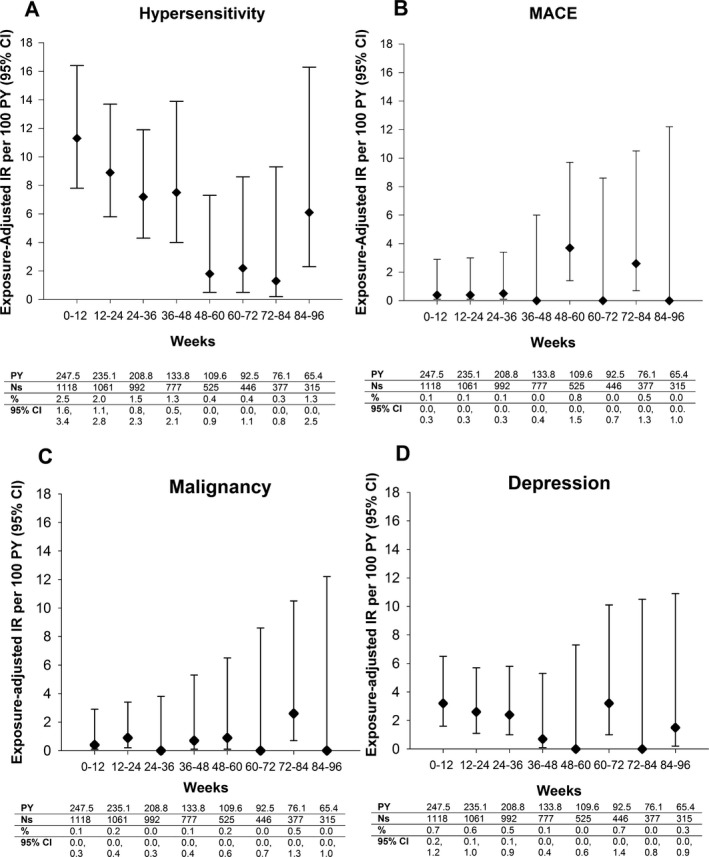
Exposure‐adjusted incidence rate of TEAEs at 12‐week intervals (week 0 to week 96) in the all ixekizumab‐treated data set (SPIRIT‐P1, SPIRIT‐P2, SPIRIT‐P3). A, Hypersensitivity. B, Major adverse cardiovascular events (MACE) (Clinical Events Committee–adjudicated). C, Malignancy. D, Depression‐related. The 95% CIs for the IRs are from likelihood ratio test of treatment effect from the Poisson regression model. AEs were coded using Medical Dictionary for Regulatory Activities version 19.1. Values are from a binomial model. See Figure 1 for other definitions.
Inflammatory bowel disease. Placebo‐controlled. No cases of IBD, ulcerative colitis, or Crohn's disease were explicitly reported (Table 3). Supplementary Listing 4, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.23738/abstract, shows preferred terms for inflammatory bowel disease. However, in reviewing other reported terms potentially representing IBD, 1 patient (in the group receiving ixekizumab every 2 weeks) who had no history of IBD developed an anal abscess and an anal fistula. These were SAEs but did not lead to discontinuation of ixekizumab treatment. The abscess resolved, and the patient underwent surgery for the fistula. The sponsor assessed that this patient had IBD.
All ixekizumab‐treated. One case (IR 0.1 [95% CI 0.0, 0.5]/100 patient‐years) each of CD and UC were reported. These were SAEs but did not lead to discontinuation of ixekizumab treatment. Although the patient with Crohn's disease was reported as a new case, that patient had a history of irritable bowel syndrome. Of the 12 patients with preexisting conditions or a history of illness suggestive of IBD, none experienced disease exacerbation during ixekizumab treatment, and none used steroids during these studies for prior conditions related to IBD, but 1 patient used mesalazine.
