

Abstract
Papillary thyroid carcinoma (PTC) is most common among all thyroid cancers. Multiple genomic alterations occur in PTC, and gene rearrangements are one of them. Here we screened 14 tumors for novel fusion transcripts by RNA‐Seq. Two samples harboring RET/PTC1 and RET/PTC3 rearrangements were positive controls whereas the remaining ones were negative regarding the common PTC alterations. We used Sanger sequencing to validate potential fusions. We detected 2 novel potentially oncogenic transcript fusions: TG‐FGFR1 and TRIM33‐NTRK1. We detected 4 novel fusion transcripts of unknown significance accompanying the TRIM33‐NTRK1 fusion: ZSWIM5‐TP53BP2, TAF4B‐WDR1, ABI2‐MTA3, and ARID1B‐PSMA1. Apart from confirming the presence of RET/PTC1 and RET/PTC3 in positive control samples, we also detected known oncogenic fusion transcripts in remaining samples: TFG‐NTRK1, ETV6‐NTRK3, MKRN1‐BRAF, EML4‐ALK, and novel isoform of CCDC6‐RET.
Keywords: papillary thyroid carcinoma, rearrangement, RNA‐Seq, transcript fusion
1. INTRODUCTION
Papillary thyroid carcinoma (PTC) is most common among all thyroid cancers. The most common driver alterations in PTC are point mutations in BRAF and RAS genes (KRAS, HRAS, and NRAS) and rearrangements of the RET gene (RET/PTC rearrangements). According to The Cancer Genome Atlas (TCGA) study, these alterations occur in 59.7%, 13%, and 6.3% of the PTC cases, respectively, and the total prevalence of gene rearrangements is 15%.1
Multiple gene rearrangements occur in PTC, and RET/PTC are the most prevalent ones.1 RET/PTC rearrangements are associated with younger age and radiation exposure.2 As documented in the previous studies, detection of these alterations in cytological specimens may be helpful in improving the accuracy of the diagnosis of PTC.3 Multiple gene rearrangements can be screened using the molecular test ThyroSeq; these rearrangements entail a high risk of cancer.4 However, it is worth noting that gene rearrangements can also be found in some benign thyroid diseases.5
In recent years, our knowledge about genomic rearrangements and transcript fusions in PTC has widely expanded, mostly thanks to RNA‐Seq, what led to the detection of numerous novel gene rearrangements in PTC.1, 6, 7, 8, 9, 10, 11 The most important was the detection of ETV6‐NTRK3, which occurs in 2%‐14.5% of PTC patients.8 In PTCs, the most common 5′ partner in gene rearrangements are RET, BRAF, NTRK3, THADA, PPARG, NTRK1, and ALK.1 All of them, except THADA, code for proteins with tyrosine kinase domains.
Our study aimed to detect novel transcript fusions in PTC to expand the knowledge about genetic alterations in this malignancy.
2. MATERIALS AND METHODS
2.1. Samples
Fresh‐frozen material from 14 PTCs was used in this study. Surgical procedures were performed in Maria Sklodowska‐Curie Institute—Oncology Center, Gliwice Branch. The study was approved by the Bioethics Committee of Maria Sklodowska‐Curie Institute—Oncology Center, Gliwice Branch. Informed consent was obtained from all patients.
The samples were selected from a group, in which common PTC mutations have been already analyzed: point mutations—BRAF V600E and mutations in codons 12, 13, and 61 of HRAS, NRAS, and KRAS genes with Sanger sequencing and rearrangements—PAX8‐PPARG, RET/PTC1, and RET/PTC3 with quantitative real‐time polymerase chain reaction (qRT‐PCR). Among 14 samples selected for RNA‐Seq experiment, there was 1 sample positive for RET/PTC1 rearrangement and 1 sample positive for RET/PTC3 rearrangement. The remaining 12 samples were negative for BRAF V600E mutation, HRAS, NRAS, KRAS hotspot mutations, PAX8‐PPARG, RET/PTC1 and RET/PTC3 rearrangements, with the exception of a few cases in which not all mutations were evaluated due to sample availability limitations (details in Supporting Information Table S1).
There were 3 males and 11 females in our study group diagnosed with classical (10 cases) and follicular (4 cases) PTC variants. Young patients were preferred during sample selection: the mean age at diagnosis was 24 years, with a median of 26 years (range: 13–40 years). The mean and median tumor diameters were 17 mm and 15 mm, respectively (range 10–34 mm). Four PTCs were multifocal, 5 with capsule invasion, and 1 with vascular invasion. Lateral neck lymph node metastases were present in 7 patients. Neither local recurrence nor distant metastases were present in any patient from the study group. The histopathological characteristics of the tumors are given in Table 1.
Table 1.
Histopathological characteristics of 14 PTC samples included in this study
| Sample | Sex | Age (years) | Histology | Tumor diameter (mm) | Multifocality | Capsule invasion | Metastasis to lateral neck lymph nodes | Vascular invasion |
|---|---|---|---|---|---|---|---|---|
| NIS164 | F | 29 | Classic | 34 | Multifocal | Yes | No | No |
| NIS203 | M | 13 | Classic | 32 | Multifocal | Yes | Yes | Yes |
| NIS207 | F | 16 | Classic | 15 | Multifocal | Yes | No | No |
| NIS280 | F | 19 | Follicular | 10 | Unifocal | No | No | No |
| PTC006 | M | 32 | Classic | 14 | Unifocal | No | No | No |
| PTC100 | F | 25 | Follicular | 15 | Multifocal | Yes | Yes | No |
| PTC102 | F | 15 | Classic | 20 | Unifocal | No | Yes | No |
| PTC106 | F | 17 | Classic | 11 | Unifocal | No | No | No |
| PTC113 | F | 23 | Classic | 18 | Unifocal | No | Yes | No |
| PTC131 | F | 29 | Classic | 15 | Unifocal | No | Yes | No |
| PTC135 | F | 29 | Follicular | 14 | Unifocal | No | Yes | No |
| PTC174 | M | 27 | Classic | 15 | Unifocal | No | No | No |
| PTC18 | F | 40 | Follicular | 10 | Unifocal | No | No | No |
| PTC181 | F | 29 | Classic | 16 | Unifocal | Yes | Yes | No |
2.2. RNA‐Seq
To detect novel fusion transcripts in PTC, we performed paired‐end RNA‐Seq on 12 PTC samples that were negative regarding most common PTC genetic alterations (BRAF V600E mutation, mutations in codons 12, 13, and 61 of HRAS, NRAS, and KRAS genes, PAX8‐PPARG, RET/PTC1, and RET/PTC3). We also performed paired‐end RNA‐Seq experiment on 1 case with RET/PTC1 and 1 case with RET/PTC3 as positive controls.
Total RNA was extracted from homogenized frozen tissue using Mini Kits (Qiagen GmbH, Hilden, Germany). RNA quantity was measured by NanoDrop ND‐1000 (Thermo Scientific, Wilmington, DE) minispectrophotometer whereas its quality was estimated by Agilent 2100 using RNA 6000 Nano Assay (Agilent Technologies, Santa Clara, CA). Only high‐quality RNA (RNA Integrity Number > 6.5) was used. Sequencing libraries were prepared with the TruSeq RNA Sample Preparation Kit v2 SetA (Illumina Inc., San Diego, CA), following the manufacturer's protocol.
