

ABSTRACT
Background
No prospective study of patients with Parkinson's disease (PD) has investigated the appearance of vertical gaze abnormalities, a feature suggestive of progressive supranuclear palsy (PSP).
Objective
To identify, within a cohort of patients with an initial diagnosis of PD, those who developed vertical gaze abnormalities during a 4‐year follow‐up, and to investigate the performance of new imaging biomarkers in predicting vertical gaze abnormalities.
Methods
A total of 110 patients initially classified as PD and 74 controls were enrolled. All patients underwent clinical assessment at baseline and every year up to the end of the follow‐up. The pons/midbrain area ratio 2.0 and the Magnetic Resonance Parkinsonism Index 2.0 were calculated.
Results
After 4‐year follow‐up, 100 of 110 patients maintained the diagnosis of PD, whereas 10 PD patients (9.1%) developed vertical gaze abnormalities, suggesting an alternative diagnosis of PSP‐parkinsonism. At baseline, the Magnetic Resonance Parkinsonism Index 2.0 was the most accurate biomarker in differentiating PD patients who developed vertical gaze abnormalities from those who maintained an initial diagnosis of PD. At the end of follow‐up, both of these biomarkers accurately distinguished PSP‐parkinsonism from PD.
Conclusions
Our results demonstrate that a number of patients with an initial diagnosis of PD developed vertical gaze abnormalities during a 4‐year follow‐up, and the diagnosis was changed from PD to PSP‐parkinsonism. In PD patients, baseline Magnetic Resonance Parkinsonism Index 2.0 showed the best performance in predicting the clinical evolution toward a PSP‐parkinsonism phenotype, enabling PSP‐parkinsonism patients to be identified at the earliest stage of the disease for promising disease‐modifying therapies. © 2019 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: magnetic resonance parkinsonism index, magnetic Resonance Parkinsonism Index 2.0, pons/midbrain area ratio 2.0, progressive supranuclear palsy‐parkinsonism, vertical gaze abnormalities
To date, the diagnosis of Parkinson's disease (PD) remains primarily clinical.1, 2 However, the accuracy of clinical diagnosis of PD is not satisfactory, particularly in early stages of the disease where the typical clinical signs are not yet fully present.2, 3, 4 Several studies have demonstrated that clinical diagnosis of PD may change after a follow‐up of a few years.5, 6 In clinic‐based studies on PD, the most frequent misdiagnoses involved atypical parkinsonisms, particularly multiple system atrophy or progressive supranuclear palsy (PSP).2, 5, 6 Recent evidence has highlighted the greater difficulty in differentiating patients with PD from those affected by PSP with parkinsonism (PSP‐P).7, 8, 9, 10 The clinical presentation of PSP‐P resembles idiopathic PD to such an extent that the 2 disorders are difficult to distinguish early on.7, 8, 9, 10 Recently, the Movement Disorder Society (MDS)–endorsed PSP Study Group11 published new diagnostic criteria for clinical diagnosis of PSP‐Richardson's (PSP‐RS) and the PSP variants, such as PSP‐P. Diagnosis of probable PSP‐RS was based on the presence of vertical gaze abnormalities (VGA) and early postural instability, whereas probable PSP‐P was characterized by VGA associated with levodopa‐responsive or levodopa‐resistant parkinsonism.11 VGA usually are the most specific neurological signs for differentiating patients with PSP‐P from those with PD8; however, these clinical features may occur later or never in PSP‐P patients.7, 8, 9, 10 The absence of VGA makes it difficult to differentiate between patients with PSP‐P and those with PD,8 suggesting that the true prevalence of PSP‐P may be underestimated.
Magnetic resonance (MR) imaging measurements have been shown to be useful for diagnosing PSP.12, 13 The MR Parkinsonism Index (MRPI) has proven to be an accurate biomarker for diagnosing patients with PSP14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26 and for predicting the clinical evolution toward PSP phenotypes of patients affected by undetermined parkinsonism27, 28 or the appearance of vertical supranuclear gaze palsy (VSGP) in patients with PSP‐P.29 Overall, this biomarker is highly accurate in diagnosing PSP‐RS12 but shows a low sensitivity in distinguishing between patients with PSP‐P and those with PD.18, 26, 30 Recently, we demonstrated that a new version of MRPI that included the measurement of third ventricle width (MRPI 2.0) was highly accurate in differentiating PSP‐P patients from PD.26 To date, to our knowledge, no prospective study has investigated in vivo the appearance of clinical features of PSP‐P in patients initially diagnosed as PD. In the current study, we used a large sample of patients with an initial clinical diagnosis of PD who were clinically and radiologically followed for 4 years to evaluate the appearance of VGA without early postural instability, a phenotype strongly suggestive of PSP‐P.11 We also investigated whether MRPI 2.0 measurements were able to predict in patients with initial diagnosis of PD the evolution toward a clinical phenotype of PSP‐P.
Methods
Baseline Evaluation
Patients
We consecutively recruited 128 patients with PD and 74 sex‐ and age‐matched healthy control participants between April 2010 and May 2013. Clinical diagnosis of PD was made by one of the authors (M.M.) with >10 years of experience in movement disorders. Patients were included if they reached the probable (disease duration ≥3 years, levodopa responsiveness and presence of 3 of 4 cardinal features of PD) or possible (disease duration <3 years, 2 of 4 cardinal features of PD) degree of diagnostic certainty for PD.31 For each patient, a complete medical history and neurological examination with accurate clinical evaluation of ocular movements and postural instability were performed, motor impairment was scored using the Unified Parkinson's Disease Rating Scale–Motor Examination (UPDRS‐ME)32 in off‐state (off medications overnight), and disease severity rated using the Hoehn and Yahr rating scale.33 An asymmetry index was calculated according to the procedure recently described.34 We used the MMSE35 to assess cognitive performance in all patients. Levodopa response was assessed both in the off‐state (off medications overnight) and 2 hours after drug administration and was considered positive if it was ≥30% on the UPDRS‐ME score (moderate 30%‐50%; good >50%‐70%; excellent >70%).36 Exclusion criteria were history of neuroleptic use within the past 6 months, evidence of atypical clinical features suggestive of parkinsonisms, evidence of normal striatal uptake in dopamine transporter iodine123‐N‐ω‐fluoropropyl‐2β‐carbomethoxy‐3β‐(4‐iodophenyl) tropane (I‐123 FP‐CIT) single‐photon emission computed tomography (SPECT), evidence of normal pressure hydrocephalus suggested by abnormal MR signs such as reduced callosal angle and enhanced Evans Index,37 and evidence on MR imaging scan of vascular lesions such as lacunar infarctions in the basal ganglia and/or subcortical vascular lesions with diffuse periventricular signal alterations. None of the control participants had a history of neurologic, psychiatric, or other major medical illnesses. All study procedures and ethical aspects were approved by the institutional review board (Magna Graecia University review board, Catanzaro, Italy). In addition, written informed consent was obtained from all those participants examined as part of the study.
