

Abstract
KIF1Bβ, a member of the kinesin superfamily of motor proteins, is a haploinsufficient tumor suppressor mapped to chromosome 1p36.2, which is frequently deleted in neural crest–derived tumors, including neuroblastoma and pheochromocytoma. While KIF1Bβ acts downstream of the nerve growth factor (NGF) pathway to induce apoptosis, further molecular functions of this gene product have largely been unexplored. In this study, we report that KIF1Bβ destabilizes the morphological structure of mitochondria, which is critical for cell survival and apoptosis. We identified YME1L1, a mitochondrial metalloprotease responsible for the cleavage of the mitochondrial GTPase OPA1, as a physical interacting partner of KIF1Bβ. KIF1Bβ interacted with YME1L1 through its death‐inducing region, as initiated the protease activity of YME1L1 to cleave the long forms of OPA1, resulting in mitochondrial fragmentation. Overexpression of YME1L1 promoted apoptosis, while knockdown of YME1L1 promoted cell growth. High YME1L1 expression was significantly associated with a better prognosis in neuroblastoma. Furthermore, in NGF‐deprived PC12 cells, KIF1Bβ and YME1L1 were upregulated, accompanied by mitochondrial fragmentation and apoptotic cell death. Small interfering RNA–mediated knockdown of either protein alone, however, remarkably inhibited the NGF depletion–induced apoptosis. Our findings indicate that tumor suppressor KIF1Bβ plays an important role in intrinsic mitochondria–mediated apoptosis through the regulation of structural and functional dynamics of mitochondria in collaboration with YME1L1. Dysfunction of the KIF1Bβ/YME1L1/OPA1 mechanism may be involved in malignant biological features of neural crest–derived tumors as well as the initiation and progression of neurodegenerative diseases.
Keywords: KIF1Bβ, mitochondrial fragmentation, neuroblastoma, tumor suppressor, YME1L1
1. INTRODUCTION
Cell survival and apoptosis are related to morphological changes in mitochondria.1, 2 Mitochondrial morphology is the consequence of equilibrium between two states, fusion, and fission in both healthy and dying cells.3, 4 Mitochondrial fusion results in long tubular mitochondria that are extensively interconnected to form web‐like networks encompassing the whole cell. In mammalian cells, mitochondrial fusion requires the co‐ordinated reorganization of the outer membrane and the inner membrane managed by three dynamin family GTPases, such as mitofusin 1 (Mfn1), Mfn2, and OPA1.3, 5, 6 Mfn1/2 localized in the mitochondrial outer membrane are involved in early steps in the process of membrane fusion. OPA1 is associated with the inner membrane and is essential for the inner membrane fusion. Contrary to mitochondrial fusion, mitochondrial fission drives extensive fragmentation practically simultaneous with apoptosis accompanied by the release of cytochrome c. Protein‐regulating mitochondrial fragmentation include dynamin‐related protein 1 (Drp1) and Fis1.1 Drp1 is a dynamin‐related GTPase primarily existing in the cytoplasm but partially associates into foci of the mitochondrial outer membrane where fragmentation occurs. Fis1, which is found on the surface of the outer membrane, is not localized specifically in mitochondrial scission sites. Despite the previous excellent reports describing these mitochondrial proteins that regulate mitochondrial morphology, the molecular mechanism underlying the dynamics of the mitochondrial network is largely unexplored.
KIF1B is the kinesin superfamily motor protein, which has been shown to transport mitochondria.7 Recently, we and other investigators reported that KIF1Bβ is a tumor suppressor mapping to chromosome 1p36.2 and is responsible for induction of apoptosis in neuroblastoma and pheochromocytoma.8, 9, 10 Loss of 1p36 is often seen in unfavorable neuroblastoma,11 and loss‐of‐function mutations in KIF1Bβ have been identified in neuroblastoma, pheochromocytoma, and medulloblastoma,10 indicating that KIF1Bβ is one of the pathogenic targets for these diseases. In addition, a loss‐of‐function mutation in the motor domain of KIF1Bβ is a genetic cause of human peripheral neuropathy, Charcot‐Marie‐Tooth disease type 2A (CMT2A).12 Interestingly, mutations in Mfn2, one of Mfn described above, have also been detected in patients with CMT2A in whom no mutation in KIF1Bβ is recognized.13 These results strongly suggest the possibility that KIF1Bβ, one of the major molecular motors of microtubule‐based intracellular transport, might have genetic and functional interactions with mitochondria.
In this study, we investigated the potential involvement of KIF1Bβ in the process of morphological alteration in mitochondria. We provide evidence that KIF1Bβ regulates mitochondrial fission in co‐operation with a mitochondrial metalloprotease YME1L1, to induce mitochondrial apoptosis.
2. MATERIALS AND METHODS
2.1. Plasmid constructs
Construction of the plasmid encoding full‐length human KIF1Bβ has been described previously.10 The KIF1Bβ‐GFP deletion construct (KIF1BβΔ3‐GFP) was produced by polymerase chain reaction (PCR)‐based amplification. Full‐length human YME1L1 was amplified using a complementary DNA (cDNA) template purchased from Invitrogen (No. 2961446; Carlsbad, CA). The YME1L1 fragment was cloned into the KpnI and NheI restriction sites of pcDNA3.1/Myc‐His B (Invitrogen) using the following primers: (sense) 5′‐CTAGCTAGCTAGGCCATGTTTTCCTTGTCGAGC; (antisense) 5′‐GGGGTACCCTCACTTCCAACTTTTTC.
2.2. Cell culture and transfection
Human cervical carcinoma HeLa (Kyoto) cells and neuroblastoma NB‐1 and SH‐SY5Y cells were grown in Dulbecco's modified Eagle's medium (DMEM) or Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum (FBS). Transient DNA and small interfering RNA (siRNA) transfection were performed using FuGENE HD transfection reagent (Roche Applied Science, Penzberg, Germany) or Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer's instruction. YME1L1‐specific and nontargeting control siRNAs were purchased from Invitrogen.