Oligo(dT) magnetic beads Agencourt Ampure XP (Beckman Coulter Inc. Brea, CA) were used to isolate poly(A) RNA from the total RNA samples. The mRNA was fragmented by heating at 94°C for 8 minutes. First‐strand cDNA was synthesized using random hexamer primers for 10 minutes at 25°C, 50 minutes at 42°C, and 15 minutes at 70°C. After the synthesis of the first strand, dNTPs, DNA Polymerase I and RNaseH were added to synthesize second‐strand cDNA for 1 hour at 16°C. The ends of double‐stranded cDNA were repaired by using End Repair Mix. A single “A” nucleotide was added to the 3′ ends of the cDNA molecules and the fragments were ligated to the paired‐end adapters. The purified cDNA was amplified by 15 cycles of PCR for 10 seconds at 98°C, 30 seconds at 60°C, and 30 seconds at 72°C using PCR primers. The quality of the resulting sequencing libraries were determined on a High Sensitivity DNA Kit using an Agilent 2100 Bioanalyser (Agilent Technologies, Santa Clara, CA) and concentration of the libraries was determined on Qubit (Invitrogen, Carlsbad, CA). The mRNASeq libraries were sequenced on a HiSeq1500 device (Illumina Inc., San Diego, CA) to generate 2×120 or 2×106 bp paired‐end reads.
2.3. RNA‐Seq data analysis
Read's quality was assessed using FastQC version 0.9.3.12 Raw FASTQ data were trimmed and filtered using Prinseq‐lite version 0.20.4, and only high‐quality reads were used in the further analysis.13
Fusion transcripts detection was performed using three bioinformatics tools in order to achieve a high sensitivity: TopHat‐Fusion (TopHat version 2.0.10),14 ChimeraScan version 0.4.5,15 and SnowShoes‐FTD version 2.0 Build 37.16 Fusion transcripts detected with these tools were further filtered with in‐house tools in order to filter out false positive findings (details provided in supplementary methods). Genome version GRCh37/hg19 was used in all these analyses.
The list of fusion transcripts present in normal samples was obtained from the paper published by Babiceanu et al.17
2.4. Validation of detected transcript fusions with Sanger sequencing
Validation of novel transcript fusions was performed with the use of Sanger's direct sequencing method on the 3130xl Genetic Analyzer (Life Technologies, Carlsbad, CA) with ABI PRISM 1.1 BigDye Terminator Cycle Sequencing Ready Reaction Kit (Life Technologies). Before sequencing, RNA (200 ng) was converted to cDNA with the Omniscript RT Kit (Qiagen GmbH). The reaction was carried out for 1 hour at 37°C in a volume of 20 μL using a mixture of 1× concentrated buffer (2 μL; Omniscript RT Kit), 5× concentrated dNTPs (2 μL; Omniscript RT Kit), 4 U/μl RT‐O polymerase (1 μL; Omniscript RT Kit), 50 μM random nonamers (1.6 μL), and 1× concentrated RiboLock RNAse inhibitor (1 μL; Fermentas Thermo Fisher Scientific, Waltham, MA). Obtained cDNA was used as a template in PCR reactions, in which each amplicon was amplified using specific primers designed with the Primer3 Input software available on the website http://frodo.wi.mit.edu/ (for primer sequences and annealing temperatures used in PCR reactions, see Supporting Information Table S2). Each amplicon contained sequences of both rearranged genes. The PCR products were visualized by electrophoresis in a 2% agarose gel in the presence of bromide ethidium (0,3 μL/mL), cleaned with Exo I (volume 0.6 μL; concentration 10 U/μl; Life Technologies) and Sap (volume 0.6 μL; concentration 2 U/μl; Boehring Manheim GmbH, Germany; Life Technologies) enzymes mixture according to manufacturer's recommendations and then sequenced as described earlier.
3. RESULTS
3.1. Detection of fusion transcripts by RNA‐Seq
In order to detect novel fusion transcripts in PTC, we performed paired‐end RNA‐Seq experiment on 14 PTC tumor samples. After filtering and trimming of raw reads, we obtained an average of 13.5 million read pairs in each sample. The read length was 120 bp in 6 samples and 50 bp in 8 samples (Supporting Information Table S3).
We detected 28 fusion events by TopHat‐Fusion, 96 by ChimeraScan, and 34 by SnowShoes‐FTD (Supporting Information Tables S4‐S6). In total, we detected 126 fusion events. Seventy‐three of them were read‐throughs or were detected in normal samples as depicted by Babiceanu et al17 and these were beyond our interest (Supporting Information Table S7). The remaining 53 fusion transcripts, which were not read‐throughs and not detected in normal samples, were further manually inspected (Supporting Information Table S8).
We manually selected candidate fusion transcripts potentially tumorigenic, which involved genes with known cancer‐associated functions. We also selected all candidate fusion transcripts that were detected by more than one program. Final list consisted of 18 transcript fusions (Table 2), found in 11 of 14 tumor samples.
Table 2.
List of candidate fusion transcripts detected in PTC with RNA‐Seq method
| Sample | 5′ Chromosome | 3′ Chromosome | 5′ Breakpoint | 3′ Breakpoint | 5′ Symbol | 3′ Symbol | 5′ Entrez | 3′ Entrez | Exon boundary Fusiona | Sanger validation results | Comment | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | NIS164 | chr8 | chr8 | 134 145 904 | 38 277 253 | TG | FGFR1 | 7038 | 2260 | Yes | Positive | Novel potentially oncogenic fusion transcript |
| 2 | NIS164 | chr8 | chr8 | 38 279 315 | 134 146 920 | FGFR1 | TG | 2260 | 7038 | Yes | Positive | Reciprocal of novel fusion transcript |
| 3 | NIS203 | chr10 | chr10 | 51 582 939 | 43 612 032 | NCOA4 | RET | 8031 | 5979 | Yes | ‐ | Positive control (RET/PTC3) |
| 4 | NIS207 | chr3 | chr1 | 100 455 548 | 156 844 363 | TFG | NTRK1 | 10 342 | 4914 | Yes | ‐ | Known oncogenic fusion transcript |
| 5 | NIS207 | chr3 | chr1 | 100 455 560 | 156 844 363 | TFG | NTRK1 | 10 342 | 4914 | Yes | ‐ | Known oncogenic fusion transcript |
| 6 | PTC100 | chr12 | chr15 | 12 006 495 | 88 576 276 | ETV6 | NTRK3 | 2120 | 4916 | Yes | ‐ | Known oncogenic fusion transcript |
| 7 | PTC102 | chr10 | chr10 | 61 665 880 | 43 612 032 | CCDC6 | RET | 8030 | 5979 | Yes | ‐ | Positive control (RET/PTC1) |
| 8 | PTC106 | chr10 | chr10 | 61 554 231 | 43 612 032 | CCDC6 | RET | 8030 | 5979 | Yes | Positive | Novel isoform of oncogenic fusion transcript |
| 9 | PTC113 | chr7 | chr7 | 140 158 807 | 140 481 493 | MKRN1 | BRAF | 23 608 | 673 | Yes | ‐ | Known oncogenic fusion transcript |
| 10 | PTC131 | chr1 | chr1 | 114 952 806 | 156 845 312 | TRIM33 | NTRK1 | 51 592 | 4914 | Yes | Positive | Novel potentially oncogenic fusion transcript |
| 11 | PTC131 | chr1 | chr1 | 114 952 806 | 156 846 192 | TRIM33 | NTRK1 | 51 592 | 4914 | Yes | Negative | ‐ |
| 12 | PTC131 | chr1 | chr1 | 45 671 428 | 223 972 016 | ZSWIM5 | TP53BP2 | 57 643 | 7159 | Yes | Positive | Novel fusion transcript |
| 13 | PTC131 | chr18 | chr4 | 23 847 587 | 10 080 625 | TAF4B | WDR1 | 6875 | 9948 | Yes | Positive | Novel fusion transcript |
| 14 | PTC131 | chr2 | chr2 | 204 245 107 | 42 867 313 | ABI2 | MTA3 | 10 152 | 57 504 | Yes | Positive | Novel fusion transcript |
| 15 | PTC131 | chr6 | chr11 | 157 150 555 | 14 540 587 | ARID1B | PSMA1 | 57 492 | 5682 | Yes | Positive | Novel fusion transcript |
| 16 | PTC135 | chr12 | chr15 | 12 006 495 | 88 576 276 | ETV6 | NTRK3 | 2120 | 4916 | Yes | ‐ | Known oncogenic fusion transcript |
| 17 | PTC174 | chr2 | chr2 | 42 522 656 | 29 446 394 | EML4 | ALK | 27 436 | 238 | Yes | ‐ | Known oncogenic fusion transcript |
| 18 | PTC181 | chr12 | chr15 | 12 006 495 | 88 576 276 | ETV6 | NTRK3 | 2120 | 4916 | Yes | ‐ | Known oncogenic fusion transcript |
Exon boundary fusion is a fusion transcript in which both sides of the junction are known exon boundaries of the parental genes.