MR Imaging Protocol
All patients and controls performed a brain MRI with a 3 T‐MR750 General Electric scanner and an 8‐channel head coil according to a recently described procedure.26
Morphometric Measurements
Automated measurements of the pons/midbrain area ratio (P/M) and the MRPI were performed according to the recently described procedure (http://mrpi.unicz.it).38
The new version of these biomarkers (P/M 2.0 and MRPI 2.0), which included the measurement of third ventricle (3rdV) width, was calculated according to the procedure described in a recent study26 and reported in the supplementary materials.
Follow‐Up Evaluation
After clinical–radiological characterization at baseline, all patients were clinically assessed by the same physician every year for an observational period of 4 years. At each annual clinical evaluation, the patients underwent a neurological examination with accurate clinical evaluation of ocular movements and postural instability. Clinical assessments were performed with the UPDRS‐ME in the off‐state (off medications overnight) and using the Hoehn and Yahr rating scale and MMSE test. At the end of the 4‐year follow‐up period, all patients repeated the clinical evaluation, levodopa acute test, and MR imaging examination with the same protocol as the baseline.
The degree of diagnostic certainty for PD31 was assessed every year in all patients, and the classification of the patients as possible or probable PD was modified accordingly. Because the modality of clinical assessment of ocular motor dysfunction and postural instability did not differ substantially between previous39 and new criteria,11 all patients without postural instability within 3 years after the disease onset who developed VGA during the follow‐up were reclassified according to the recent diagnostic criteria for probable PSP‐P.11 Thus, the following 2 levels of VGA were considered: slow velocity of vertical saccades and VSGP. Slowness of vertical greater than horizontal saccadic eye movements was considered as the criterion for slow velocity of vertical saccades (O2 level).11 A clear limitation of the range of voluntary gaze in the vertical more than in the horizontal plane, affecting both up‐ and downgaze, were considered as criterion for VSGP (O1 level).11 A clinical diagnosis of probable PSP‐P was thus suspected when the patient developed VGA (O1 or O2) associated with levodopa‐responsive (A3 level) or levodopa‐resistant (A2 level) parkinsonism, without early postural instability within 3 years after the disease onset.11 The progression of the disease was evaluated for UPDRS‐ME and for P/M, MRPI, P/M 2.0, and MRPI 2.0 as the percentage difference from baseline to follow‐up values.
Statistical Analysis
Differences in sex and clinical features distribution were assessed by Fisher's exact test. McNemar's test was used to assess changes in dopaminergic responsiveness from baseline to follow‐up evaluation. The Shapiro‐Wilk test was used to check for normality before comparing continuous variables. Baseline demographic, clinical, and radiological data were compared using either the Student's t test or the Mann–Whitney U test. Baseline to follow‐up comparisons were performed either by the paired t test or by the Wilcoxon signed rank test.
A mixed‐model analysis of variance was performed to assess the effect of time (baseline, follow‐up) as the within factor and status (PD, PSP‐P) as the between factor and their mixed effect on MRPI, MRPI 2.0, P/M, and P/M 2.0. With age‐related variables, an analysis of covariance was performed to control for the age effect. All tests were 2‐tailed and the α level was set at P < .05. When comparing radiological measures, P values were corrected according to Bonferroni. We assessed sensitivity, specificity, positive predictive value, negative predictive value, and diagnostic accuracy of baseline and follow‐up measurements of P/M, P/M 2.0, MRPI, and MRPI 2.0 in differentiating patients who maintained an initial diagnosis of PD from those who developed VGA. Optimal cut‐off levels were defined as the values with the highest sum of sensitivity and specificity on the receiver operating characteristic curves. To assess intrarater and interrater reliability, intraclass correlation coefficients were calculated. Analyses were performed using R (R for Unix/Linux, version 3.1.1, the R Foundation for Statistical Computing, Vienna, Austria).
Results
Demographic, clinical, and neuroimaging data of patients with PD and controls at baseline evaluation are listed in Table 1. Among the 128 PD patients initially enrolled, 18 patients were excluded from the study (4 patients died before the end of the follow‐up; 5 patients dropped out; 7 patients developed atypical clinical features suggestive of multiple system atrophy [early orthostatic fall in blood pressure and loss of levodopa response], and 2 patients developed clinical features suggestive of dementia with Lewy bodies [early cognitive decline with fluctuating confusion and hallucinations]). Of 40 patients with an initial diagnosis of possible PD who showed a levodopa responsiveness lower than 30% at baseline, 19 developed a good levodopa response at the end of follow‐up (Table 1 and Table 2). These patients at baseline had less than 2 years of disease duration and had not had an adequate trial of levodopa or dopamine agonists. At baseline, 21 of 40 patients with possible PD and all patients (n = 70) with probable PD showed levodopa responsiveness (moderate, n = 51; good, n = 30; excellent, n = 10). At baseline, 9 of 10 patients who developed VGA during the follow‐up showed a moderate response to levodopa, whereas only 1 patient had a good levodopa response.
Table 1.