Other adverse events of special interest. Placebo‐controlled. No cases of CEC‐confirmed MACE were reported in any treatment group. Malignancies developed in 2 ixekizumab‐treated patients (0.4%) and 0 placebo‐treated patients (Table 3), including 1 basal cell carcinoma and 1 prostate cancer (SAE causing discontinuation of treatment). Eight (1.8%) ixekizumab‐treated patients (3 [1.3%] placebo) experienced ≥1 depression‐related event (Table 3). None were SAEs. One ixekizumab‐treated patient discontinued treatment because of depression; this patient (in the group receiving ixekizumab every 2 weeks) had a history of depression and had been treated with venlafaxine.
All ixekizumab‐treated. No TEAEs of uveitis were reported. Nine patients (IR 0.7 [95% CI 0.3–1.3]/100 patient‐years) had CEC‐confirmed MACE (2 vascular deaths, 3 nonfatal myocardial infarctions, 4 nonfatal strokes). The MACE incidence did not increase with longer ixekizumab exposure (Figure 2B). Nine patients (IR 0.7 [95% CI 0.3–1.3]/100 patient‐years) developed ≥1 malignancy; among these 9 patients, 5 had non‐melanoma skin cancer. The remaining 4 patients had breast cancer (n = 2), prostate cancer (n = 1), and papillary thyroid cancer (n = 1); these were SAEs causing discontinuation of treatment. There was no increase in the malignancy rate with longer ixekizumab exposure (Figure 2C). There was no evidence of an increase in depression‐related events over time (Figure 2D). The incidence of depression‐related events was 1.7 (95% CI 1.2–2.6)/100 patient‐years. None of these events was serious, and only the ixekizumab‐treated patient discontinued treatment due to depression. No suicides or self‐injury–related behaviors were reported. No patient met the laboratory criteria for potential drug‐induced liver injury.
Discussion
In the current study, we report safety results for 1,373.4 patient‐years of ixekizumab exposure in 1,118 patients with PsA. The rates of TEAEs and SAEs did not increase with longer ixekizumab exposure. During the placebo‐controlled period, patients in the ixekizumab group experienced higher rates of overall TEAEs and individual types of TEAEs (serious infections, mucocutaneous Candida infection, hypersensitivities [non‐anaphylactic], and ISRs) than the placebo group. These results are consistent with the known safety profile of ixekizumab in patients with moderate‐to‐severe psoriasis 9.
The use of immunomodulation therapies may be associated with an increased risk of infection 20, 21, 22. As shown in the current study, during ixekizumab treatment, the rates of patients with infections did not increase with longer ixekizumab exposure, and the rate of serious infections was low (IR 1.2 [95% CI 0.7–1.9]/100 patient‐years). For comparison, among patients with confirmed PsA, the rates of serious infection per 100 patient‐years were 1.06 in those treated with ustekinumab, 2.83 in patients treated with infliximab, 2.58 in those treated with adalimumab/etanercept, and 1.63 in those treated with nonbiologics 22. TEAEs of infections were upper respiratory tract and other common types of infection.
Opportunistic infections were limited to oral and esophageal Candida infections (as expected, based on the known role of IL‐17A in host defense against these infections) 10, 11, 12, 13 and localized herpes zoster. The lack of deep organ or bloodstream Candida infections is consistent with published cases of IL‐17 deficiency 10, 11, 13. Two serious Candida infections (esophageal) were reported, but these events did not lead to ixekizumab discontinuation, nor did any other Candida infection. Candida infections resolved or were being treated at the time of database locking. There were no reports of endemic mycoses, invasive aspergillosis, or other deep fungal infections. Several patients had a TEAE of latent TB, as detected through protocol‐required yearly testing; there were no cases of active TB.
Because IL‐17A antagonism is involved in neutrophil trafficking and granulopoiesis 23, the observed decreases in neutrophil counts with ixekizumab versus placebo were expected; however, no patient had grade 4 neutropenia. Grade 2 and grade 3 neutropenia were not temporally associated with infections. Neutropenia rates did not increase with longer ixekizumab exposure. Patients with PsA who were enrolled in ixekizumab treatment studies were allowed to be treated with concomitant cDMARDs. Conventional DMARDs such as methotrexate can cause bone marrow suppression 24, which may have contributed to the neutropenia observed in ixekizumab‐treated patients; however, the presence of cDMARDs does not explain differences between the ixekizumab‐treated patients and the placebo‐treated patients.