The following novel transcript fusions were detected with RNA‐Seq method: TG‐FGFR1, FGFR1‐TG, two isoforms of TRIM33‐NTRK1, ARID1B‐PSMA1, TAF4B‐WDR1, ABI2‐MTA3, ZSWIM5‐TP53BP2, and the novel isoform of CCDC6‐RET. RET/PTC1 and RET/PTC3 fusion transcripts were found, as expected, in positive control samples. We also detected known oncogenic fusion transcripts: TFG‐NTRK1, ETV6‐NTRK3 (in three samples), MKRN1‐BRAF, and EML4‐ALK.
3.2. Validation of fusion transcripts
We performed validation by direct Sanger sequencing for all 9 novel fusion transcripts. We confirmed the existence of 8/9 fusions: TG‐FGFR1, FGFR1‐TG, one isoform of TRIM33‐NTRK1 (with a breakpoint in chr1:114952806‐chr1:156845312), ARID1B‐PSMA1, TAF4B‐WDR1, ABI2‐MTA3, ZSWIM5‐TP53BP2 as well as the novel isoform of CCDC6‐RET (Figures 1 and 2; Supporting Information Figures S1‐S6). We did not confirm the second TRIM33‐NTRK1 fusion isoform, with a breakpoint in chr1:114952806‐chr1:156846192. When we used primers designed for that TRIM33‐NTRK1 isoform, we did not observe the expected sequence.
Figure 1.
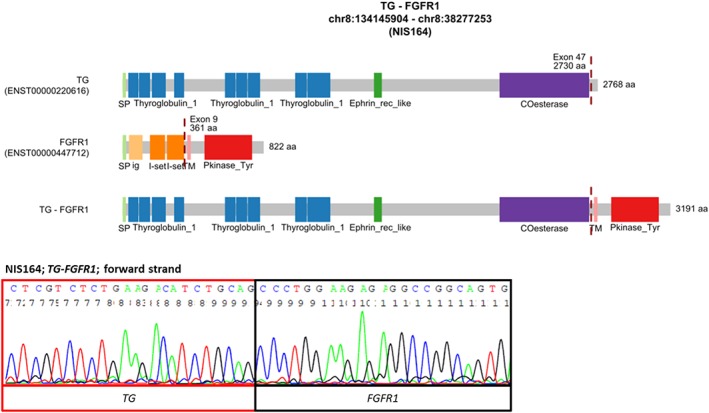
The novel TG‐FGFR1 fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: COesterase, Carboxylesterase family; Ephrin_rec_like, Putative ephrin‐receptor like; ig, Immunoglobulin domain; I‐set, Immunoglobulin I‐set domain; Pkinase_Tyr, Protein tyrosine kinase; SP, signal peptide; Thyroglobulin_1, Thyroglobulin type‐1 repeat; TM, transmembrane region [Color figure can be viewed at wileyonlinelibrary.com]
Figure 2.
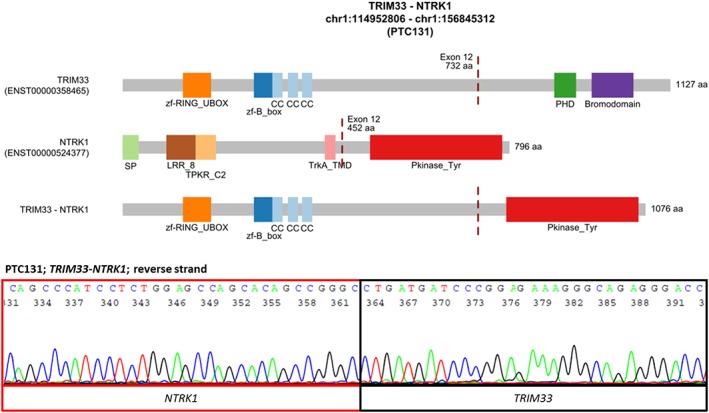
The novel TRIM33‐NTRK1 fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: Bromodomain, Bromodomain; CC, coiled‐coil region; LRR_8, Leucine rich repeat; PHD, PHD‐finger; Pkinase_Tyr, Protein tyrosine kinase; SP, signal peptide; TPKR_C2, Tyrosine‐protein kinase receptor C2 Ig‐like domain; TrkA_TMD, Tyrosine kinase receptor A trans‐membrane domain; zf‐B_box, B‐box zinc finger; zf‐RING_UBOX, RING‐type zinc‐finger [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Novel fusion transcripts
We detected 2 novel in‐frame fusion transcripts, which are potential driver alterations: TG‐FGFR1 and TRIM33‐NTRK1.
We identified a novel fusion of thyroglobulin (TG) and fibroblast growth factor receptor 1 (FGFR1) in sample NIS164 (Figure 1). The sample was a multifocal classic PTC tumor, 34 mm in diameter, with capsule invasion, no metastasis to lateral neck lymph nodes, and no vascular invasion. The patient was a 29‐year‐old female. TG and FGFR1 genes are localized at 8q24 and 8p11. TG‐FGFR1 juxtaposes exons 1‐47 of TG (ENST00000220616) to exons 9‐18 of FGFR1 (ENST00000447712). The fusion protein is predicted to include 1‐2730 amino acids (AA) of TG and 361‐822 AA of FGFR1. All domains encoded by TG and the whole protein kinase domain encoded by FGFR1 are retained in this fusion. In NIS164 sample harboring TG‐FGFR1, also a reciprocal fusion FGFR1‐TG was detected, which fuses exons 1‐8 of FGFR1 to exon 48 of TG (Supporting Information Figure S1).
We identified a novel fusion of tripartite motif containing 33 (TRIM33) and neurotrophic receptor tyrosine kinase 1 (NTRK1) in PTC131 sample (Figure 2). The sample was a unifocal classic PTC tumor, 15 mm in diameter, with metastasis to lateral neck lymph nodes, no capsule invasion, and no vascular invasion. The patient was a 29‐year‐old female. The TRIM33 and NTRK1 genes are localized at 1p13 and 1q23. TRIM33‐NTRK1 juxtaposes exons 1‐12 of TRIM33 (ENST00000358465) to exons 12‐17 of NTRK1 (ENST00000524377). The fusion protein is predicted to include 1‐732 AA of TRIM33 and 452‐796 AA of NTRK1. The whole tyrosine kinase domain encoded by NTRK1 is retained in fusion.