Clinicoradiologic data of patients initially classified as Parkinson's disease and controls at baseline evaluation.
| Clinicoradiologic Data | Parkinson's disease | Controls | P valuea |
|---|---|---|---|
| n | 110 | 74 | – |
| Sex, no. M/F | 73/37 | 49/25 | 1b |
| Probable/possible PD level | 70/40 | – | – |
| Age at examination, y, mean ± standard deviation (range) | 62.9 ± 8.1 (42‐80) | 64.3 ± 8.5 (50‐83) | .40c |
| Age at disease onset, y, mean ± standard deviation (range) | 58.5 ± 8.0 (40‐77) | – | – |
| Disease duration, y, mean ± standard deviation (range) | 4.4 ± 3.9 (1‐24) | – | – |
| Clinical features | |||
| Rest tremor, n (%) | 77 (70.0) | – | – |
| Asymmetric motor symptoms, n (%) | 74 (67.2) | – | – |
| MMSE score, median value (range) | 27 (16‐30) | 28 (24‐30) | .005c |
| UPDRS‐ME score, median value (range) | 21 (8‐44) | – | – |
| H‐Y score, median value (range) | 2 (1.5‐3) | – | – |
| Levodopa responsiveness, n (%)d | 91 (82.7) | – | – |
| Brain MRI measurements, mean ± standard deviation (range) | |||
| P/M | 3.87 ± 0.6 (2.67‐5.31) | 3.60 ± 0.4 (2.81‐4.55) | .01e |
| P/M 2.0 | 0.58 ± 0.2 (0.18‐1.19) | 0.52 ± 0.2 (0.15‐0.99) | 1e |
| MRPI | 9.15 ± 1.4 (6.19‐11.93) | 8.84 ± 1.4 (6.25‐12.12) | 1e |
| MRPI 2.0 | 1.37 ± 0.5 (0.40‐2.71) | 1.27 ± 0.5 (0.43‐2.28) | 1e |
F, female; M, male; H‐Y, Hoehn and Yahr rating scale; P/M, pons/midbrain area ratio; MRPI, Magnetic Resonance Parkinsonism Index.
Clinicoradiological comparisons at baseline between all patients with initial diagnosis of Parkinson's disease and controls.
Fisher's exact test.
Mann–Whitney U test.
Number (percentage) of patients who showed a clinical improvement of 30% or greater in comparison with that detected in off state.
Mann–Whitney U test with Bonferroni correction.
Table 2.
Clinicoradiologic data of patients who maintained initial diagnosis of Parkinson's disease (PD) at the end of follow‐up and of patients with initial diagnosis of PD at baseline who developed features (vertical gaze abnormalities) of progressive supranuclear palsy‐parkinsonism (PSP‐P) during the follow‐up
| Clinicoradiologic Data | PD | PSP‐P | ||||
|---|---|---|---|---|---|---|
| Baseline | 4‐year follow‐up | P valuea | Baseline | 4‐year follow‐up | P valueb | |
| n | 100 | 100 | \ | 10 | 10 | \ |
| Sex, no. M/F | 64/36 | 64/36 | \ | 9/1 | 9/1 | \ |
| Probable/possible PD level | 60/40 | 100/0 | \ | 10/0 | \ | \ |
| Age at examination, y, mean ± standard deviation (range) | 62.4 ± 8.0 (42‐80) | 66.4 ± 8.0 (46‐84) | \ | 69.0 ± 5.9 (61‐79) | 73.0 ± 5.9 (65‐83) | \ |
| Age at disease onset, y, mean ± standard deviation (range) | 58.1 ± 8.2 (40‐77) | 58.1 ± 8.2 (40‐77) | \ | 62.6 ± 5.7 (54‐70) | 62.6 ± 5.7 (54‐70) | \ |
| Disease duration, y, mean ± standard deviation (range) | 4.3 ± 3.9 (1‐24) | 8.3 ± 3.9 (5‐28) | \ | 6.4 ± 2.3 (3‐9) | 10.4 ± 2.3 (7‐13) | \ |
| Clinical features | ||||||
| Rest tremor, n (%) | 72 (72.0) | 73 (73.0) | 1c | 5 (50) | 1 (10) | .13c |
| Asymmetric motor symptoms, n (%)d | 69 (69.0) | 58 (58.0) | .003c | 5 (50) | 2 (20) | .25c |
| Vertical gaze abnormalities, n (%) | 0 (0) | 0 (0) | \ | 0 (0) | 10 (100) | .004c |
| MMSE score, median value (range) | 27 (16‐30) | 25 (13‐29) | <.001e | 27 (25‐30) | 20.5 (14‐26) | .009e |
| UPDRS‐ME score, median value (range) | 20 (8‐44) | 29.5 (18‐57) | <.001e | 27 (20‐29) | 37 (31‐43) | .006e |
| H‐Y score, median value (range) | 2 (1.5‐3) | 2 (2‐5) | .051e | 2 (2‐2) | 3 (2‐4) | .01e |
| Levodopa responsiveness, n (%)f | 81 (81.0) | 100 (100) | <.001c | 10 (100) | 1 (10) | .008c |
| Brain MRI measurements, mean ± standard deviation (range) | ||||||
| P/M | 3.78 ± 0.5 (2.67‐5.04) | 3.89 ± 0.5 (2.67‐5.08) | .77g | 4.71 ± 0.4 (4.17‐5.31) | 5.85 ± 0.8 (4.87‐7.19) | .001h |
| P/M 2.0 | 0.54 ± 0.2 (0.18‐0.93) | 0.59 ± 0.2 (0.23‐0.98) | .57g | 1.03 ± 0.1 (0.87‐1.19) | 1.37 ± 0.3 (1.01‐1.97) | .004h |
| MRPI | 8.98 ± 1.4 (6.19‐11.68) | 9.26 ± 1.4 (6.37‐11.87) | .40g | 10.81 ± 0.8 (9.89‐11.93) | 15.06 ± 1.4 (12.90‐18.10) | <.001h |
| MRPI 2.0 | 1.27 ± 0.4 (0.40‐2.10) | 1.40 ± 0.4 (0.60‐2.08) | .37g | 2.36 ± 0.2 (2.13‐2.71) | 3.51 ± 0.6 (2.88‐4.70) | <.001h |
PD, patients who maintained initial diagnosis of PD at the end of follow‐up; PSP‐P, patients with initial diagnosis of PD at baseline who developed vertical gaze abnormalities during the follow‐up; F, female; M, male; H‐Y, Hoehn and Yahr rating scale; P/M, pons/midbrain area ratio; MRPI, Magnetic Resonance Parkinsonism Index.
Clinicoradiological comparisons between baseline and follow‐up in PD patients.
Clinicoradiological comparisons between baseline and follow‐up in group PSP‐P patients.
McNemar's test.
Percentage of patients with UPDRS motor asymmetry index >2 points.
Wilcoxon signed rank test.
Number (percentage) of patients who showed a clinical improvement of 30% or greater in comparison with that detected in the off state. Of 40 patients with initial diagnosis of possible PD, 19 showed at baseline a levodopa responsiveness lower than 30%. These patients who have not had an adequate trial with levodopa o dopamine agonists developed a good response to levodopa at the end of follow‐up.
Repeated‐measures analysis of covariance, effect of time (baseline, follow‐up), with Bonferroni correction.
Paired t test with Bonferroni correction.