2.3. Neuronal apoptosis assay
Rat pheochromocytoma PC12 cells were cultured in DMEM medium supplemented with 10% horse serum (HS) and 5% FBS on collagen IV–coated dishes. The cells were treated with nerve growth factor (NGF; DMEM supplemented with 1% HS and 100 ng/mL NGF [N6009; Sigma, St. Louis, MO]) for 6 days, followed by NGF withdrawal (DMEM supplemented with anti‐NGF antibody [N6655; Sigma]).
2.4. Fluorescent microscopy
MitoTracker Red CMXRos (M7512; Invitrogen) and an anti‐cytochrome c antibody (6H2.B4; BD Pharmingen, Franklin Lakes, NJ) were used to detect mitochondria. The details are described in the Supporting Information.
2.5. Flow cytometry
Cells were collected and washed with ice cold phosphate‐buffered saline. Cells were treated with 500 µg/mL of RNase A (Sigma) and subsequently stained with 50 µg/mL of propidium iodide (Sigma) with 0.2% Triton X‐100 for 15 minutes at 37°C and were then analyzed by a fluorescence‐activated cell sorting (FACS) calibur flow cytometer (Beckton Dickinson, Franklin Lakes, NJ).
2.6. Mitochondrial membrane potential
JC‐1 probe (ImmunoChemistry Technologies, Bloomington, MN) was used to measure ΔΨm, according to the manufacturer's instructions. The red fluorescence of JC‐1, which represents ΔΨm, was quantified using an FACS flow cytometer.
2.7. Subcellular fractionation
Cells were harvested and resuspended in 500 μL of fractionation buffer (20 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM ethylenediaminetetraacetic acid, 1 mM ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid, and 250 mM sucrose). After sonication and centrifugation at 800g for 5 minutes, the resulting supernatant was then centrifuged at 10 000g for an additional 15 minutes. Supernatants and pellets from the final centrifugation step comprised the cytoplasmic and mitochondrial fractions, respectively.
2.8. Yeast two‐hybrid screening assay
For yeast two‐hybrid assays, the death‐inducing region (DIR) of KIF1Bβ was used as bait, and the fetal brain cDNA library (Matchmaker 3; Clontech, Mountain View, CA) was used as a prey. Yeast cells were cotransformed with both plasmids, and positive clones were chosen from 1 × 105 colonies using a blue‐white selection procedure, in accordance with the manufacturer's instructions.
2.9. Glutathione S‐transferase pull‐down assay
The glutathione S‐transferase GST‐YME1L1 fusion protein was purified using Glutathione Sepharose 4B beads (GE Healthcare, Chicago, IL). The DIR of KIF1Bβ was radiolabelled in vitro using the TnT T7 Quick Coupled transcription/translation system (Promega, Madison, WI) in the presence of [35S] methionine and then incubated with either mock or GST‐YME1L1 fusion proteins. The pull‐down assay was performed as described previously.14
2.10. Immunoblotting and immunoprecipitation
Immunoblotting and immunoprecipitation assays were performed as described previously.15 The details are described in the Supporting Information.
2.11. Reverse transcriptase PCR and real‐time quantitative RT‐PCR
Total RNA was extracted from 101 neuroblastoma clinical samples using TRIzol reagent (Invitrogen), according to the manufacturer's protocol. The reverse transcription reaction was performed using the SuperScript II reverse transcriptase (Invitrogen). Real‐time quantitative PCR (SYBR Green PCR) was conducted using an ABI Prism 7700 sequence detection system (Perkin‐Elmer Applied Biosystems, Foster City, CA), as described in the Supporting Information.
2.12. Statistical analysis
Statistical analyses were performed using Student's t tests. Kaplan‐Meier analysis was performed to evaluate overall survival. A P value less than 0.05 was considered to be significant.
3. RESULTS
3.1. KIF1Bβ induces mitochondrial fragmentation
We first examined whether the overexpression of KIF1Bβ could affect mitochondrial morphology. Such morphological changes in mitochondria were conventionally quantified by measuring the length of mitochondria (Figure S1). In our experiment, mitochondria show a mixture of granular and filamentous forms with an average length of 2.6 μm in HeLa cells under normal condition (Figure 1A). Intriguingly, the overexpression of KIF1Bβ induced the enrichment of granulated structures in mitochondria, an indicator of mitochondrial fragmentation, whereas such morphological changes were not observed in the cells overexpressed with KIF1Bα, the alternative splicing variant of KIF1B in the C‐terminal region,10 suggesting that KIF1Bβ but not KIF1Bα might have an ability to regulate the mitochondrial morphology. To further confirm mitochondrial fragmentation mediated by KIF1Bβ, we used the lipophilic mitochondrial JC‐1 fluorescent probe to measure membrane potential (ΔΨm), as an indicator of structural maintenance in the mitochondrial membrane.16, 17 Consistently, the overexpression of KIF1Bβ decreased red fluorescence value of the lipophilic mitochondrial JC‐1 probe, indicating that KIF1Bβ lowered ΔΨm (Figure 1B). These results signify that KIF1Bβ induces disruption of mitochondrial structure. Meanwhile, we found that KIF1Bβ‐mediated fragmentation in mitochondria was accompanied by apoptosis. FACS analysis showed that the number of cells in sub‐G0/G1 phases was remarkably increased in KIF1Bβ‐expressed HeLa cells, as compared with mock‐treated ones (Figure 1C). Also the cleavage of poly ADP‐ribose polymerase (PARP), a classical maker of apoptosis, was also observed in those cells (Figure 1C). Moreover, subcellular fractionation revealed that the overexpression of KIF1Bβ released cytochrome c from mitochondrial to cytoplasmic fractions (Figure 1D), indicating the induction of mitochondrial apoptosis. Taken together, our findings demonstrate that KIF1Bβ induces apoptosis through mediating mitochondrial fragmentation.
Figure 1.