The higher frequency of TEAEs in the ixekizumab group compared with the placebo group was mainly attributable to a higher frequency of patients with ≥1 ISR. Although ISRs were common during the placebo‐controlled period, the frequencies of patients with ISRs decreased substantially with longer ixekizumab exposure. ISRs were well tolerated and did not typically lead to discontinuation. Likewise, the incidence of discontinuations due to hypersensitivity was only 0.4 (95% CI 0.2–1.0)/100 patient‐years; anaphylaxis was not reported. Aggregate findings for patients with treatment‐emergent antidrug antibody positivity do not support a clear relationship between hypersensitivity and immunogenicity and between ISRs and immunogenicity.
There are potential concerns regarding new or exacerbated IBD in patients treated with IL‐17A blockers 14, 15. In the current study, the incidences of IBD (including Crohn's disease and ulcerative colitis) were low (IR 0.1 [95% CI 0.0–0.5]/100 patient‐years) for both Crohn's disease and ulcerative colitis and IR 0.1 (95% CI 0.0–0.6)/patient years for unspecified IBD. Similarly, the incidences of patients with other AESIs, including depression, confirmed MACE, and malignancies, were low and did not increase substantially with longer ixekizumab exposure. Additionally, the types of MACE did not change with longer ixekizumab exposure, and the incidence of MACE was consistent with background rates among patients with PsA 25, 26. No self‐injury–related events or suicides were reported.
A strength of this study is inclusion of 2 data sets. The placebo‐controlled period data set allows direct comparison of ixekizumab with placebo for 24 weeks, whereas the all ixekizumab‐treated data set is larger with an aggregate exposure of 1,373.4 patient‐years, thus allowing detection of less frequent AEs and allowing assessment of IRs of AEs over time. Limitations include the lack of a comparator group past 24 weeks and incomplete patient follow‐up at the database locks used for the purpose of this study; updated data will be reported periodically.
In conclusion, ixekizumab‐treated patients had higher rates of overall TEAEs, serious infections, mucocutaneous Candida infection, hypersensitivities (non‐anaphylactic), and ISRs compared with patients treated with placebo. However, the safety profile of ixekizumab in patients with PsA is consistent with the mechanism of IL‐17A antagonism and with the known safety profile of ixekizumab in the treatment of patients with plaque psoriasis 7, 8, 9.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Mease had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Mease, Moriarty, Adams, Xu.
Acquisition of data. Mease, Roussou, Burmester, Goupille, Nash.
Analysis and interpretation of data. Mease, Burmester, Goupille, Gottlieb, Moriarty, Benichou, Adams, Xu, Nash.
Role of the study sponsor
Eli Lilly and Company facilitated the study design, performed the statistical analyses, provided writing assistance for the manuscript, and reviewed and approved the manuscript prior to submission. The authors independently collected the data, interpreted the results, and had the final decision to submit the manuscript for publication. Writing assistance was provided by Matthew Hufford, PhD (Eli Lilly and Company) and Lori Kornberg, PhD (Syneos Health, Raleigh, NC). Publication of this article was not contingent upon approval by Eli Lilly and Company.
Supporting information
Acknowledgments
The authors would like to thank the patients, their families, and the study personnel who participated in these clinical trials. In addition, the authors would like to thank Lotus Mallbris, MD, PhD (Eli Lilly) for her critical review of this manuscript and Sandra Garces, MD, PhD (Eli Lilly) for reviewing the revised manuscript.
Supported by Eli Lilly and Company.