At baseline, P/M 2.0, MRPI, and MRPI 2.0 values of PD patients were not different from values of the controls with the exception of the P/M values, which were significantly higher in the PD patients than in controls (Table 1). Table 2 shows baseline and follow‐up clinical and imaging data of the patients who maintained the initial diagnosis of PD up to the end of the follow‐up (PD; n = 100) and of those who developed VGA during the follow‐up, and changed diagnosis from PD to PSP‐P (PSP‐P; n = 10). All of these 10 patients developed slowness of vertical saccades during the follow‐up; 5 of these 10 patients showed VSGP at the end of follow‐up. Patients with VSGP showed higher imaging biomarkers values (P/M, mean ± SD: 6.01 ± 0.9; P/M 2.0, mean ± SD: 1.40 ± 0.2; MRPI, mean ± SD: 15.68 ± 1.5; MRPI 2.0, mean ± SD: 3.69 ± 0.6) than those with slowness of vertical saccades (P/M, mean ± SD: 5.69 ± 0.8; P/M 2.0, mean ± SD: 1.33 ± 0.4; MRPI, mean ± SD: 14.44 ± 1.1; MRPI 2.0, mean ± SD: 3.33 ± 0.7). In addition, 9 of 10 PD patients who developed VGA lost the response to levodopa during the follow‐up (Table 2). All patients who developed VGA during the follow‐up had at least 3 years of parkinsonism without history of postural instability and normal pull‐test at baseline (Table 2). In the PD patients, there were no significant differences in morphometric measures between baseline and follow‐up, whereas in patients with PSP‐P, all morphometric values were significantly worse at follow‐up than at baseline (Table 2). In both PD and PSP‐P, clinical variables significantly worsened between baseline and follow‐up (Table 2).
The between‐group comparison showed that all morphometric measures (P/M, P/M 2.0, MRPI, and MRPI 2.0) were significantly higher in patients with PSP‐P than in those with PD, both at baseline and at follow‐up (Table 2 and Supplementary Table 1). MRPI 2.0 values showed no overlap between patients with PD and patients with PSP‐P at both evaluations (Fig. 1). Most clinical variables showed a slight significant difference at baseline and a marked difference at follow‐up between the 2 patient groups (Table 2 and Supplementary Table 1).
Figure 1.
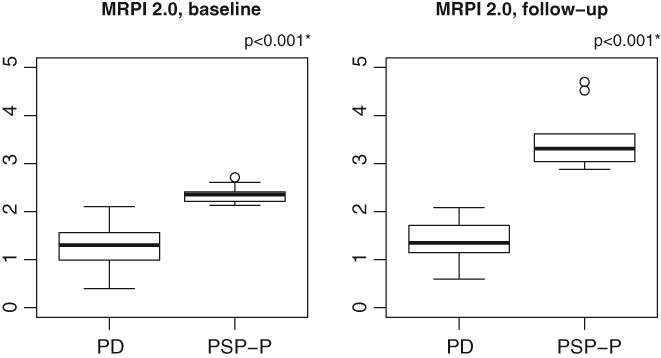
Box plots of baseline and follow‐up Magnetic Resonance Parkinsonism Index 2.0 (MRPI 2.0) measurements in patients who maintained diagnosis of Parkinson's disease at the end of follow‐up (PD) and in those who developed clinical signs of progressive supranuclear palsy‐parkinsonism during the follow‐up (PSP‐P). Vertical solid lines (whiskers) show lower and upper values. Box stretches from lower hinge (25th percentile) to upper hinge (75th percentile). The median is shown as a line across each box. *Two‐sample t test with Bonferroni correction.
When status–time (disease follow‐up) interaction was considered, there were significant differences between PD patients and PSP‐P patients for all morphometric measurements (P < .001), thus showing that disease progression was more marked in patients with PSP‐P than in those with PD (Fig. 2A). Figure 2B shows for each biomarker (MRPI, MRPI 2.0, P/M, P/M 2.0) the percentage difference from baseline to follow‐up in patients with PSP‐P.
Figure 2.
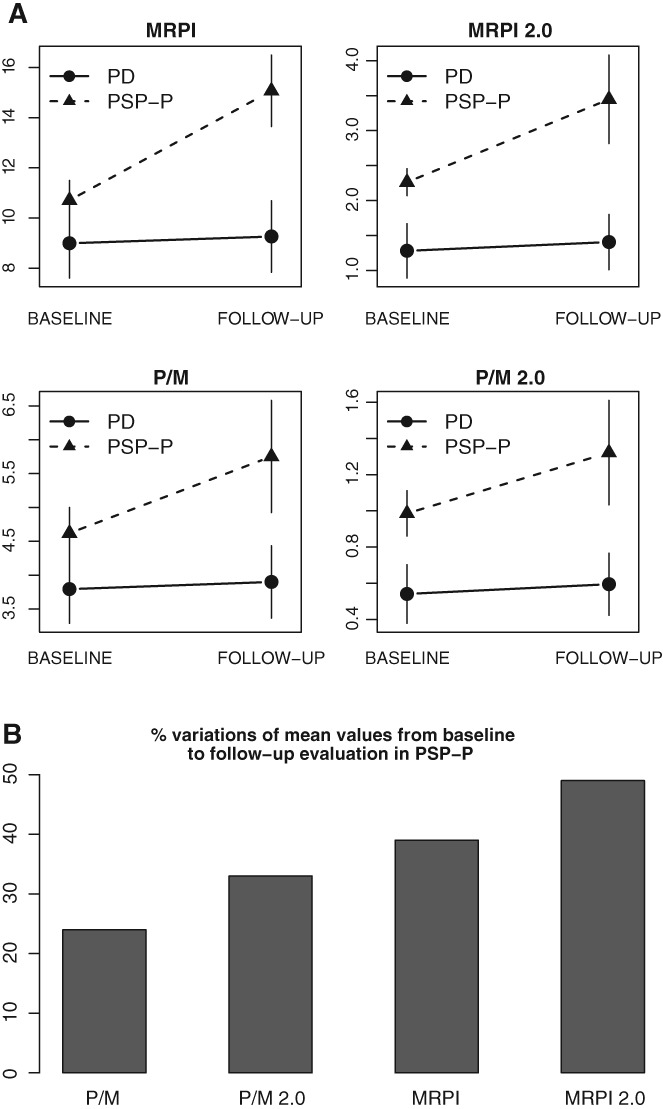
(A) Mixed‐model (time, status) effects of morphometric magnetic resonance measurements (P/M, P/M 2.0, MRPI, and MRPI 2.0), after controlling for age. Effect of MRPI: status (PD vs PSP‐P), P < .001; time (baseline vs follow‐up), P = .49; status × time, P < .001. Effect of MRPI 2.0: status, P < .001; time, P = .56; status × time, P < .001. Effect of P/M: status, P < .001; time, P = .39; status × time, P < .001. Effect of P/M 2.0: status, P < .001; time, P = .70; status × time, P = .002. (B) Percentage variations in P/M, P/M 2.0, MRPI, and MRPI 2.0 mean values from baseline to follow‐up evaluation in PSP‐P patients. P/M, pons/midbrain area ratio; MRPI, magnetic resonance parkinsonism index; Parkinson's disease (PD), patients who maintained diagnosis of PD at the end of follow‐up; progressive supranuclear palsy‐parkinsonism (PSP‐P), patients who developed clinical signs of PSP‐P during the follow‐up.
Table 3 shows the performances of P/M, P/M 2.0, MRPI, and MRPI 2.0 measurements in differentiating patients with PD from those with PSP‐P at baseline and follow‐up. At baseline, MRPI 2.0 showed the highest accuracy (100%) in predicting the appearance of VGA in all patients (n = 10) with initial diagnosis of PD who later changed diagnosis from PD to PSP‐P. At the end of follow‐up, MRPI 2.0, MRPI, and P/M 2.0 showed an excellent performance (accuracy, 100%) in differentiating PSP‐P patients from those with PD (Table 3). Baseline clinical variables showed a lower accuracy than imaging biomarkers in predicting VGA in PD patients with initial diagnosis of PD (Supplementary Table 2). In addition, at baseline, MRPI 2.0 performance was highly accurate in differentiating the PSP‐P group from the non‐PSP‐P group (PD, multiple system atrophy, and dementia with Lewy bodies patients; Supplementary Table 3). There was an excellent correlation between intrarater (intraclass correlation coefficients: 3rdV width, 0.993; frontal horns (FH) width, 0.991; 3rdV/FH, 0.995) and interrater (intraclass correlation coefficients: 3rdV width, 0.982; FH width, 0.987; 3rdV/FH, 0.989) agreement.
Table 3.
Performances of P/M, P/M 2.0, MRPI, and MRPI 2.0 measurements in differentiating patients with Parkinson's disease from patients with progressive supranuclear palsy‐parkinsonism at baseline and follow‐up
| Performance Measures | Baseline, PD vs PSP‐P | 4‐year follow‐up, PD vs PSP‐P | ||||||
|---|---|---|---|---|---|---|---|---|
| P/M | P/M 2.0 | MRPI | MRPI 2.0 | P/M | P/M 2.0 | MRPI | MRPI 2.0 | |
| Cutoff value | ≥ 4.34a | ≥ 0.87a | ≥ 9.89a | ≥ 2.13a | ≥ 4.87a | ≥ 1.01a | ≥ 12.90a | ≥ 2.88a |
| Sensitivity, % | 90.0 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Specificity, % | 87.0 | 99.0 | 72.0 | 100 | 96.0 | 100 | 100 | 100 |
| PPV, % | 40.9 | 90.9 | 26.3 | 100 | 71.4 | 100 | 100 | 100 |
| NPV, % | 98.9 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Accuracy, % | 87.3 | 99.1 | 74.6 | 100 | 96.4 | 100 | 100 | 100 |
PD, patients who maintained initial diagnosis of PD at the end of follow‐up; PSP‐P, patients with initial diagnosis of PD at baseline who developed vertical gaze abnormalities during the follow‐up; P/M, pons/midbrain area ratio; MRPI, Magnetic Resonance Parkinsonism Index; PPV, positive predictive value; NPV, negative predictive value.
Optimal cutoff values were determined using receiver operating characteristics curve analysis.
Discussion
The results of our study show that a number of patients initially classified as PD developed VGA during a 4‐year follow‐up. It is worth noting that at baseline only MRPI 2.0 predicted the development of VGA in a percentage (9.1%) of patients with an initial diagnosis of PD with 100% sensitivity and specificity, whereas at the end of follow‐up all imaging biomarkers accurately differentiated patients who had developed features of PSP‐P from those who remained classifiable as PD.
To date, no studies have investigated patients initially classified as PD who might have developed clinical features of PSP‐P, such as VGA over time. In the current study, we clinically and radiologically followed a large group of patients who fulfilled the diagnostic criteria for possible or probable PD31 to look for the appearance of VGA, which is highly suggestive of PSP, during a long follow‐up period (4 years). At the end of the 4‐year follow‐up, the majority of PD patients (100 of 110 patients; 90.9%) had not developed atypical clinical features, and each of these patients reached a probable degree of diagnostic certainty for PD.31
However, a small percentage of patients (10 of 110 patients; 9.1%) who did not show early postural instability within 3 years after the disease onset developed VGA during the 4‐year follow‐up, thus indicating the evolution toward a PSP‐P phenotype.11 In particular, at the end of follow‐up, 5 of 10 patients showed slowness of vertical saccades, and 5 had VSGP. When we applied the recent MDS criteria for PSP,11 all of these 10 patients were classified as probable PSP‐P (5 were classified as O1‐A2, 4 as O2‐A2, and 1 as O2‐A3). Thus, the revision of the initial diagnosis of PD can occur during a follow‐up, and changes in diagnosis of PD are most commonly a result of the development of additional atypical features suggesting parkinsonisms.5, 6 Our findings are consistent with previous studies5, 6 that showed that a number of patients initially diagnosed as having PD were later found to have an alternative diagnosis as a result of the development of atypical clinical features. A recent review and meta‐analysis2 showed that the overall quality of clinical diagnosis of PD was inadequate, even in tertiary centers with movement disorders experts, thus suggesting the need for easily accessible biomarkers of disease to support clinical diagnosis in vivo.
In the past few years, MRPI has been widely proposed to support clinicians in the difficult task of differentiating clinically patients with PSP from those with PD or those with other parkinsonisms.14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26 A recent study12 from the MDS‐endorsed group for PSP stated that the P/M and MRPI were the most reliable biomarkers for diagnosing PSP‐RS both in the early and late stages of the disease. However, the use of MRPI in clinical practice has been limited because the manual calculation of this combined measurement is operator dependent and time consuming. A large multicenter study in Italy recently introduced a new accurate fully automated method for MRPI calculation that does not require the manual segmentation of brain regions involved in the calculation of the index.38
To date, only a few studies have focused on evaluating the accuracy of MRPI for differentiation of patients with PSP‐P from those with PD.18, 26, 30 Some authors reported a low sensitivity of this biomarker in differentiating PSP‐P from PD,18, 30 a finding confirmed by a study in a larger cohort of PSP‐P patients.26 Recently, we demonstrated that a new version of MRPI (MRPI 2.0) was highly accurate in differentiating patients with PSP‐P from those with PD and controls.26 This version included the measurement of the 3rdV width in the calculation, a brain structure that has been reported to be enlarged in patients with PSP.26, 40
Consistent with our previous data,14, 17, 23, 26, 38 in the current study no patients with initial diagnosis of PD showed abnormal morphometric MR measurements (P/M 2.0, MRPI, and MRPI 2.0) at baseline evaluation in comparison with controls with the exception of P/M, which showed higher values in PD than in controls, a finding probably a result of the slight reduction of the midbrain area that may be observed in PD patients.41
At baseline evaluation, P/M, P/M 2.0, and MRPI values of PSP‐P patients were significantly higher than values of patients with PD, with overlapping values between the 2 groups. In contrast, baseline MRPI 2.0 values had no overlap between PD and PSP‐P patients (Fig. 1), and this biomarker showed the best performance with an accuracy of 100% in differentiating patients who maintained initial diagnosis of PD from those who changed diagnosis from PD to PSP‐P. MRPI had the worst performance (accuracy, 74.6%), whereas P/M (accuracy, 87.3%) and P/M 2.0 (accuracy, 99.1%) showed an intermediate performance in differentiating PD patients from patients with PSP‐P.
Our findings demonstrate the usefulness of these new imaging biomarkers, and specifically of the MRPI 2.0, in predicting the development of VGA and the clinical evolution towards PSP phenotypes in patients with the initial diagnosis of PD. These biomarkers could help clinicians identify very early those patients who may change the initial diagnosis from PD to PSP‐P, thus impacting on patient counseling, prognosis, and therapeutic strategies. On this basis, the use of accurate biomarkers may enable clinical trials to be performed at early stages of disease when new therapies are most likely to be efficacious. Further longitudinal studies in larger cohorts are warranted.42
At the end of the follow‐up period, MRPI, P/M 2.0, and MRPI 2.0 accurately differentiated (sensitivity and specificity of 100%) patients with PSP‐P from those with PD without an overlap of individual values. This result highlights the usefulness of these biomarkers also in supporting the clinical diagnosis of PSP‐P. Unlike our previous study,26 which showed a better performance of MRPI 2.0 than MRPI (96.6% vs 88.5%, respectively) in differentiating PSP‐P from PD, in the current study both biomarkers had a very high accuracy (100%) in differentiating between these 2 patient groups at the end of follow‐up. This discrepancy in MRPI performance can be the result of the smaller sample size of PSP‐P analyzed in the current study than that (34 PSP‐P patients) evaluated in the previous study.
In all patients who maintained a PD phenotype, all morphometric values did not vary significantly between the baseline and the end of the follow‐up period, whereas these values increased significantly in the same period of time in patients who changed the initial diagnosis from PD to PSP‐P. When the interaction between time (baseline vs follow‐up) and status (PD vs PSP‐P) was considered, we found that PD patients who developed signs of PSP‐P showed a faster course of the disease during the follow‐up period than patients who did not have a change in the initial diagnosis of PD. This more rapid disease progression rate was reflected in the change of morphometric values at 4‐year follow‐up when patients with PSP‐P were scored significantly worse (P < .001) than those with PD (Table 2 and Fig. 2A). Moreover, in the PSP‐P group, MRPI 2.0 values increased by 49.0% at the end of follow‐up when compared with baseline, whereas the worsening of the other biomarkers values was smaller with values progressively increasing from 24.0% (P/M) to 33% (P/M 2.0) up to 39.0% (MRPI). The UPDRS‐ME scores showed a similar increase between baseline and follow‐up (37.0%) to that observed with morphometric measures, thus indicating a close relationship between the extent of clinical worsening and the increase in imaging biomarkers values. By contrast, in patients who maintained initial diagnosis of PD, the morphometric values did not change significantly between baseline and follow‐up, whereas the clinical scales worsened in the same period of time, thus suggesting that the current imaging biomarkers do not reflect disease progression in PD.
To the best of our knowledge, this is the first study to assess the relationship between changes in MRI morphometric values and the progression rate of PSP‐P during a 4‐year follow‐up. Although this result may be of great interest for the evaluation of future therapies on the progression of the disease using reliable, objective, and operator‐independent MRI measurements, the small sample size limits the general applicability of our findings.
There are some limitations to this study. First, the lack of a neuropathological confirmation of the diagnosis could lead to a misclassification of patients. However, our clinical diagnoses for PD and PSP‐P were performed according to consensus diagnostic criteria,11, 31 and all patients included in our study were evaluated in a standard fashion by one of the authors (M.M.) who had more than 10 years of experience in the diagnosis of movement disorders. Although new clinical diagnostic criteria for PD have been recently proposed43 and very recently validated,44 these criteria have not been used in our patients because the patient recruitment ended before the publication of the new diagnostic criteria. Second, the abnormalities of vertical saccades may occasionally be present both in aging and in vascular parkinsonism. However, no patients had brain vascular lesions, and all patients who developed VGA had high MRPI values, whereas MRPI was reported to be within normal values in older participants41 and vascular parkinsonism.45 Third, VGA can also appear very late during the natural history of PSP‐P.7, 8, 9, 10 Thus, it is possible that some patients with a probable diagnosis of PD could develop VGA later, requiring a longer follow‐up. Fourth, the sample size of PD patients who had a change in the initial diagnosis from PD to PSP‐P during the follow‐up (10 of 110 PD patients) was small, and it cannot be concluded with certainty that MRPI 2.0, which showed the best performance in our cohort, will have a similar good performance in a different group of individuals. Further longitudinal studies in independent larger cohorts are needed to confirm our findings. Finally, our patients underwent an MR imaging examination with a 3 T scanner, which is not always available in movement disorder clinics. However, previous studies on MRPI using a 3 T scanner38, 46 showed similar results to those obtained with scanners at a lower magnetic field for MRPI measurements in patients with PD and PSP.
In conclusion, the results of our study demonstrate that a number of patients with an initial clinical diagnosis of PD developed VGA without early postural instability during a follow‐up period of 4 years, thus allowing the initial diagnosis to be refined from PD to PSP‐P. MRPI 2.0 was the most accurate biomarker, more powerful than clinical variables, in predicting the development of signs of PSP‐P in patients with an initial clinical diagnosis of PD.
Authors' Roles
1. Research project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript: A. Writing of the first draft, B. Review and Critique
A.Q.: 1A, 1B, 1C, 2A, 2C, 3A, 3B
M.M.: 1B, 1C, 3A, 3B
B.V.: 2B, 2C
S.N.: 1C
E.P.: 1C
U.S.: 1C
M.C.: 1B
V.V.: 1C
A.Q.: 1C, 3A, 3B
G.B.: 1C
C.S.: 1C
G.N.: 1C
G.A.: 1C
R.N.: 1C
F.N.: 1C
M.S.: 1C
Full financial disclosures for the previous 12 months
Nothing to report.
Supporting information
Supplementary Table S1 Comparison of clinicoradiologic data of patients who maintained initial diagnosis of Parkinson's disease (PD) at the end of follow‐up, and of patients who developed features vertical gaze abnormalities of progressive supranuclear palsy‐parkinsonism (PSP‐P) during the follow‐up.
Supplementary Table S2 Performances of demographic data, UPDRS‐ME, Hoehn‐Yahr and MMSE score values in differentiating patients with Parkinson's disease from patients with progressive supranuclear palsy‐parkinsonism.
Supplementary Table S3 Performances of P/M, P/M 2.0, MRPI and MRPI 2.0 measurements in differentiating the group of patients with Parkinson's disease, multiple system atrophy, and dementia with Lewy bodies from patients with progressive supranuclear palsy‐parkinsonism
Appendix S1. Supporting information
Acknowledgment
The authors thank the patients for their participation in the study.
Relevant conflicts of interests/financial disclosures: Nothing to report.
References
- 1. Poewe W, Seppi K, Tanner CM et al. Parkinson disease. Nat Rev Dis Primers 2017;3:17013 10.1038/nrdp.2017.13.3. [DOI] [PubMed] [Google Scholar]
- 2. Rizzo G, Copetti M, Arcuti S, Martino D, Fontana A, Logroscino G. Accuracy of clinical diagnosis of Parkinson disease: a systematic review and meta‐analysis. Neurology 2016;86:566‐576. [DOI] [PubMed] [Google Scholar]
- 3. Hughes AJ, Daniel SE, Ben‐Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 2002;125:861‐870. [DOI] [PubMed] [Google Scholar]
- 4. Joutsa J, Gardberg M, Röyttä M, Kaasinen V. Diagnostic accuracy of parkinsonism syndromes by general neurologists. Parkinsonism Relat Disord 2014;20:840‐844. [DOI] [PubMed] [Google Scholar]
- 5. Caslake R, Moore JN, Gordon JC, Harris CE, Counsell C. Changes in diagnosis with follow‐up in an incident cohort of patients with parkinsonism. J Neurol Neurosurg Psychiatry 2008;79:1202‐1207. [DOI] [PubMed] [Google Scholar]
- 6. Jankovic J, Rajput AH, McDermott MP, Perl DP. The evolution of diagnosis in early Parkinson disease. Parkinson Study Group. Arch Neurol 2000;57:369‐372. [DOI] [PubMed] [Google Scholar]
- 7. Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009;8:270‐279. [DOI] [PubMed] [Google Scholar]
- 8. Williams DR, Lees AJ. What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy‐parkinsonism (PSP‐P)? Mov Disord 2010; 25:357‐362. [DOI] [PubMed] [Google Scholar]
- 9. Boxer AL, Yu JT, Golbe LI, Litvan I, Lang AE, Höglinger GU. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol 2017;16:552‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord 2014;29:1758‐1766. [DOI] [PubMed] [Google Scholar]
- 11. Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord 2017;32:853‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Whitwell JL, Höglinger GU, Antonini A, et al. Radiological biomarkers for diagnosis in PSP: where are we and where do we need to be? Mov Disord 2017;32:955‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heim B, Krismer F, De Marzi R, Seppi K. Magnetic resonance imaging for the diagnosis of Parkinson's disease. J Neural Transm 2017;124:915‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Quattrone A, Nicoletti G, Messina D, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology 2008;246:214‐221. [DOI] [PubMed] [Google Scholar]
- 15. Hussl A, Mahlknecht P, Scherfler C, et al. Diagnostic accuracy of the magnetic resonance parkinsonism index and the midbrain‐to‐pontine area ratio to differentiate progressive supranuclear palsy from Parkinson's disease and the Parkinson variant of multiple system atrophy. Mov Disord 2010;25:2444‐2449. [DOI] [PubMed] [Google Scholar]
- 16. Lehéricy S, Hartmann A, Lannuzel A, et al. Magnetic resonance imaging lesion pattern in Guadeloupean parkinsonism is distinct from progressive supranuclear palsy. Brain 2010;133:2410‐2425. [DOI] [PubMed] [Google Scholar]
- 17. Morelli M, Arabia G, Salsone M, et al. Accuracy of magnetic resonance parkinsonism index for differentiation of progressive supranuclear palsy from probable or possible Parkinson disease. Mov Disord 2011;26:527‐533. [DOI] [PubMed] [Google Scholar]
- 18. Longoni G, Agosta F, Kostić VS, et al. MRI measurements of brainstem structures in patients with Richardson's syndrome, progressive supranuclear palsy‐parkinsonism, and Parkinson disease. Mov Disord 2011;26:247‐255. [DOI] [PubMed] [Google Scholar]
- 19. Jones N. Movement disorders: imaging differentiates progressive supranuclear palsy from Parkinson disease. Nat Rev Neurol 2011;7:186. [DOI] [PubMed] [Google Scholar]
- 20. Constantinides VC, Paraskevas GP, Velonakis G, Toulas P, Stamboulis E, Kapaki E. MRI planimetry and magnetic reasonance parkinsonism index in the differential diagnosis of patients with parkinsonism. Am J Neuroradiol 2018; 39:1047‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zanigni S, Calandra‐Buonaura G, Manners DN, et al. Accuracy of MR markers for differentiating progressive supranuclear palsy from Parkinson's disease. Neuroimage Clin 2016;11:736‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sankhla CS, Patil KB, Sawant N, Gupta S. Diagnostic accuracy of Magnetic Resonance Parkinsonism Index in differentiating progressive supranuclear palsy from Parkinson's disease and controls in Indian patients. Neurol India 2016;64:239‐245. [DOI] [PubMed] [Google Scholar]
- 23. Nigro S, Morelli M, Arabia G, et al. Magnetic Resonance Parkinsonism Index and midbrain to pons ratio: which index better distinguishes progressive supranuclear palsy patients with a low degree of diagnostic certainty from patients with Parkinson disease? Parkinsonism Relat Disord 2017;41:31‐36. [DOI] [PubMed] [Google Scholar]
- 24. Quattrone A, Nigro S. MRI measures of brainstem in Parkinsonian syndromes: where we stand and where need to go. Mov Disord 2017;32:1261. [DOI] [PubMed] [Google Scholar]
- 25. Nizamani WM, Mubarak F, Barakzai MD, Ahmed MS. Role of magnetic resonance planimetry and magnetic resonance parkinsonism index in discriminating Parkinson's disease and progressive supranucler palsy: a retrospective study based on 1.5 and 3 T MRI. Int J Gen Med 2017;10:375‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quattrone A, Morelli M, Vescio B, et al. A new MR imaging index for differentiation of progressive supranuclear palsy‐parkinsonism from Parkinson's disease. Parkinsonism Relat Disord 2018;54:3‐8. [DOI] [PubMed] [Google Scholar]
- 27. Morelli M, Arabia G, Novellino F, et al. MRI measurements predict PSP in unclassifiable parkinsonisms: a cohort study. Neurology 2011;77:1042‐1047. [DOI] [PubMed] [Google Scholar]
- 28. Mangesius S, Hussl A, Krismer F, et al. MR planimetry in neurodegenerative parkinsonism yields high diagnostic accuracy for PSP. Parkinsonism Relat Disord 2018;46:47‐55. [DOI] [PubMed] [Google Scholar]
- 29. Quattrone A, Morelli M, Williams DR et al. MR parkinsonism index predicts vertical supranuclear palsy in patients with PSP‐parkinsonism. Neurology 2016;87:1266‐1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Agosta F, Pievani M, Svetel M, et al. Diffusion tensor MRI contributes to differentiate Richardson's syndrome from PSP‐parkinsonism. Neurobiol Aging 2012;33:2817‐2826. [DOI] [PubMed] [Google Scholar]
- 31. Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol 1999;56:33‐39. [DOI] [PubMed] [Google Scholar]
- 32. Fahn S, Elton RL. Unified Parkinson's Disease Rating Scale In: Fahn S, Marsden CD, Calne D, Goldstein M, eds. Recent Developments in Parkinson's Disease. Florham Park, NJ: MacMillan Healthcare Information; 1987:153‐163. [Google Scholar]
- 33. Hoehn MM, Yahr MD. Parkinsonism: onset, progression, and mortality. Neurology 1967;17:427‐442. [DOI] [PubMed] [Google Scholar]
- 34. Schrefler C, Seppi K, Mair KJ, et al. Left hemispheric predominance of nigrostriatal dysfunction in Parkinson's disease. Brain 2012;135:3348‐3354. [DOI] [PubMed] [Google Scholar]
- 35. Folstein MF, Folstein SE, McHugh PR. "Mini‐mental state:" a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189‐198. [DOI] [PubMed] [Google Scholar]
- 36. Hughes AJ, Colosimo C, Kleedorfer B, Daniel SE, Lees AJ. The dopaminergic response in multiple system atrophy. J Neurol Neurosurg Psychiatry 1992;55:1009‐1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miskin N, Patel H, Franceschi AM, et al. Diagnosis of normal‐pressure hydrocephalus: use of traditional measures in the era of volumetric MR imaging. Radiology 2017;285:197‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nigro S, Arabia G, Antonini A, et al. Magnetic Resonance Parkinsonism Index: diagnostic accuracy of a fully automated algorithm in comparison with the manual measurement in a large Italian multicentre study in patients with progressive supranuclear palsy. Eur Radiol 2017;27:2665‐2675. [DOI] [PubMed] [Google Scholar]
- 39. Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele‐Richardson‐Olszewski syndrome): report of the NINDS‐SPSP international workshop. Neurology 1996;47:1‐9. [DOI] [PubMed] [Google Scholar]
- 40. Messina D, Cerasa A, Condino F, et al. Patterns of brain atrophy in Parkinson's disease, progressive supranuclear palsy and multiple system atrophy. Parkinsonism Relat Disord 2011;17:172‐176. [DOI] [PubMed] [Google Scholar]
- 41. Morelli M, Arabia G, Messina D, et al. Effect of aging on magnetic resonance measures differentiating progressive supranuclear palsy from Parkinson's disease. Mov Disord 2014;29:488‐495. [DOI] [PubMed] [Google Scholar]
- 42. Krismer F, Seppi K. Diagnosis of PSP‐P: Can a newly developed MRPI make the difference? Parkinsonism Relat Disord 2018;54:1‐2. [DOI] [PubMed] [Google Scholar]
- 43. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30:1591‐1601. [DOI] [PubMed] [Google Scholar]
- 44. Postuma RB, Poewe W, Litvan I, et al. Validation of the MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2018;33:1601‐1608. [DOI] [PubMed] [Google Scholar]
- 45. Mostile G, Nicoletti A, Cicero CE et al. Magnetic resonance parkinsonism index in progressive supranuclear palsy and vascular parkinsonism. Neurol Sci 2016;37:591‐595. [DOI] [PubMed] [Google Scholar]
- 46. Mangesius S, Krismer F, Gizewski ER, et al. 1.5 versus 3 Tesla magnetic resonance planimetry in neurodegenerative parkinsonism. Mov Disord 2016;31:1925‐1927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1 Comparison of clinicoradiologic data of patients who maintained initial diagnosis of Parkinson's disease (PD) at the end of follow‐up, and of patients who developed features vertical gaze abnormalities of progressive supranuclear palsy‐parkinsonism (PSP‐P) during the follow‐up.
Supplementary Table S2 Performances of demographic data, UPDRS‐ME, Hoehn‐Yahr and MMSE score values in differentiating patients with Parkinson's disease from patients with progressive supranuclear palsy‐parkinsonism.
Supplementary Table S3 Performances of P/M, P/M 2.0, MRPI and MRPI 2.0 measurements in differentiating the group of patients with Parkinson's disease, multiple system atrophy, and dementia with Lewy bodies from patients with progressive supranuclear palsy‐parkinsonism
Appendix S1. Supporting information