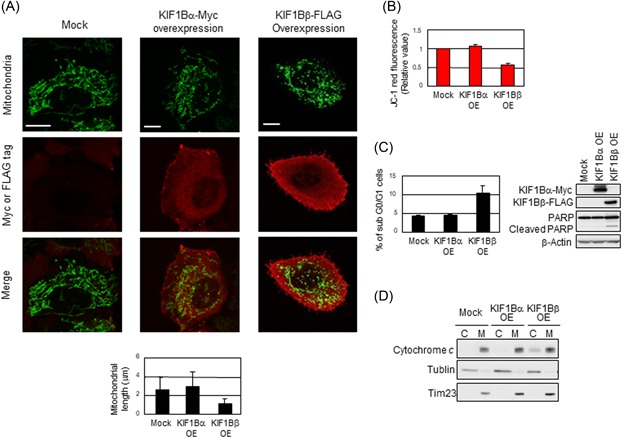
Overexpression of KIF1Bβ induces mitochondrial fragmentation. A, Overexpression of KIF1Bβ results in mitochondrial fragmentation. HeLa cells were transfected with empty vector (Mock), KIF1Bα‐Myc, or KIF1Bβ‐FLAG expression vectors. After 48 hours, mitochondrial morphology was observed by indirect immunofluorescence using the BacMam‐GFP mitochondrial probe (green), Myc or FLAG tag antibodies (red). Scale bar, 25 µm. Average mitochondrial length is shown in a bar plot (n = 100). Data are presented as means ± SD (B) KIF1Bβ overexpression decreases membrane potential (ΔΨm). The average values of JC‐1 red fluorescence are shown as the normalized values by the value obtained from control. *P < 0.05 (n = 3). C, Overexpression of KIF1Bβ induces apoptosis. Cells in (A) were analyzed by flow cytometry (left panel) for the sub‐G0/G1 population. Cleavage of PARP was detected by Western blot analysis experiments using whole cell lysates (right panel). D, KIF1Bβ overexpression induces cytochrome c release from mitochondria to cytoplasm. Cells in (A) were fractionated into cytoplasmic (C) and mitochondrial (M) fractions and was subjected to immunoblotting. PARP, poly ADP‐ribose polymerase [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Deficiency in KIF1Bβ yields mitochondrial fusion
We next investigated the effect of deficiency in KIF1Bβ on mitochondrial morphology, utilizing NB‐1 cell line derived from a primary neuroblastoma sample.18 This cell line, harboring homozygous deletion at chromosomal region 1p36, is a null deletion mutant strain in regard to KIF1Bβ.9, 18 SH‐SY5Y, another neuroblastoma‐derived cell line, as well as HeLa cells expressing KIF1Bβ were used as KIF1Bβ‐positive controls. Among these cell lines, only NB‐1 exhibited spontaneous fusion in mitochondria (Figure S2A). However, such elongated structures reverted into the granulated pattern with the add‐back of KIF1Bβ, but not its variant KIF1Bα (Figure 2A), accompanied by a sharp decrease in ΔΨm (Figure 2B), further supporting our finding that KIF1Bβ plays a role in mitochondrial fragmentation. Because mitochondrial fusion renders the cells resistant to stress stimulation,2 we examined cellular stress response in the presence or absence of KIF1Bβ under the treatment with doxorubicin. Expectedly, naïve NB‐1 cells showed resistance to doxorubicin treatment, as indicated by less number of sub‐G0/G1 cells compared with KIF1Bβ‐positive cell lines (Figure S2B). The resistance of NB‐1 cells was largely attenuated with the add‐back of KIF1Bβ (Figure 2C).
Figure 2.
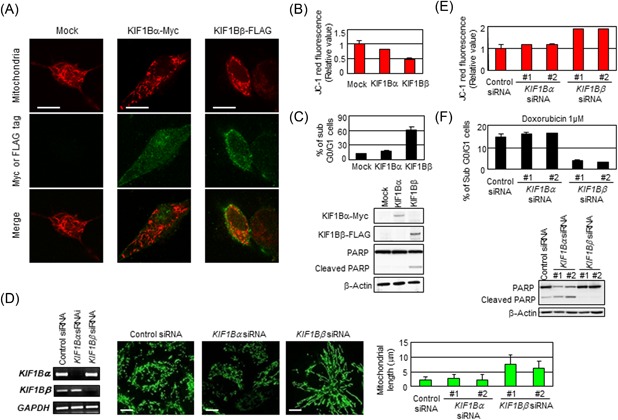
Deficiency in KIF1Bβ yields mitochondrial fusion. A, Add‐back of KIF1Bβ reverts spontaneous mitochondrial fusion in NB‐1 cells. NB‐1 cells were transfected with empty vector (Mock), KIF1Bα‐Myc, or KIF1Bβ‐FLAG expression vectors. After 48 hours, mitochondria are visualized by MitoTrackerRed CMXRos. B, Add‐back of KIF1Bβ decreases membrane potential. The average values of JC‐1 red fluorescence are measured in NB‐1 cells and shown as the normalized values by the value obtained from control. *P < 0.05 (n = 3). C, Add‐back of KIF1Bβ attenuates drug resistance. NB‐1 cells transfected with KIF1Bα or KIF1Bβ were treated with doxorubicin. Apoptosis was analyzed by flow cytometry and immunoblotting. D, Knockdown of KIF1Bβ results in mitochondrial fusion. HeLa cells were transfected with a scramble siRNA (control) or two kinds of specific siRNAs against KIF1Bα or KIF1Bβ for 48 hours. Knockdown efficiency was confirmed by semiquantitative RT‐PCR (left panel). Morphological changes in mitochondria were observed by indirect immunofluorescence using the BacMam‐GFP mitochondrial probe (green). Scale bar, 25 µm. The average mitochondrial length was measured (right panel, n = 100). E, Knockdown of KIF1Bβ increases membrane potential ΔΨm. The values of JC‐1 red fluorescence in the cells in (D) were measured and shown as the normalized values by the value obtained from control. F, Knockdown of KIF1Bβ inhibits apoptosis induced by doxorubicin. After the siRNA knockdown, HeLa cells were treated with 1 µM doxorubicin for 48 hours. Apoptosis was analyzed by flow cytometry and immunoblotting. RT‐PCR, real‐time polymerase chain reaction; siRNA, small interfering RNA [Color figure can be viewed at wileyonlinelibrary.com]
On the other hand, in HeLa cells expressing KIF1Bβ, siRNA‐mediated knockdown of KIF1Bβ apparently resulted in elongation of mitochondria, while knockdown of KIF1Bα did not show a major change in mitochondrial morphology (Figures 2D and S3). Also, knockdown of KIF1Bβ increased mitochondrial membrane potential ΔΨm (Figure 2E), suggesting mitochondrial fusion.19 Correspondingly, knockdown of KIF1Bβ inhibited apoptosis in response to doxorubicin treatment in HeLa cells (Figure 2F). Taken together, these findings support our view that KIF1Bβ may induce apoptosis through mediating mitochondrial fragmentation.
3.3. The DIR of KIF1Bβ is essential for mitochondrial fragmentation
We previously reported that KIF1Bβ has a DIR in the C‐terminal domain, which is capable of inducing apoptosis, while KIF1Bα does not have such region.9 To examine whether this region was involved in fragmentation in mitochondria, we constructed a deletion mutant of KIF1Bβ lacking the C‐terminal half including the DIR, namely KIF1BβΔ3 (Figure 3A). The overexpression of full‐length KIF1Bβ‐GFP in HeLa cells increased the number of sub‐G0/G1 cells and cleavage of PARP, while KIF1BβΔ3‐GFP did not, confirming that the DIR is capable of inducing apoptosis (Figure 3A). Utilizing these expression vectors, mitochondrial morphology in transfected cells was observed. Whereas full‐length KIF1Bβ facilitates fragmentation of mitochondria, and the overexpression of KIF1BβΔ3‐GFP did not significantly change the morphology (Figure 3B). In accordance with this, the addition of KIF1BβΔ3‐GFP did not nullify spontaneous fusion in NB‐1 cells, in sharp contrast to that of full‐length KIF1Bβ (Figure S2D). These results proved that the DIR of KIF1Bβ is required to induce mitochondrial fragmentation.
Figure 3.
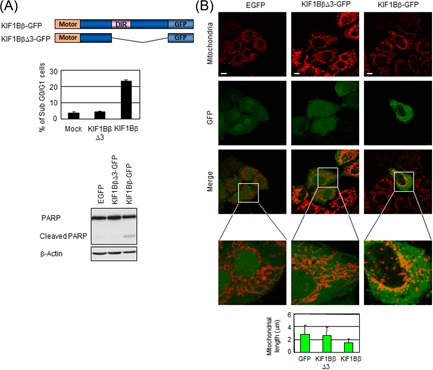
The death‐inducing region of KIF1Bβ is essential for mitochondrial fragmentation. A, The death‐inducing region is essential for apoptosis induction. Schematic representation of GFP‐tagged KIF1Bβ full‐length and its death‐inducing region deletion mutant KIF1BβΔ3 is shown (top panel). HeLa cells were transfected with EGFP (control), KIF1BβΔ3‐GFP, or KIF1Bβ‐GFP for 48 hours and then subjected to flow cytometry (lower left panel) and immunoblotting (lower right panel). B, The overexpression of KIF1BβΔ3 does not induce mitochondrial fragmentation. EGFP, KIF1BβΔ3‐GFP, or KIF1Bβ‐GFP (green) was overexpressed in HeLa cells. Mitochondria were visualized by the BacMam‐RFP mitochondrial probe (red). Scale bar, 25 µm. Average mitochondrial length is shown in a bar plot (n = 100). DIR, death‐inducing region; EGFP, enhanced green fluorescent protein; Motor, motor domain; PARP, poly ADP‐ribose polymerase [Color figure can be viewed at wileyonlinelibrary.com]
3.4. The mitochondrial metalloprotease YME1L1 physically interacts with KIF1Bβ
To better understand the mechanism of KIF1Bβ‐mediated mitochondrial apoptosis, we sought to identify potential interacting partners of this protein. Toward this end, a yeast two‐hybrid screening assay was performed, using the DIR of KIF1Bβ as bait.9 From this screen, YME1L1 was identified as a protein that physically interacts with the DIR of KIF1Bβ. YME1L1 is a mitochondrial metalloprotease involved in the cleavage of OPA1, a protein that localizes to the inner mitochondrial membrane.20, 21, 22 GST pull‐down (Figure 4A) and immunoprecipitation (Figure 4B) assays were also performed to confirm the interaction between YME1L1 and KIF1Bβ, both in vitro and in vivo. GST‐tagged YME1L1 successfully affinity‐precipitated a radiolabeled in vitro translation product of the DIR of KIF1Bβ, as well as full‐length KIF1Bβ. In addition, GST‐tagged KIF1Bβ (full‐length) could successfully affinity‐precipitate YME1L1 (Figure 4A). Immunoprecipitation assays also demonstrated a physical interaction between FLAG‐tagged KIF1Bβ and Myc‐tagged YME1L1. Moreover, the interaction of YME1L1 with endogenous KIF1Bβ was detected using an anti‐YME1L1 antibody (Figure 4B). These results suggest that the mitochondrial metalloprotease YME1L1 physically interacts with KIF1Bβ.
Figure 4.
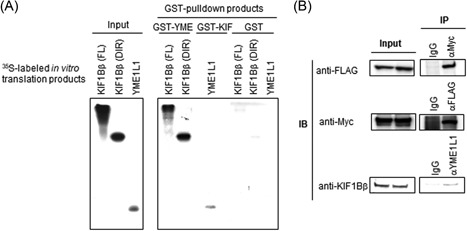
Mitochondrial metalloprotease YME1L1 interacts with KIF1Bβ. A, Physical interaction between the death‐inducing region of KIF1Bβ and YME1L1 in vitro. [35S] labeled KIF1Bβ death‐inducing region (DIR), full‐length KIF1Bβ, or YME1L1 was incubated with recombinant GST‐YME1L1, GST‐KIF1Bβ, or GST negative control. GST proteins were analyzed by Western blot analysis and autoradiography. B, Physical interaction between KIF1Bβ and YME1L1 in vivo. Lysates from HeLa cells cotransfected with expression plasmids encoding KIF1Bβ‐FLAG and YME1L1‐Myc were used in immunoprecipitation (IP) assays using the indicated antibodies, followed by immunoblotting. Endogenous KIF1Bβ was immunoprecipitated using a YME1L1‐specific antibody and detected with an anti‐KIF1B antibody (bottom row)
3.5. YME1L1 overexpression induces apoptosis, while YME1L1 deficiency promotes cell growth
We then asked whether YME1L1 shares similar cellular functions to those of KIF1Bβ. Given that YME1L1 functions in the apoptotic pathway with KIF1Bβ, we wondered whether YME1L1 exhibited tumor suppressive activity because KIF1Bβ acts as a tumor suppressor in neuroblastoma. To ascertain this, we measured YME1L1 expression levels in 101 primary samples of neuroblastoma by quantitative real‐time PCR and analyzed the data using a log‐rank test. High YME1L1 expression was statistically associated with a better prognosis (P = 0.046), while low YME1L1 expression was associated with a poor rate of survival (Figure 5A). Furthermore, similar to KIF1Bβ, overexpression of YME1L1 induced apoptosis, as demonstrated by PARP cleavage (Figure 5B). Conversely, siRNA‐mediated knockdown of YME1L1 promoted cell growth (Figure 5C). Taken together, these results suggest that YME1L1 exhibits tumor suppressive activity, facilitating mitochondrial apoptosis. Therefore, KIF1Bβ may have a specific role at mitochondria, where YME1L1 plays a central role in apoptosis.
Figure 5.
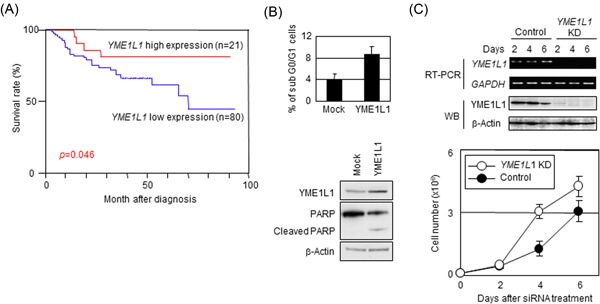
Overexpression of YME1L1 induces apoptosis, while the deficiency in YME1L1 facilitates cell growth. A, Cumulative survival curves for neuroblastoma patients (n = 101). Kaplan‐Meier survival curves for patients with neuroblastoma categorized according to YME1L1 expression. B, Overexpression of YME1L1 induces apoptosis. HeLa cells were transfected with either empty vector (Mock) or the YME1L1 expression vector and were then analyzed by flow cytometry (top panel) and immunoblotting (bottom panel). C, Knockdown of YME1L1 facilitates cell growth. 1 × 105 HeLa cells were treated with siRNAs (control or YME1L1 siRNA), and the cell number was then counted every day for a total of 6 days (bottom panel) in triplicate. siRNA knockdown efficacy was confirmed by RT‐PCR and immunoblotting (top panel). *P < 0.05. PARP, poly ADP‐ribose polymerase; siRNA, small interfering RNA; RT‐PCR, real‐time polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
3.6. KIF1Bβ promotes mitochondrial fragmentation through binding YME1L1
We next sought to investigate the molecular mechanism underlying KIF1Bβ‐mediated mitochondrial fragmentation via interaction with YME1L1. For this purpose, HeLa cells were transfected with KIF1Bβ, YME1L1, or both expression vectors, and then the mitochondrial morphology was examined. The ratio of fragmented cells to nonfragmented cells was the highest in cotransfected cells, suggesting that cotransfection of KIF1Bβ and YME1L1 induced the most severe fragmentation in mitochondria, compared with single transfection (Figure 6A). In addition, cotransfection yielded the lowest value in ΔΨm and the highest number of sub‐G0/G1 population (Figure 6B). These results suggest that KIF1Bβ co‐ordinates with YME1L1 to promote mitochondrial fragmentation and apoptosis.
Figure 6.
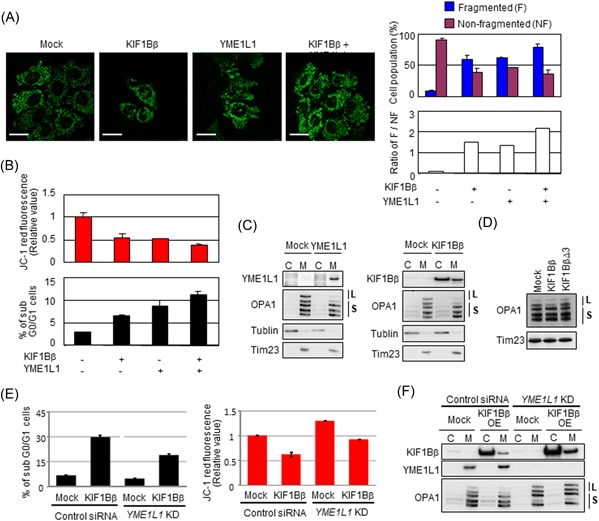
KIF1Bβ promotes mitochondrial fragmentation through binding YME1L1. A, Coexpression of KIF1Bβ and YME1L1 promotes mitochondrial fragmentation. HeLa cells were transfected with KIF1Bβ, YME1L1 or both, and mitochondrial morphologies were visualized with BacMam‐GFP mitochondrial probe (green). Cell population (%) harboring fragmented mitochondria and nonfragmented mitochondria, as well as their ratios (white bars), are also shown (n = 100). B, Coexpression of KIF1Bβ and YME1L1 induces apoptosis. Cells in (A) were used for mitochondrial membrane potential ΔΨm (top panel) and FACS (bottom panel) analyses. C, Overexpression of either YME1L1 or KIF1Bβ induces the cleavage of the long forms of OPA1. Cells transfected with each expression vector were used for subcellular fractionation. D, The death‐inducing region of KIF1Bβ is necessary to cleave the long forms of OPA1. HeLa cells were transfected with full‐length KIF1Bβ or KIF1BβΔ3 expression vectors for 48 hours and mitochondrial fractions were prepared for immunoblotting. It should be noted that the largest form of OPA1 (top band) occasionally receives spontaneous cleavage, and thus it disappears regardless of experimental conditions. E, Depletion of YME1L1 protects from KIF1Bβ‐induced cell death and decreases membrane potential ΔΨm. KIF1Bβ was overexpressed in YME1L1‐knocked down HeLa cells. These cells and control cells were used for FACS analyses (left panel) and mitochondrial membrane potential ΔΨm (right panel). F, Overexpression of KIF1Bβ in the absence of YME1L1 does not alter long forms of OPA1. The cells used in (E) were subjected to Western blot analysis. C, cytoplasm; FACS, fluorescence‐activated cell sorting; L, long‐form OPA1; M, mitochondria; S, short form OPA1 [Color figure can be viewed at wileyonlinelibrary.com]
YME1L1 has been identified as a mitochondrial metalloprotease that cleaves mitochondrial protein OPA1.19, 20, 21 We further analyzed the proteolysis of OPA1 catalyzed by YME1L1 in these cells. OPA1 is a dynamin‐related GTPase responsible for the control of mitochondrial fusion.21, 22 Alternative splicing and proteolysis of OPA1 yield five bands, including long forms (L‐OPA1; Figure 6C, upper two bands indicated by L) and short forms (S‐OPA1; lower three bands indicated by S).23 It has been demonstrated that OPA1 must be present in both long and short forms for fusion to proceed with the balance of those forms being maintained by constitutive processing.24 In contrast, the cleavage of L‐OPA1 triggers mitochondrial fission.25 As expected, the overexpression of YME1L1 decreased L‐OPA1 (Figure 6C; left panel). Interestingly, KIF1Bβ overexpression also decreased L‐OPA1 (Figure 6C, right panel), whereas the overexpression of KIF1BβΔ3 failed (Figure 6D), indicating that the DIR of KIF1Bβ is necessary for OPA1 cleavage by YME1L1. Taken together, these results suggest that KIF1Bβ might stimulate the protease activity of YME1L1 by physical interaction through the DIR, resulting in OPA1 cleavage that leads to mitochondrial fragmentation.
We also checked whether depletion of YME1L1 protects from KIF1Bβ‐induced cell death. We overexpressed KIF1Bβ in YME1L1‐knocked down cells and checked the sub‐G0/G1 fraction (Figure 6E, left panel). The sub‐G0/G1 fraction in KIF1Bβ overexpressed control siRNA–treated cells was 29.6% (second row in Figure 6E). And this was decreased to 18.8% in KIF1Bβ overexpressed YME1L1‐knocked down cells (fourth row in Figure 6E). This result shows that the depletion of YME1L1 protected cells from KIF1Bβ‐induced cell death. The mitochondrial membrane potential ΔΨm also recovered with the depletion of YME1L1 (Figure 6E, right panel). Surprisingly, the L‐OPA1 was not cleaved in YME1L1‐depleted cells with KIF1Bβ overexpression (Figure 6F).
In addition, we measured the expression levels of OPA1 in the same sample set of 101 primary neuroblastomas. Opposite to the YME1L1 expression, higher expression levels of OPA1 in patients with neuroblastoma were closely associated with a low survival rate (Figure S5), implying that dysfunction of KIF1Bβ/YME1L1/OPA1 might contribute to the malignant behaviors of neuroblastoma.
3.7. NGF depletion upregulates KIF1Bβ and YME1L1 in PC12 cells leading to apoptosis
KIF1Bβ has been identified as a downstream molecule of the NGF pathway to induce apoptosis. To further investigate the functional role of KIF1Bβ/YME1L1 during NGF depletion–induced apoptosis, we used rat pheochromocytoma‐derived PC12 cell line, a model system to mimic programmed cell death during neuronal development, to examine the expressions of KIF1Bβ and YME1L1 during NGF deprivation–induced apoptosis. The PC12 cell line was cultured in the presence of NGF for 6 days followed by continuous incubation with NGF (NGF incubation) or removal of NGF (NGF depletion). Under our experimental conditions, NGF addition induced neuronal differentiation (data not shown) and subsequent NGF depletion resulted in robust induction of apoptosis (Figure 7A), which was accompanied by mitochondrial fragmentation (Figure 7B). Consistently, NGF depletion invoked the upregulation of both KIF1Bβ and YME1L1 at protein levels at 12 hours after NGF withdrawal in a time‐dependent manner (Figure 7A and 7C), indicating that KIF1Bβ and YME1L1 are involved in the initiation and progression of NGF deprivation–induced apoptosis. On the other hand, NGF depletion–induced apoptotic cell death was strikingly inhibited by knockdown mediated by two kinds of siRNA specifically against KIF1Bβ or YME1L1 in PC12 cells (Figure 7D), supporting our notion that KIF1Bβ/YME1L1‐mediated mitochondrial fragmentation plays a critical role in NGF deprivation–induced apoptosis.
Figure 7.
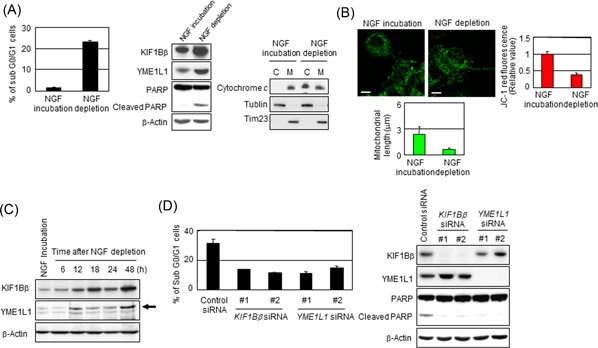
NGF depletion activates KIF1Bβ and YME1L1 in PC12 cells leading to apoptosis. A, NGF depletion results in apoptotic cell death in PC12 cells. PC12 cells were treated with NGF for 6 days and then were incubated with or without NGF for additional 2 days. Cells were subjected to FACS or immunoblotting analyses. B, NGF depletion promotes mitochondrial fragmentation in PC12 cells. Mitochondrial morphology in the cells in (A) was observed by indirect immunofluorescence utilizing BacMam‐GFP (green). The mitochondrial length was measured, and average values are shown (n = 100). Membrane potential was quantified by the JC‐1 red fluorescence measurement assay and was shown as the normalized values by the value obtained from NGF‐treated cells. *P < 0.05 (n = 3). C, NGF depletion activates KIF1Bβ and YME1L1 in PC12 cells. At indicated time points, cells were collected and subjected for immunoblotting. Endogenous rat YME1L1 is indicated by the arrowhead. D, Knockdown of either KIF1Bβ or YME1L1 prevents apoptosis mediated by NGF depletion. At 6 days after incubation with NGF, NGF was depleted from culture medium. Simultaneously, the knockdown of KIF1Bβ or YME1L1 utilizing independent siRNAs for each gene was performed for 2 days. Cells were used for FACS or immunoblotting analyses. C, cytoplasm; FACS, fluorescence‐activated cell sorting; M, mitochondria; siRNA, small interfering RNA [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
In this study, we report for the first time that KIF1Bβ, a downstream molecule of the NGF pathway, physically interacts with mitochondrial metalloprotease YME1L1 utilizing its DIR to promote the proteolysis of OPA1 catalyzed by YME1L1, resulting in mitochondrial fragmentation and consequent mitochondrial apoptosis. These findings explain how KIF1Bβ affects cell fate through the alteration of mitochondrial morphology, which plays an important role in NGF deprivation–induced apoptosis. Dysfunction of the KIF1Bβ/YME1L1/OPA1 mechanism may be involved not only in NGF‐responsive nervous system diseases but also in mitochondrial morphological aberration–associated diseases.
We found that the DIR of KIF1Bβ was essential for its function in the regulation of mitochondrial morphology (Figure 3B). Using this region as bait, we identified YME1L1 as a binding partner of KIF1Bβ. The overexpression of YME1L1 led the induced cleavage of the long‐form OPA1 (Figure 6C), which has been regarded to be related to the mitochondrial fragmentation.24, 25 Intriguingly, full‐length KIF1Bβ but not KIF1BβΔ3, a deletion mutant lacking the DIR, decreased the long forms of OPA1 (Figure 6C and 6D). Moreover, in KIF1Bβ null NB‐1 cells that express YME1L1 and OPA1 (data not shown), mitochondria exhibited spontaneous fusion pattern (Figure S2A), which was reversed to granulated structures following the add‐back of KIF1Bβ (Figure 2A). On the basis of these findings, it is plausible that KIF1Bβ is essential for the YME1L1's protease activity. Binding of KIF1Bβ to YME1L1 via its DIR probably might initiate the protease activity of YME1L1 for cleavage of the long‐form OPA1, resulting in mitochondrial fragmentation as well as consequent apoptosis. KIF1Bβ is a motor protein localized mainly in the cytoplasm,9 whereas YME1L1 is a mitochondrial protein localized at the inner membrane.20 Interaction of KIF1Bβ with YME1L1 suggests a possibility that KIF1Bβ might promote the transportation of YME1L1 into mitochondria. However, the amount of YME1L1 existing in mitochondria did not significantly change regardless of the protein level of KIF1Bβ (data not shown), implying that KIF1Bβ might not play an active role in the transportation of YME1L1. The mechanism underlying how KIF1Bβ regulates the YME1L1 activity needs to be investigated deeply.
Our findings provide a critical clue to link mitochondrial morphology and cancer biology. KIF1Bβ has been identified as a downstream target of the NGF pathway.9, 10 Both KIF1Bβ and YME1L1 were upregulated at the earlier time after NGF deprivation in PC12 cells (Figure 7A and 7C). Furthermore, knockdown of KIF1Bβ and YME1L1 largely attenuated NGF withdrawal–induced apoptosis (Figure 7D). These results reveal that the regulation of KIF1Bβ/YME1L1/OPA1 on mitochondrial morphology plays a critical role in NGF‐dependent program cell death. Recently, Li et al26 also found that KIF1Bβ controls mitochondria morphology. They found that KIF1Bβ activates calcineurin (CN) and that KIF1Bβ affects mitochondrial dynamics through CN‐dependent dephosphorylation of Drp1, causing mitochondrial fission and apoptosis. Their findings are similar to our findings that KIF1Bβ plays an important role in mitochondrial morphology and cell apoptosis.
In neuroblastoma, one of the neural crest–derived tumors, some tumors exhibit NGF dependence for survival and differentiation, and these tumors exhibit high expressions of NGF receptors TrkA and P75NTR.27 In these tumors, NGF deficiency induces spontaneous regression and is associated with a more favorable outcome. However, tumors lacking NGF dependence often possess aggressive features and are associated with poorer prognosis. Notably, loss of heterogeneity at the KIF1Bβ locus is frequently observed in advanced neuroblastoma, which is accompanied by decreased KIF1Bβ expression.10 We found that high YME1L1 expression was associated with a better overall survival in neuroblastoma (Figure 5A). In addition, the expressions of YME1L1 and OPA1 appear to be oppositely associated with the survival rate of patients with neuroblastoma in our sample set (Figures 5A and S4). These findings suggest that dysfunction of the KIF1Bβ/YME1L1 complex may facilitate the escape of tumor cells from NGF deprivation–induced apoptosis, contributing to their malignant behavior and, consequently, unfavorable prognosis in neuroblastoma. Supportively, add‐back of KIF1Bβ in KIF1Bβ null neuroblastoma cell line NB‐1 induced mitochondrial fragmentation and remarkably attenuated the resistance to doxorubicin treatment (Figure S2). Besides our results that YME1L1 has an ability to inhibit cell growth and to induce apoptosis (Figure 5B and 5C), it has been reported that OPA1 has an antiapoptotic function and knockdown of OPA1 induced the release of cytochrome c and apoptosis.28, 29 In addition to neuroblastoma, loss‐of‐function mutations in KIF1Bβ have been identified in other neural crest–derived tumors, including pheochromocytoma and medulloblastoma.11 Further investigation of the mechanism underlying KIF1Bβ/YME1L1‐mediated regulation of mitochondrial biology should provide additional insights into our understanding of the oncogenesis and malignant progression of these tumors as well as the development of effective therapeutic strategies.
Aberrant mitochondrial morphology is one of the major characteristics in neurodegenerative disease. For instance, mitochondrial fission is frequently observed in Alzheimer's and Parkinson's diseases.30, 31 In Alzheimer's disease, amyloid‐β localizes in mitochondria and causes fragmentation.32 Recent genetic studies in flies suggested that Pink1 and Parkin act to promote mitochondrial fragmentation in hereditary Parkinson's disease.30, 33, 34 Indeed, defects in mitochondrial proteins have been identified as the cause of neurodegenerative diseases. Defective OPA1 induces optic atrophy.32, 35 KIF1Bβ, as well as Mfn2, is the genetic cause of CMT2A.13, 16 On the basis of our findings in this study, we propose that the dysfunction of KIF1Bβ/YME1L1/OPA1 may be involved in the occurrence and progression of neurodegenerative disorders.
Further detailed investigation on the molecular mechanism underlying the regulation of KIF1Bβ/YME1L1/OPA1 on mitochondrial morphology as well as identification of other potential associated molecules will be necessary for deeper understanding the role of mitochondria in cancers and neurodegenerative diseases. These approaches will be expected to throw light on the development of a novel therapeutic strategy against these diseases.
CONFLICT OF INTERESTS
The authors declare that they have no conflict of interests.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank Dr Kiyohiro Ando for valuable discussions during the early stages of experimental design. This work was supported in part by a Grant‐in‐Aid from the Ministry of Health, Labor, and Welfare for the Third Term Comprehensive Control Research for Cancer, a Grant‐in‐Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology, and by the Practical Research for Innovative Cancer Control from the Japan Agency for Medical Research and Development (AMED) and the Takeda Science Foundation, Japan.
Ando K, Yokochi T, Mukai A, et al. Tumor suppressor KIF1Bβ regulates mitochondrial apoptosis in collaboration with YME1L1. Molecular Carcinogenesis. 2019;58:1134‐1144. 10.1002/mc.22997
References
REFERENCES
- 1. Lee Y, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001‐5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tondera D, Grandemange S, Jourdain A, et al. Slp‐2 is required for stress‐induced mitochondrial hyperfusion. EMBO J. 2009;28:1589‐1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet. 2005;14(Spec No. 2):R283‐R289. [DOI] [PubMed] [Google Scholar]
- 4. Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858‐862. [DOI] [PubMed] [Google Scholar]
- 7. Nangaku M, Sato‐Yoshitake R, Okada Y, et al. KIF1B, a novel microtubule plus end‐directed monomeric motor protein for transport of mitochondria. Cell. 1994;79:1209‐1220. [DOI] [PubMed] [Google Scholar]
- 8. Nagai M, Ichimiya S, Ozaki T, et al. Identification of the full‐length KIAA0591 gene encoding a novel kinesin‐related protein which is mapped to the neuroblastoma suppressor gene locus at 1p36.2. Int J Oncol. 2000;16:907‐916. [DOI] [PubMed] [Google Scholar]
- 9. Munirajan AK, Ando K, Mukai A, et al. KIF1Bβ functions as a haploinsufficient tumor suppressor gene mapped to chromosome 1p36.2 by inducing apoptotic cell death. J Biol Chem. 2008;283:24426‐24434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schlisio S, Kenchappa RS, Vredeveld LCW, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008;22:884‐893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caron H. Allelic loss of chromosome 1 and additional chromosome 17 material are both unfavourable prognostic markers in neuroblastoma. Med Pediatr Oncol. 1995;24:215‐221. [DOI] [PubMed] [Google Scholar]
- 12. Zhao C, Takita J, Tanaka Y, et al. Charcot‐marie‐tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587‐597. [DOI] [PubMed] [Google Scholar]
- 13. Kijima K, Numakura C, Izumino H, et al. Mitochondrial GTPase mitofusin 2 mutation in charcot‐marie‐tooth neuropathy type 2A. Hum Genet. 2005;116:23‐27. [DOI] [PubMed] [Google Scholar]
- 14. Koida N, Ozaki T, Yamamoto H, et al. Inhibitory role of plk1 in the regulation of p73‐dependent apoptosis through physical interaction and phosphorylation. J Biol Chem. 2008;283:8555‐8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamada C, Ozaki T, Ando K, et al. RUNX3 modulates DNA damage‐mediated phosphorylation of tumor suppressor p53 at Ser‐15 and acts as a co‐activator for p53. J Biol Chem. 2010;285:16693‐16703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cossarizza A, Baccaranicontri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J‐aggregate forming lipophilic cation 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazolcarbocyanine iodide (JC‐1). Biochem Biophys Res Commun. 1993;197:40‐45. [DOI] [PubMed] [Google Scholar]
- 17. Reers M, Smith TW, Chen LB. J‐aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30:4480‐4486. [DOI] [PubMed] [Google Scholar]
- 18. Ohira M, Kageyama H, Mihara M, et al. Identification and characterization of a 500‐kb homozygously deleted region at 1p36.2‐p36.3 in a neuroblastoma cell line. Oncogene. 2000;19:4302‐4307. [DOI] [PubMed] [Google Scholar]
- 19. Legros F, Lombès A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. 2002;13:4343‐4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coppola M, Pizzigoni A, Banfi S, Bassi MT, Casari G, Incerti B. Identification and characterization of YME1L1, a novel paraplegin‐related gene. Genomics. 2000;66:48‐54. [DOI] [PubMed] [Google Scholar]
- 21. Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin‐like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966‐2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ehses S, Raschke I, Mancuso G, et al. Regulation of OPA1 processing and mitochondrial fusion by m‐AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187:1023‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Head B, Griparic L, Amiri M, Gandre‐Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959‐966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li S, Fell SM, Surova O, et al. The 1p36 tumor suppressor KIF 1Bβ is required for calcineurin activation, controlling mitochondrial fission and apoptosis. Dev Cell. 2016;36(2):164‐178. [DOI] [PubMed] [Google Scholar]
- 27. Nakagawara A. The NGF story and neuroblastoma. Med Pediatr Oncol. 1998;31:113‐115. [DOI] [PubMed] [Google Scholar]
- 28. Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J Biol Chem. 2005;280:35742‐35750. [DOI] [PubMed] [Google Scholar]
- 29. Olichon A, Baricault L, Gas N, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743‐7746. [DOI] [PubMed] [Google Scholar]
- 30. Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA. 2008;105:14503‐14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448‐452. [DOI] [PubMed] [Google Scholar]
- 32. Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA. 2008;105:1638‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang Y, Ouyang Y, Yang L, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA. 2008;105:7070‐7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alexander C, Votruba M, Pesch UEA, et al. OPA1, encoding a dynamin‐related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211‐215. [DOI] [PubMed] [Google Scholar]
- 35. Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin‐related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207‐210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information