Dr. Mease has received research grants from AbbVie, Amgen, Bristol‐Myers Squibb, Celgene, Janssen, Eli Lilly and Company, Novartis, Pfizer, Sun Pharmaceutical and UCB Pharma and consulting fees, speaking fees, and/or honoraria from AbbVie, Amgen, Eli Lilly and Company, and Novartis (more than $10,000 each). Dr. Roussou has received research grants from Pfizer, UCB Pharma, Merck, and Eli Lilly and Company. Dr. Burmester has received honoraria (less than $10,000 each) and research support from AbbVie, Pfizer, Roche, UCB Pharma, Merck Sharp & Dohme, Bristol‐Myers Squibb, Sanofi, Novartis, Eli Lilly, and Company and Janssen. Dr. Goupille has received consulting fees from AbbVie, Biogaran, Bristol‐Myers Squibb, Celgene, Eli Lilly and Company, Janssen, MSD, Novartis, Pfizer, Roche‐Chugai, and UCB Pharma (less than $10,000 each). Dr. Gottlieb has received consulting fees from Janssen, AbbVie, UCB Pharma, and Eli Lilly and Company (more than $10,000 each), and from Celgene, Bristol‐Myers Squibb, Beiersdorf, Novartis, Incyte, Dr. Reddy's Laboratories, Valeant, Demira, Allergan, and Sun Pharmaceutical (less than $10,000 each). She has received research/educational grants from UCB Pharma, Eli Lilly and Company, Novartis, Janssen, and Incyte. Drs. Moriarty, Benichou, Adams, and Xu own stock in Eli Lilly and Company. Dr. Nash has received honoraria (less than $10,000 each) and research support from Pfizer, Roche, UCB Pharma, Merck Sharp & Dohme, Bristol‐Myers Squibb, Sanofi, Novartis, Eli Lilly, AbbVie, and Janssen.
References
- 1. Raychaudhuri SP. Role of IL‐17 in psoriasis and psoriatic arthritis. Clin Rev Allergy Immunol 2013;44:183–93. [DOI] [PubMed] [Google Scholar]
- 2. McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti‐interleukin‐17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2015;386:1137–46. [DOI] [PubMed] [Google Scholar]
- 3. Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab inhibition of interleukin‐17A in patients with psoriatic arthritis. N Engl J Med 2015;373:1329–39. [DOI] [PubMed] [Google Scholar]
- 4. Liu L, Lu J, Allan BW, Tang Y, Tetreault J, Chow CK, et al. Generation and characterization of ixekizumab, a humanized monoclonal antibody that neutralizes interleukin‐17A. J Inflamm Res 2016;9:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mease PJ, van der Heijde D, Ritchlin CT, Okada M, Cuchacovich RS, Shuler CL, et al. Ixekizumab, an interleukin‐17A specific monoclonal antibody, for the treatment of biologic‐naive patients with active psoriatic arthritis: results from the 24‐week randomised, double‐blind, placebo‐controlled and active (adalimumab)‐controlled period of the phase III trial SPIRIT‐P1. Ann Rheum Dis 2017;76:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nash P, Kirkham B, Okada M, Rahman P, Combe B, Burmester GR, et al. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24‐week randomised, double‐blind, placebo‐controlled period of the SPIRIT‐P2 phase 3 trial. Lancet 2017;389:2317–27. [DOI] [PubMed] [Google Scholar]
- 7. Gordon KB, Blauvelt A, Papp KA, Langley RG, Luger T, Ohtsuki M, et al. Phase 3 trials of ixekizumab in moderate‐to‐severe plaque psoriasis. N Engl J Med 2016;28:345–56. [DOI] [PubMed] [Google Scholar]
- 8. Griffiths CE, Reich K, Lebwohl M, van de Kerkhof P, Paul C, Menter A, et al. Comparison of ixekizumab with etanercept or placebo in moderate‐to‐severe psoriasis (UNCOVER‐2 and UNCOVER‐3): results from two phase 3 randomised trials. Lancet 2015;386:541–51. [DOI] [PubMed] [Google Scholar]