In the sample PTC131, apart from the novel TRIM33‐NTRK1 fusion, we detected 4 other novel fusion transcripts of unknown significance: ZSWIM5‐TP53BP2, TAF4B‐WDR1, ABI2‐MTA3, and ARID1B‐PSMA1 (Supporting Information Figures S2‐S5). All 4 fusions apart from ABI2‐MTA3 are in‐frame. ABI2‐MTA3 is out‐of‐frame and a premature stop codon occurs in 10th codon after the breakpoint.
We also found a novel in‐frame isoform of the known oncogenic fusion CCDC6‐RET in PTC106 sample (Supporting Information Figure S6). In the novel isoform, exons 1‐8 of CCDC6 (ENST00000263102) are fused to exons 12‐20 of RET (ENST00000355710), in contrary to the most common CCDC6‐RET rearrangement encompassing over 98% of cases, in which exon 1 of CCDC6 is fused to exons 12‐20 of the RET gene (Supporting Information Figure S6).18 The whole protein kinase domain encoded by RET is retained in the novel fusion isoform.
3.4. Known fusion transcripts
Using RNA‐Seq, NCOA4‐RET (RET/PTC1) and CCDC6‐RET (RET/PTC3) were found, as expected, in positive control samples. In the remaining samples, we also detected in‐frame fusion transcripts already reported in the literature: two isoforms of TFG‐NTRK1, ETV6‐NTRK3 (in 3 samples), MKRN1‐BRAF, and EML4‐ALK (Supporting Information Figures S7‐S11). In all of them, 3′ partner encodes tyrosine kinase domain, and the whole domain is retained in the predicted fusion protein.
In both the TFG‐NTRK1 isoforms that we detected, a fusion between exon 6 of TFG (ENST00000418917 or ENST00000240851) and exon 10 of NTRK1 (ENST00000524377) was present. The two isoforms differed in the length of exon 6 of the TFG gene, which is 129 bp in TFG variant ENST00000418917 and 141 bp in TFG variant ENST00000240851 (Supporting Information Figures S7, S8). In ETV6‐NTRK3, the fusion between exon 4 of ETV6 (ENST00000396373) and exon 14 of NTRK3 (ENST00000394480) was present (Supporting Information Figure S9). We detected ETV6‐NTRK3 in 3 samples, and it was the most prevalent alteration in our group. Two of three samples harboring ETV6‐NTRK3 were follicular variant of PTC. In MKRN1‐BRAF, the fusion between exon 4 of MKRN1 (ENST00000255977) and exon 11 of BRAF (ENST00000288602) was present (Supporting Information Figure S10). In EML4‐ALK, the fusion between exon 13 of EML4 (ENST00000318522) and exon 20 of ALK (ENST00000389048) was present (Supporting Information Figure S11).
4. DISCUSSION
In this study, RNA‐Seq was used to determine the presence of transcript fusions in PTCs. Fourteen PTC samples were examined, negative for the most common point mutations of the BRAF and RAS genes, and PAX8‐PPARG rearrangements, with only 2 harboring RET/PTC1 and RET/PTC3 transcript fusions, treated as the positive controls. Among the analyzed samples novel fusion transcripts were found in two samples: TG‐FGFR1 and TRIM33‐NTRK1, 7 demonstrated known fusion transcripts (ETV6‐NTRK3 in 3 samples, TFG‐NTRK1, EML4‐ALK, MKRN1‐BRAF, and novel isoform of CCDC6‐RET) and in 3 PTCs no fusion transcripts were detected. The tumor with TRIM33‐NTRK1 also carried 4 other transcript fusions of unknown significance: ARID1B‐PSMA1, TAF4B‐WDR1, ABI2‐MTA3, and ZSWIM5‐TP53BP2.
TG‐FGFR1 is a novel potentially oncogenic fusion transcript. TG is a glycoprotein homodimer produced predominantly by the thyroid gland. Only 1 case of TG fusion has been described so far in the literature: TG‐THADA.1 FGFR1, in turn, is a member of the FGFR family, which activation by mutations, amplification, or translocations plays roles in cancer initiation and development.19 A number of FGFR1, FGFR2, and FGFR3 rearrangements was identified in different cancers, including bladder cancer, breast cancer, head and neck cancer, lung squamous cell carcinoma, and thyroid cancer.20, 21, 22 The TG‐FGFR1 fusion transcript found in our PTC sample encodes a tyrosine kinase domain, which, when activated by TG, transmits the activation signal to the downstream effectors. It suggests that the TG‐FGFR1 may be responsible for cancer initiation and progression. The expression level of TG‐FGFR1 is driven by the promoter of TG, a gene with high expression in the thyroid, which may result in an aberrant overexpression of TG‐FGFR1. One of the mechanisms that switches on the kinase domain in the fusion proteins is the dimerization by one of the domains present in the partner protein.23 It has been shown that the cholinesterase‐like domain located in C‐terminal part of TG is responsible for dimerization.24 This domain is preserved in the TG‐FGFR1 fusion protein and it may cause the FGFR1 domains to dimerize, resulting in activation of FGFR1 tyrosine kinase in the absence of ligands. The same sample harbored in addition a reciprocal fusion FGFR1‐TG.
A second novel fusion detected in our PTC samples was the TRIM33‐NTRK1 rearrangement. TRIM33 encodes a tripartite motif containing 33, a transcriptional corepressor, also known as RFG7. This gene has been demonstrated to create a fusion with the RET gene (TRIM33‐RET) in radiation‐induced thyroid carcinomas.25 Similarly, NTRK1 is also a known fusion partner gene in PTCs. NTRK1 rearrangements occur in up to 13% of PTCs (12% in the Polish population).1, 26, 27, 28, 29 The TRIM33‐NTRK1 fusion leads to activation of NTRK1 tyrosine kinase domain, which in turn activates downstream effectors. The mechanism of tyrosine kinase activation in TRIM33‐NTRK1 may be similar to that in the TRIM33‐RET fusion protein. TRIM33 encodes a coiled‐coil domain that allows ligand‐independent dimerization of the chimeric protein and activation of the truncated RET receptor in TRIM33‐RET.30 The TRIM33‐NTRK1 fusion, similarly to TG‐FGFR1 rearrangement, may be a potential oncogene in PTC development. However, only in vitro functional studies can assess the role of the novel fusion proteins TG‐FGFR1 and TRIM33‐NTRK1 in PTC pathogenesis.
An additional 4 rearrangements (ZSWIM5‐TP53BP2, TAF4B‐WDR1, ABI2‐MTA3, and ARID1B‐PSMA1) were found in the PTC sample harboring the TRIM33‐NTRK1 rearrangement. These additional aberrations have not been described in any other cancer, and only ABI2 and ARID1B have been involved in fusion genes. It is hence difficult to define the role of these fusions in tumorigenesis. It is possible that these fusions are a consequence of genomic instability and are secondary phenomena.
ABI2 is a KMT2A translocation partner in acute myeloid leukemia.31 ABI2, being a functional homologue of ABI1, is known as an ABL1 regulator and is considered a tumor suppressor due to its inhibitory function in ABL1 signaling. The MTA3 gene is a member of the metastasis‐associated protein family, identified as key regulators of the epithelial‐mesenchymal transition process and E‐cadherin expression.32 MTA3 has been described to be under‐expressed in some malignancies, including breast cancer, ovarian cancer, gastroesophageal junction adenocarcinoma or endometrial cancer, and even as a suppressor of metastases in these tumors.33, 34, 35 Shan et al36 described decreased MTA3 expression in glioma and its association with prognosis, which suggests that MTA3 is a suppressor gene in this malignancy. Interestingly, the ABI2‐MTA3 fusion, found in our set of PTC samples, is out‐of‐frame and a premature stop codon occurs in 10th codon after the breakpoint, which may lead to silencing of MTA3 expression. As regards the remaining 3 additional alterations, ZSWIM5‐TP53BP2, TAF4B‐WDR1, and ARID1B‐PSMA1, they all represent in‐frame fusions.
Fusion gene partners of the TAF4B‐WDR1 are the TATA‐box binding protein associated factor 4b (TAF4B), involved in initiation of transcription of genes by RNA polymerase II, and WD repeat domain 1 (WDR1) involved in protein–protein interactions due to WD domains. WDR1 plays a crucial role in cytokinesis and cell migration and may be important in the ability of cancer cells to proliferate and invade surrounding tissues.37, 38 Overexpression of WDR1 was reported in different cancers, including breast cancer, ovarian carcinoma, and thyroid neoplasia.39, 40, 41
The fusion transcript ZSWIM5‐TP53BP2 is made of zinc finger SWIM‐type containing 5 gene (ZSWIM5) and tumor protein TP53 binding protein 2 (TP53BP2, also known as ASPP2). TP53BP2 is a member of the ASPP (apoptosis‐stimulating protein of p53) family of TP53 interacting proteins, involved in apoptosis and cell growth regulation. It has been demonstrated that TP53BP2 plays a role as a tumor suppressor42 via interactions between Ank/SH3 domains, present in TP53BP2, and numerous partner proteins like TP53, NFKB1, and BCL2.43 The fusion transcript, detected by us, retained the SH3 domain; however, it does not have ankyrin (Ank) repeats. Lack of these domains may inhibit TP53BP2 tumor suppressor functions.
The last fusion, accompanying the TRIM33‐NTRK1 rearrangement, is the ARID1B‐PSMA1 fusion. ARID1B (AT‐rich interaction domain 1B) encodes a protein that is a component of the SWI/SNF chromatin remodeling complex, which may play a role in cell‐cycle activation. Tumor suppressor activity of ARID1B has been demonstrated in vitro in pancreatic cancer cells.44 Moreover, deletions and mutations of this gene have been reported in hepatocellular carcinoma, childhood neuroblastoma, PTC, and other types of cancer.45 ARID1B has also been identified as an additional ZNF384 fusion partner in pediatric acute lymphoblastic leukemia.46 PSMA1 (proteasome subunit alpha 1), in turn, was shown to be up‐regulated in a number of cancers.47
We also detected novel in‐frame isoform of the known oncogenic fusion CCDC6‐RET, which similarly to other RET rearrangements, also retained the RET tyrosine kinase domain leading to RET activation.
In our group of samples, we also found 4 oncogenic fusions already reported in the literature: TFG‐NTRK1, ETV6‐NTRK3, MKRN1‐BRAF, and EML4‐ALK. TFG‐NTRK1 was previously reported in only a few PTC cases.27, 48, 49 However, it was not reported in other cancers.18, 50 The longer of two isoforms detected in our study, TFG (exon 6 of ENST00000240851)—NTRK1 (exon 10 of ENST00000524377), has been already reported in PTC.1, 51 ETV6‐NTRK3 was the most prevalent alteration in PTC set analyzed by us, as it was found in 3 samples. According to the literature, the ETV6‐NTRK3 occurs in 2%‐14.5% of PTC patients.8 It also occurs in cancers of the salivary gland, kidney, and other tissues.18, 50 Two of the three samples harboring ETV6‐NTRK3 were follicular variants of PTCs. This is in agreement with recent findings that most post‐Chernobyl PTCs in which ETV6‐NTRK3 was identified were classified as follicular variant of PTC.6, 8 The isoform detected in our study, which juxtaposes exon 4 of ETV6 (ENST00000396373) and exon 14 of NTRK3 (ENST00000394480), has been reported in PTC1, 6, 8, 51 and gastrointestinal stromal tumor.52 MKRN1‐BRAF has been reported in a few cases of PTC.1, 7 It was also described to be present in anaplastic thyroid cancer, pilocytic astrocytoma, head and neck neuroendocrine carcinoma, colon adenocarcinoma, and low‐grade serous ovarian cancer.53, 54, 55, 56 The isoform detected in our study, which juxtaposes exon 4 of MKRN1 (ENST00000255977) to exon 11 of BRAF (ENST00000288602), has been reported in pilocytic astrocytoma,54 colon adenocarcinoma55 and in low‐grade serous ovarian cancer.56 EML4‐ALK was reported in PTC in a number of studies.1, 9, 57, 58 It also occurs in about 7% cases of non‐small‐cell lung cancer and in other cancers.50, 59 The isoform detected in our study, which juxtaposes exon 13 of EML4 (ENST00000318522) and exon 20 of ALK (ENST00000389048) was reported in lung carcinoma59 and papillary thyroid carcinoma.9, 57
Although the number of analyzed PTC cases is small, they were carefully selected, and only young PTC patients without known somatic mutations of the BRAF and RAS genes, PAX8‐PPARG, RET/PTC1, and RET/PTC3 rearrangements were taken into consideration. We found new fusion transcripts with a potential oncogenic role and a number of known rearrangements. Our study shows that although large analyses like TCGA study gave us a lot of new data about PTC biology, still some information is missing, and further analyses are needed. There is no doubt that better understanding of molecular PTC background will open new diagnostic and therapeutic possibilities.
Supporting information
Appendix S1 : Supporting Information
Supporting Information Figure S1 Novel FGFR1‐TG fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: COesterase, Carboxylesterase family; Ephrin_rec_like, Putative ephrin‐receptor like; I‐set, Immunoglobulin I‐set domain; ig, Immunoglobulin domain; Pkinase_Tyr, Protein tyrosine kinase; SP, signal peptide; Thyroglobulin_1, Thyroglobulin type‐1 repeat; TM, transmembrane region
Supporting Information Figure S2. Novel ZSWIM5‐TP53BP2 fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: Ank_2, Ankyrin repeats (3 copies); CC, coiled‐coil region; SH3_1, SH3 domain
Supporting Information Figure S3. Novel TAF4B‐WDR1 fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: CC, coiled‐coil region; TAF4, Transcription initiation factor TFIID component TAF4 family; TAFH, NHR1 homology to TAF; WD40, WD domain, G‐beta repeat
Supporting Information Figure S4. Novel ABI2‐MTA3 fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: Abi_HHR, Abl‐interactor HHR; BAH, BAH domain; ELM2, ELM2 domain; GATA, GATA zinc finger; MTA_R1, MTA R1 domain; Myb_DNA‐binding, Myb‐like DNA‐binding domain; SH3_9, Variant SH3 domain
Supporting Information Figure S5. Novel ARID1B‐PSMA1 fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: ARID, ARID/BRIGHT DNA binding domain; BAF250_C, SWI/SNF‐like complex subunit BAF250/Osa; CC, coiled‐coil region; Proteasome, Proteasome subunit; Proteasome_A_N, Proteasome subunit A N‐terminal signature
Supporting Information Figure S6. Novel isoform of CCDC6‐RET fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: Cadherin, Cadherin domain; CC, coiled‐coil region; DUF2046, Uncharacterized conserved protein H4 (DUF2046); Pkinase_Tyr, Protein tyrosine kinase; RET_CLD1, RET Cadherin like domain 1; RET_CLD3, RET Cadherin like domain 3; RET_CLD4, RET Cadherin like domain 4; SP, signal peptide; TM, transmembrane region
Supporting Information Figure S7. Known TFG‐NTRK1 fusion event detected in PTC. Breakpoint: chr3:100455548‐ chr1:156844363. Abbreviations: CC, coiled‐coil region; LRR_8, Leucine rich repeat; PB1, PB1 domain; Pkinase_Tyr, Protein tyrosine kinase; SP, signal peptide; TPKR_C2, Tyrosine‐protein kinase receptor C2 Ig‐like domain; TrkA_TMD, Tyrosine kinase receptor A trans‐membrane domain
Supporting Information Figure S8. Known TFG‐NTRK1 fusion event detected in PTC. Breakpoint: chr3:100455560‐chr1:156844363. Abbreviations: CC, coiled‐coil region; LRR_8, Leucine rich repeat; PB1, PB1 domain; Pkinase_Tyr, Protein tyrosine kinase; SP, signal peptide; TPKR_C2, Tyrosine‐protein kinase receptor C2 Ig‐like domain; TrkA_TMD, Tyrosine kinase receptor A trans‐membrane domain
Supporting Information Figure S9. Known ETV6‐NTRK3 fusion event detected in PTC. Abbreviations: Ets, Ets‐domain; I‐set, Immunoglobulin I‐set domain; ig, Immunoglobulin domain; LRR_8, Leucine rich repeat; LRRNT, Leucine rich repeat N‐terminal domain; Pkinase_Tyr, Protein tyrosine kinase; SAM_PNT, Sterile alpha motif (SAM)/Pointed domain; SP, signal peptide; TM, transmembrane region; TPKR_C2, Tyrosine‐protein kinase receptor C2 Ig‐like domain
Supporting Information Figure S10. Known MKRN1‐BRAF fusion event detected in PTC. Abbreviations: C1_1, Phorbol esters/diacylglycerol binding domain (C1 domain); CC, coiled‐coil region; MKRN1_C, E3 ubiquitin‐protein ligase makorin‐1, C‐terminal; Pkinase_Tyr, Protein tyrosine kinase; RBD, Raf‐like Ras‐binding domain; zf‐C3HC4, Zinc finger, C3HC4 type (RING finger); zf‐CCCH_4, CCCH‐type zinc finger
Supporting Information Figure S11. Known EML4‐ALK fusion event detected in PTC. Abbreviations: CC, coiled‐coil region; Gly_rich, Glycine rich protein; HELP, HELP motif; MAM, MAM domain, meprin/A5/mu; Pkinase_Tyr, Protein tyrosine kinase; SP, signal peptide; TM, transmembrane region; WD40, WD domain, G‐beta repeat
Supporting Information Table S1. Mutations in analyzed samples detected by Sanger sequencing and qRT‐PCR. 0—negative, 1—positive, ND—not done.
Supporting Information Table S2. Primers sequences and annealing temperatures used in PCR reactions
Supporting Information Table S3. Summary of data generated in RNA‐seq experiment (after applying filtering and trimming of reads)
Supporting Information Table S4. TopHat Fusion output (after applying in‐house filters)
Supporting Information Table S5. ChimeraScan output (after applying in‐house filters)
Supporting Information Table S6. SnowShoes output
Supporting Information Table S7. Combined outputs from TopHat, ChimeraScan and SnowShoes: read‐through gene fusions and fusions occurring in normal samples (as depicted by Babiceanu et al.)
Supporting Information Table S8. Combined outputs from TopHat, ChimeraScan and SnowShoes: gene fusions that are not read‐through and do not occur in normal samples (potential rearrangements)
Pfeifer A, Rusinek D, Żebracka‐Gala J, et al. Novel TG‐FGFR1 and TRIM33‐NTRK1 transcript fusions in papillary thyroid carcinoma. Genes Chromosomes Cancer. 2019;58:558–566. 10.1002/gcc.22737
Funding information POIG.02.01.00‐00‐166/08. Śląska BIO‐FARMA. Centrum Biotechnologii, Bioinżynierii i Bioinformatyki; Polish National Center of Research and Development, Grant/Award Number: STRATEGMED2/267398/4/NCBR/2015; PL‐Grid Infrastructure; Polish National Science Center, Grant/Award Number: DEC‐2011/03/N/NZ2/03495
REFERENCES
- 1. Cancer Genome Atlas Research Network . Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Su X, Li Z, He C, Chen W, Fu X, Yang A. Radiation exposure, young age, and female gender are associated with high prevalence of RET/PTC1 and RET/PTC3 in papillary thyroid cancer: a meta‐analysis. Oncotarget. 2016;7:16716‐16730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cantara S, Capezzone M, Marchisotta S, et al. Impact of proto‐oncogene mutation detection in cytological specimens from thyroid nodules improves the diagnostic accuracy of cytology. J Clin Endocrinol Metab. 2010;95:1365‐1369. [DOI] [PubMed] [Google Scholar]
- 4. Nikiforov YE, Carty SE, Chiosea SI, et al. Impact of the multi‐gene ThyroSeq next‐generation sequencing assay on cancer diagnosis in thyroid nodules with atypia of undetermined significance/follicular lesion of undetermined significance cytology. Thyroid. 2015;25:1217‐1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guerra A, Sapio MR, Marotta V, et al. Prevalence of RET/PTC rearrangement in benign and malignant thyroid nodules and its clinical application. Endocr J. 2011;58:31‐38. [DOI] [PubMed] [Google Scholar]
- 6. Ricarte‐Filho JC, Li S, Garcia‐Rendueles MER, et al. Identification of kinase fusion oncogenes in post‐Chernobyl radiation‐induced thyroid cancers. J Clin Invest. 2013;123:4935‐4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smallridge RC, Chindris A‐M, Asmann YW, et al. RNA sequencing identifies multiple fusion transcripts, differentially expressed genes, and reduced expression of immune function genes in BRAF (V600E) mutant vs BRAF wild‐type papillary thyroid carcinoma. J Clin Endocrinol Metab. 2014;99:E338‐E347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leeman‐Neill RJ, Kelly LM, Liu P, et al. ETV6‐NTRK3 is a common chromosomal rearrangement in radiation‐associated thyroid cancer. Cancer. 2014;120:799‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kelly LM, Barila G, Liu P, et al. Identification of the transforming STRN‐ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci U S A. 2014;111:4233‐4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Costa V, Esposito R, Ziviello C, et al. New somatic mutations and WNK1‐B4GALNT3 gene fusion in papillary thyroid carcinoma. Oncotarget. 2015;6:11242‐11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoo SK, Lee S, Kim SJ, et al. Comprehensive analysis of the transcriptional and mutational landscape of follicular and papillary thyroid cancers. PLoS Genet. 2016;12:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andrews S. FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 13. Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim D, Salzberg SL. TopHat‐Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27:2903‐2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Asmann YW, Hossain A, Necela BM, et al. A novel bioinformatics pipeline for identification and characterization of fusion transcripts in breast cancer and normal cell lines. Nucleic Acids Res. 2011;39:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Babiceanu M, Qin F, Xie Z, et al. Recurrent chimeric fusion RNAs in non‐cancer tissues and cells. Nucleic Acids Res. 2016;44:2859‐2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high‐resolution. Nucleic Acids Res. 2017;45:D777‐D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18:1855‐1862. [DOI] [PubMed] [Google Scholar]
- 20. Wu Y‐M, Su F, Kalyana‐Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3:636‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parker BC, Engels M, Annala M, Zhang W. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J Pathol. 2014;232:4‐15. [DOI] [PubMed] [Google Scholar]
- 22. Wang R, Wang L, Li Y, et al. FGFR1/3 tyrosine kinase fusions define a unique molecular subtype of non‐small cell lung cancer. Clin Cancer Res. 2014;20:4107‐4114. [DOI] [PubMed] [Google Scholar]
- 23. Medves S, Demoulin J‐B. Tyrosine kinase gene fusions in cancer: translating mechanisms into targeted therapies. J Cell Mol Med. 2012;16:237‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee J, Wang X, Di Jeso B, Arvan P. The cholinesterase‐like domain, essential in thyroglobulin trafficking for thyroid hormone synthesis, is required for protein dimerization. J Biol Chem. 2009;284:12752‐12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rabes HM. Gene rearrangements in radiation‐induced thyroid carcinogenesis. Med Pediatr Oncol. 2001;36:574‐582. [DOI] [PubMed] [Google Scholar]
- 26. Bongarzone I, Fugazzola L, Vigneri P, et al. Age‐related activation of the tyrosine kinase receptor protooncogenes RET and NTRK1 in papillary thyroid carcinoma. J Clin Endocrinol Metab. 1996;81:2006‐2009. [DOI] [PubMed] [Google Scholar]
- 27. Musholt TJ, Musholt PB, Khaladj N, Schulz D, Scheumann GFW, Klempnauer J. Prognostic significance of RET and NTRK1 rearrangements in sporadic papillary thyroid carcinoma. Surgery. 2000;128:984‐993. [DOI] [PubMed] [Google Scholar]
- 28. Rabes HM, Demidchik EP, Sidorow JD, et al. Pattern of radiation‐induced RET and NTRK1 rearrangements in 191 post‐Chernobyl papillary thyroid carcinomas: biological, phenotypic, and clinical implications. Clin Res Cancer. 2000;6:1093‐1103. [PubMed] [Google Scholar]
- 29. Brzeziańska E, Karbownik M, Migdalska‐Sek M, Pastuszak‐Lewandoska D, Włoch J, Lewiński A. Molecular analysis of the RET and NTRK1 gene rearrangements in papillary thyroid carcinoma in the Polish population. Mutat Res. 2006;599:26‐35. [DOI] [PubMed] [Google Scholar]
- 30. Nikiforov YE. RET/PTC rearrangement in thyroid tumors. Endocr Pathol. 2002;13:3‐16. [DOI] [PubMed] [Google Scholar]
- 31. Coenen EA, Zwaan CM, Meyer C, et al. Abl‐interactor 2 (ABI2): a novel MLL translocation partner in acute myeloid leukemia. Leuk Res. 2012;36:e113‐e115. [DOI] [PubMed] [Google Scholar]
- 32. Toh Y, Nicolson GL. The role of the MTA family and their encoded proteins in human cancers: molecular functions and clinical implications. Clin Exp Metastasis. 2009;26:215‐227. [DOI] [PubMed] [Google Scholar]
- 33. Fearon ER. Connecting estrogen receptor function, transcriptional repression, and E‐cadherin expression in breast cancer. Cancer Cell. 2003;3:307‐310. [DOI] [PubMed] [Google Scholar]
- 34. Dong H, Guo H, Xie L, et al. The metastasis‐associated gene MTA3, a component of the Mi‐2/NuRD transcriptional repression complex, predicts prognosis of gastroesophageal junction adenocarcinoma. PLoS One. 2013;8:e62986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bruning A, Juckstock J, Blankenstein T, Makovitzky J, Kunze S, Mylonas I. The metastasis‐associated gene MTA3 is downregulated in advanced endometrioid adenocarcinomas. Histol Histopathol. 2010;25:1447‐1456. [DOI] [PubMed] [Google Scholar]
- 36. Shan S, Hui G, Hou F, et al. Expression of metastasis‐associated protein 3 in human brain glioma related to tumor prognosis. Neurol Sci. 2015;36:1799‐1804. [DOI] [PubMed] [Google Scholar]
- 37. Kato A, Kurita S, Hayashi A, Kaji N, Ohashi K, Mizuno K. Critical roles of actin‐interacting protein 1 in cytokinesis and chemotactic migration of mammalian cells. Biochem J. 2008;414:261‐270. [DOI] [PubMed] [Google Scholar]
- 38. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 39. Haslene‐Hox H, Oveland E, Woie K, Salvesen HB, Wiig H, Tenstad O. Increased WD‐repeat containing protein 1 in interstitial fluid from ovarian carcinomas shown by comparative proteomic analysis of malignant and healthy gynecological tissue. Biochim Biophys Acta. 2013;1834:2347‐2359. [DOI] [PubMed] [Google Scholar]
- 40. Izawa S, Okamura T, Matsuzawa K, et al. Autoantibody against WD repeat domain 1 is a novel serological biomarker for screening of thyroid neoplasia. Clin Endocrinol (Oxf). 2013;79:35‐42. [DOI] [PubMed] [Google Scholar]
- 41. Kim D‐H, Bae J, Lee JW, et al. Proteomic analysis of breast cancer tissue reveals upregulation of actin‐remodeling proteins and its relevance to cancer invasiveness. Proteomics Clin Appl. 2009;3:30‐40. [DOI] [PubMed] [Google Scholar]
- 42. Van Hook K, Wang Z, Chen D, et al. DeltaN‐ASPP2, a novel isoform of the ASPP2 tumor suppressor, promotes cellular survival. Biochem Biophys Res Commun. 2017;482:1271‐1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rotem S, Katz C, Benyamini H, et al. The structure and interactions of the proline‐rich domain of ASPP2. J Biol Chem. 2008;283:18990‐18999. [DOI] [PubMed] [Google Scholar]
- 44. Khursheed M, Kolla JN, Kotapalli V, et al. ARID1B, a member of the human SWI/SNF chromatin remodeling complex, exhibits tumour‐suppressor activities in pancreatic cancer cell lines. Br J Cancer. 2013;108:2056‐2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vengoechea J, Carpenter L, Zarate YA. Papillary thyroid cancer in a patient with interstitial 6q25 deletion including ARID1B. Am J Med Genet A. 2014;164A:1857‐1859. 164. [DOI] [PubMed] [Google Scholar]
- 46. Shago M, Abla O, Hitzler J, Weitzman S, Abdelhaleem M. Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr Blood Cancer. 2016;63:1915‐1921. [DOI] [PubMed] [Google Scholar]
- 47. Li Y, Huang J, Sun J, et al. The transcription levels and prognostic values of seven proteasome alpha subunits in human cancers. Oncotarget. 2017;8:4501‐4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Greco A, Mariani C, Miranda C, et al. The DNA rearrangement that generates the TRK‐T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled‐coil domain. Mol Cell Biol. 1995;15:6118‐6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mencinger M, Panagopoulos I, Andreasson P, Lassen C, Mitelman F, Aman P. Characterization and chromosomal mapping of the human TFG gene involved in thyroid carcinoma. Genomics. 1997;41:327‐331. [DOI] [PubMed] [Google Scholar]
- 50. Mitelman F, Johansson B, Mertens F. Mitelman database of chromosome aberrations and gene fusions in cancer. http://cgap.nci.nih.gov/Chromosomes/Mitelman. Published 2018.
- 51. Hu X, Wang Q, Tang M, et al. TumorFusions: an integrative resource for cancer‐associated transcript fusions. Nucleic Acids Res. 2018;46:D1144‐D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brenca M, Rossi S, Polano M, et al. Transcriptome sequencing identifies ETV6 – NTRK3 as a gene fusion involved in GIST. J Pathol. 2016;238:543‐549. [DOI] [PubMed] [Google Scholar]
- 53. Kasaian K, Wiseman SM, Walker BA, et al. The genomic and transcriptomic landscape of anaplastic thyroid cancer: implications for therapy. BMC Cancer. 2015;15:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jones DTW, Hutter B, Jäger N, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ross JS, Wang K, Chmielecki J, et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer. 2016;138:881‐890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grisham RN, Sylvester BE, Won H, et al. Extreme outlier analysis identifies occult mitogen‐ activated protein kinase pathway mutations in patients with low‐grade serous ovarian cancer. J Clin Oncol. 2015;33:4099‐4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hamatani K, Mukai M, Takahashi K, Hayashi Y, Nakachi K, Kusunoki Y. Rearranged anaplastic lymphoma kinase (ALK) gene in adult‐onset papillary thyroid cancer amongst atomic bomb survivors. Thyroid. 2012;22:1153‐1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Demeure MJ, Aziz M, Rosenberg R, Gurley SD, Bussey KJ, Carpten JD. Whole‐genome sequencing of an aggressive BRAF wild‐type papillary thyroid cancer identified EML4‐ALK translocation as a therapeutic target. World J Surg. 2014;38:1296‐1305. [DOI] [PubMed] [Google Scholar]
- 59. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature. 2007;448:561‐566. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 : Supporting Information
Supporting Information Figure S1 Novel FGFR1‐TG fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: COesterase, Carboxylesterase family; Ephrin_rec_like, Putative ephrin‐receptor like; I‐set, Immunoglobulin I‐set domain; ig, Immunoglobulin domain; Pkinase_Tyr, Protein tyrosine kinase; SP, signal peptide; Thyroglobulin_1, Thyroglobulin type‐1 repeat; TM, transmembrane region
Supporting Information Figure S2. Novel ZSWIM5‐TP53BP2 fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: Ank_2, Ankyrin repeats (3 copies); CC, coiled‐coil region; SH3_1, SH3 domain
Supporting Information Figure S3. Novel TAF4B‐WDR1 fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: CC, coiled‐coil region; TAF4, Transcription initiation factor TFIID component TAF4 family; TAFH, NHR1 homology to TAF; WD40, WD domain, G‐beta repeat
Supporting Information Figure S4. Novel ABI2‐MTA3 fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: Abi_HHR, Abl‐interactor HHR; BAH, BAH domain; ELM2, ELM2 domain; GATA, GATA zinc finger; MTA_R1, MTA R1 domain; Myb_DNA‐binding, Myb‐like DNA‐binding domain; SH3_9, Variant SH3 domain
Supporting Information Figure S5. Novel ARID1B‐PSMA1 fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: ARID, ARID/BRIGHT DNA binding domain; BAF250_C, SWI/SNF‐like complex subunit BAF250/Osa; CC, coiled‐coil region; Proteasome, Proteasome subunit; Proteasome_A_N, Proteasome subunit A N‐terminal signature
Supporting Information Figure S6. Novel isoform of CCDC6‐RET fusion transcript detected in PTC. The upper image shows the schematic diagram of the predicted fusion protein. The lower image shows the confirmation of the fusion transcript by direct Sanger sequencing. Abbreviations: Cadherin, Cadherin domain; CC, coiled‐coil region; DUF2046, Uncharacterized conserved protein H4 (DUF2046); Pkinase_Tyr, Protein tyrosine kinase; RET_CLD1, RET Cadherin like domain 1; RET_CLD3, RET Cadherin like domain 3; RET_CLD4, RET Cadherin like domain 4; SP, signal peptide; TM, transmembrane region
Supporting Information Figure S7. Known TFG‐NTRK1 fusion event detected in PTC. Breakpoint: chr3:100455548‐ chr1:156844363. Abbreviations: CC, coiled‐coil region; LRR_8, Leucine rich repeat; PB1, PB1 domain; Pkinase_Tyr, Protein tyrosine kinase; SP, signal peptide; TPKR_C2, Tyrosine‐protein kinase receptor C2 Ig‐like domain; TrkA_TMD, Tyrosine kinase receptor A trans‐membrane domain
Supporting Information Figure S8. Known TFG‐NTRK1 fusion event detected in PTC. Breakpoint: chr3:100455560‐chr1:156844363. Abbreviations: CC, coiled‐coil region; LRR_8, Leucine rich repeat; PB1, PB1 domain; Pkinase_Tyr, Protein tyrosine kinase; SP, signal peptide; TPKR_C2, Tyrosine‐protein kinase receptor C2 Ig‐like domain; TrkA_TMD, Tyrosine kinase receptor A trans‐membrane domain
Supporting Information Figure S9. Known ETV6‐NTRK3 fusion event detected in PTC. Abbreviations: Ets, Ets‐domain; I‐set, Immunoglobulin I‐set domain; ig, Immunoglobulin domain; LRR_8, Leucine rich repeat; LRRNT, Leucine rich repeat N‐terminal domain; Pkinase_Tyr, Protein tyrosine kinase; SAM_PNT, Sterile alpha motif (SAM)/Pointed domain; SP, signal peptide; TM, transmembrane region; TPKR_C2, Tyrosine‐protein kinase receptor C2 Ig‐like domain
Supporting Information Figure S10. Known MKRN1‐BRAF fusion event detected in PTC. Abbreviations: C1_1, Phorbol esters/diacylglycerol binding domain (C1 domain); CC, coiled‐coil region; MKRN1_C, E3 ubiquitin‐protein ligase makorin‐1, C‐terminal; Pkinase_Tyr, Protein tyrosine kinase; RBD, Raf‐like Ras‐binding domain; zf‐C3HC4, Zinc finger, C3HC4 type (RING finger); zf‐CCCH_4, CCCH‐type zinc finger
Supporting Information Figure S11. Known EML4‐ALK fusion event detected in PTC. Abbreviations: CC, coiled‐coil region; Gly_rich, Glycine rich protein; HELP, HELP motif; MAM, MAM domain, meprin/A5/mu; Pkinase_Tyr, Protein tyrosine kinase; SP, signal peptide; TM, transmembrane region; WD40, WD domain, G‐beta repeat
Supporting Information Table S1. Mutations in analyzed samples detected by Sanger sequencing and qRT‐PCR. 0—negative, 1—positive, ND—not done.
Supporting Information Table S2. Primers sequences and annealing temperatures used in PCR reactions
Supporting Information Table S3. Summary of data generated in RNA‐seq experiment (after applying filtering and trimming of reads)
Supporting Information Table S4. TopHat Fusion output (after applying in‐house filters)
Supporting Information Table S5. ChimeraScan output (after applying in‐house filters)
Supporting Information Table S6. SnowShoes output
Supporting Information Table S7. Combined outputs from TopHat, ChimeraScan and SnowShoes: read‐through gene fusions and fusions occurring in normal samples (as depicted by Babiceanu et al.)
Supporting Information Table S8. Combined outputs from TopHat, ChimeraScan and SnowShoes: gene fusions that are not read‐through and do not occur in normal samples (potential rearrangements)