- 9. Strober B, Leonardi C, Papp KA, Mrowietz U, Ohtsuki M, Bissonnette R, et al. Short‐ and long‐term safety outcomes with ixekizumab from 7 clinical trials in psoriasis: etanercept comparisons and integrated data. J Am Acad Dermatol 2017;76:432–440.e.17. [DOI] [PubMed] [Google Scholar]
- 10. Cypowyj S, Picard C, Maródi L, Casanova JL, Puel A. Immunity to infection in IL 17‐deficient mice and humans. Eur J Immunol 2012;42:2246–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gaffen SL, Hernández‐Santos N, Peterson AC. IL‐17 signaling in host defense against Candida albicans. Immunol Res 2011;50:181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peck A, Mellins ED. Precarious balance: Th17 cells in host defense. Infect Immun 2010;78:32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin‐17 immunity. Science 2011;332:65–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti‐IL‐17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double‐blind placebo‐controlled trial. Gut 2012;61:1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mozaffari S, Nikfar S, Abdollahi M. Inflammatory bowel disease therapies discontinued between 2009 and 2014. Expert Opin Investig Drugs 2015;24:949–56. [DOI] [PubMed] [Google Scholar]
- 16. Boyman O, Comte D, Spertini F. Adverse reactions to biologic agents and their medical management. Nat Rev Rheumatol 2014;10:612–27. [DOI] [PubMed] [Google Scholar]
- 17. Molinelli E, Campanati A, Ganzetti G, Offidani A. Biologic therapy in immune mediated inflammatory disease: basic science and clinical concepts. Curr Drug Saf 2016;11:35–43. [DOI] [PubMed] [Google Scholar]
- 18. Ruderman EM. Overview of safety of non‐biologic and biologic DMARDs. Rheumatology (Oxford) 2012;51:vi37–43. [DOI] [PubMed] [Google Scholar]
- 19. Van de Kerkhof PC, Griffiths CE, Reich K, Leonardi CL, Blauvelt A, Tsai TF, et al. Secukinumab long‐term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol 2016;75:83–98. [DOI] [PubMed] [Google Scholar]
- 20. Murdaca G, Spano F, Contatore M, Guastalla A, Penza E, Magnani O, et al. Infection risk associated with anti‐TNF‐α agents: a review. Expert Opin Drug Saf 2015;14:571–82. [DOI] [PubMed] [Google Scholar]
- 21. Selmi C, Ceribelli A, Naguwa SM, Cantarini L, Shoenfeld Y. Safety issues and concerns of new immunomodulators in rheumatology. Expert Opin Drug Saf 2015;14:389–99. [DOI] [PubMed] [Google Scholar]
- 22. Ritchlin CT, Gottlieb AB, Menter A, Mease PJ, Kalia S, Kerdel F, et al. Serious infections in psoriasis patients with psoriatic arthritis in the Psoriasis Longitudinal Assessment and Registry Study [abstract]. Arthritis Rheumatol 2015;67 Suppl:S1692. [Google Scholar]
- 23. Krstic A, Mojsilovic S, Jovcic G, Bugarski D. The potential of interleukin‐17 to mediate hematopoietic response. Immunol Res 2012;52:34–41. [DOI] [PubMed] [Google Scholar]
- 24. Weinblatt ME. Toxicity of low dose methotrexate in rheumatoid arthritis. J Rheumatol Suppl 1985;12 Suppl 12:35–9. [PubMed] [Google Scholar]
- 25. Li L, Hagberg KW, Peng M, Shah K, Paris M, Jick S. Rates of cardiovascular disease and major adverse cardiovascular events in patients with psoriatic arthritis compared to patients without psoriatic arthritis. J Clin Rheumatol 2015;21:405–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ogdie A, Yu Y, Haynes K, Love TJ, Maliha S, Jiang Y, et al. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population‐based cohort study. Ann Rheum Dis 2015;74:326–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coates LC, Fransen J, Helliwell PS. Defining minimal disease activity in psoriatic arthritis: a proposed objective target for treatment. Ann Rheum Dis 2010;69:48–53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials