

Abstract
This review addresses two major functions of apolipoprotein (apo) A5 including (1) its role in maintaining normal plasma levels of circulating triglyceride (TG) and (2) its role as a component of hepatic lipid droplets. ApoA5 is synthesized solely in the liver and circulating concentrations are extremely low. In the plasma, apoA5 associates with TG-rich lipoproteins and enhances TG hydrolysis and remnant lipoprotein clearance. ApoA5 loss-of-function single nucleotide polymorphisms are associated with reduced lipolysis, poor remnant clearance and concomitantly, hypertriglyceridemia. Although there have been substantial breakthroughs in understanding pathophysiology associated with secreted apoA5, there is a paucity of knowledge on the functionality of intracellular apoA5. However, recent studies indicate that overexpression of intracellular apoA5 is positively associated with accumulation of TG-rich lipid droplets in hepatocytes. It is thought that apoA5 may have a causal role in non-alcoholic fatty liver disease (NAFLD) and thus, may serve as a target for developing therapeutics for NAFLD.
Keywords: Apolipoprotein A5, hepatocytes, hypertriglyceridemia, lipid-droplets, non-alcoholic fatty liver disease, single nucleotide polymorphisms, triglyceride-rich lipoproteins
INTRODUCTION
Apolipoprotein (apo) A5 (also referred to as apoA-V) is an intriguing exchangeable apolipoprotein that was discovered in 2001 by reverse genetics [1, 2]. APOA5 localizes to the APOA1, APOC3, APOA4 gene cluster on chromosome 11q23. The gene is expressed solely in liver, giving rise to a 366 amino acid pre-protein that contains a 23 amino acid signal sequence. Despite the fact that plasma levels of mature apoA5 are extremely low [3], it plays a key role in triglyceride (TG) homeostasis. A deficiency of apoA5 in mice [1] or humans [4, 5] results in hypertriglyceridemia (HTG). In addition to regulating plasma TG levels, evidence indicates apoA5 plays a role in intracellular TG metabolism [1, 6]. Thus, this low abundance protein functions in two distinct locations, the plasma compartment and within hepatocytes. The present review examines salient features of apoA5-mediated TG regulation and its connection to HTG and non-alcoholic fatty liver disease (NAFLD).
EXTRACELLULAR FUNCTION OF APOA5
The seminal studies of Pennacchio et al. [1] revealed that transgenic mice over-expressing human apoA5 manifest decreased plasma TG while mice deficient in apoa5 had strongly elevated TG levels. In vitro and in vivo studies have revealed that apoA5 indirectly stimulates lipoprotein lipase (LPL) mediated TG hydrolysis in plasma [7]. This function involves interaction between apoA5 (as a component of TG-rich lipoproteins) and glycosylphosphatidylinositol-anchored HDL binding protein 1 (GPIHBP1). The emerging model proposes that three components (apoA5, GPIHBP1 and LPL) cluster in plasma membrane subdomains of capillary endothelial cells (EC) to form a coordinate structure that enhances lipolysis of apoC2 containing TG-rich lipoproteins, including chylomicrons and large VLDL [8, 9]. Cellular uptake of the free fatty acid products generates cholesterol-containing remnant lipoproteins that are ultimately cleared from circulation by receptor-mediated endocytosis. In addition to GPIHBP1, apoA5 has been shown to bind members of the LDL receptor family [10] and/or heparan sulfate proteoglycans [11, 12], thereby contributing to remnant lipoprotein clearance. Maintenance of plasma TG homeostasis is thus dependent not only on apoA5’s capacity to stimulate lipolysis but also on its ability to promote remnant lipoprotein clearance.
In post-prandial plasma, apoA5 is found principally on chylomicrons and VLDL. However, following TG hydrolysis, apoA5 migrates to HDL, which appears to function as a plasma reservoir for apoA5. The exceedingly low concentration of apoA5 in plasma (~100 – 250 ng/ml [3]) begs the question of how it promotes lipolysis when only an estimated 1 in ~24 VLDL particles [review, 13] possess apoA5. It is possible, that some apoA5 is bound to EC surface heparan sulfate proteoglycans, increasing the concentration of this protein that is accessible to plasma. Consistent with this, injection of heparin into APOA5 transgenic mice released additional apoA5 into plasma [14], increasing its concentration by 25 %. Although Blade et al. [15] found no evidence for apoA5 on the surface of cultured McA-RH7777 hepatoma cells, apoA5 interaction with EC, via heparan sulfate proteoglycans, remains a plausible explanation. It is currently unknown whether EC-bound apoA5 influences the interaction between GPIHBP1 and LPL to enhance the efficiency of lipoprotein-associated TG hydrolysis.
STRUCTURAL AND FUNCTIONAL CONSEQUENCES OF APOA5 MUTATIONS
ApoA5 is a hydrophobic protein with well-defined functional domains [review, 16]. The extreme C-terminus (mature sequence positions 295 – 343) facilitates lipid binding while the N-terminal domain (amino acids 1 to 146 of mature apoA5) adopts a water-soluble helix bundle conformation. The intervening central segment (residues 147 – 294) possesses a positively charged region (residues 186 – 227) that is functionally important for lipolysis [review, 17]. It is not surprising, therefore, that the Q145X truncation mutant described by Priore Oliva et al. [4], as well as the Q139X mutant described by Marcais et al. [5], are associated with severe HTG. These truncated apoA5s lack both the C-terminal lipid binding motif and the positively charged central domain required for GPIHBP1 / LDL receptor family / heparan sulfate proteoglycan binding. Unlike full-length apoA5, when expressed in McA-RH7777 cells, C-terminal truncation variants are recovered in the lipid-free fraction of conditioned medium [18]. This finding corroborates the observation of Marcais et al. [5] that the Q139X APOA5 mutation gives rise to a truncated apoA5 that localizes to the lipoprotein deficient fraction of plasma. Thus, in vivo and in vitro studies support the premise that C-terminal deletion abolishes apoA5’s affinity for lipoproteins. This loss of function results in reduced lipolysis and remnant clearance, with the subsequent manifestation of HTG.
CONSEQUENCES OF APOA5 POLYMORPHISMS
Human population studies have identified several single nucleotide polymorphisms (SNPs) that correlate with aberrant TG metabolism. The -1131T>C SNP, located in the promoter region of APOA5, results in defective transcription [19]. The -3A>G SNP, located in the Kozak sequence, affects APOA5 mRNA translation efficiency [20]. A coding SNP, c.56C>G, results in substitution of serine at position 19 by tryptophan (S19W) in the apoA5 signal sequence. Evidence suggests this SNP interferes with apoA5 secretion [21]. Genome wide association studies (GWAS) provide further support for the concept that APOA5 influences plasma TG levels [22]. Moreover, a recent whole exome sequencing study involving over 9600 subjects, revealed numerous minor APOA5 SNPs that correlate with premature myocardial infarction [23]. Despite the association between common and rare APOA5 SNPs and TG levels, the precise mechanism(s) whereby given SNPs contribute to HTG is not well understood.
One SNP recognized for its association with elevated plasma TG is the c.553G>T polymorphism wherein a Cys for Gly substitution is introduced at residue 185 of the pre-protein [19] (corresponding to residue 162 in mature apoA5). In addition to this SNP-specific cysteine, mature human apoA5 possesses a Cys at residue 204. The c.553G>T APOA5 polymorphism is prevalent in Chinese subjects [24–26] where it exhibits high allelic frequency. Heterozygous carriers of this SNP (G/T alleles) had a doubling of plasma TG while homozygous expression of the T/T allele was associated with extreme TG elevation (TG= 2,292±447 mg/dl compared to 92±38 mg/dl for G/G subjects) [26]. This SNP is associated with increased risk of cardiovascular disease and is of interest because it is likely that millions of people worldwide are carriers. Noteworthy is the observation of Pullinger et al. [26] that there was no significant difference between plasma levels of apoA5 in T/T subjects compared to G/G subjects, although LPL activity was adversely affected. Recent studies using adeno-associated virus gene transfer technology to introduce c.553G>T APOA5 into apoa5 (−/−) mice shed new light on the mechanism underlying elevated TG associated with this polymorphism [27]. When expressed in apoa5 (−/−) mice, greater than 50% of G162C apoA-V was recovered in the lipoprotein-deficient fraction of plasma wherein it formed hetero-disulfide bonds with unrelated cysteine-bearing proteins. As in the mouse model, humans carrying the T/T alleles also had the majority of apoA5 in the lipoprotein-deficient plasma fraction where the protein formed disulfide bonds with numerous cysteine-containing proteins. Commensurate with sequestration of apoA5 in the lipoprotein-deficient fraction, plasma TG levels in mice and humans were substantially elevated [27]. Furthermore, the presence of apoA5 in the lipoprotein-free fraction of plasma likely explains the observation of Pullinger et al. [26] that LPL activity was significantly reduced in T/T subjects. Results of these studies demonstrate that the introduction of a second Cys in apoA5, as a consequence of the c.553G>T SNP, results in loss of apoA5 function, with subsequent manifestation of HTG. It is not known whether hetero-disulfide bonds formed in plasma have an adverse effect on the ability of the variant apoA5 to bind to the EC surface, further contributing to reduced LPL activation. Importantly, recent exome sequence analyses verify that this SNP is a risk factor in premature myocardial infarction [23]. This analysis also revealed that, in addition to the G185C apoA5, other low frequency Cys substitution polymorphisms in APOA5 are associated with increased risk of early myocardial infarction including R245C apoA5, R282C apoA5 and R343C apoA5. A question that remains is whether the latter three variants also form hetero-disulfide bonds with extraneous plasma proteins, leading to compromised function.
INTRACELLULAR LIPID DROPLETS AND APOA5
Lipid droplets (LD) are ubiquitous organelles in plant and animal cells. Universal features of LD include a cytosolic location, spherical geometry, a hydrophobic core composed of neutral lipids (TG and cholesteryl esters) and a surface monolayer comprised of phospholipid and protein, particularly members of the perilipin family. In terms of composition and morphology, LDs are not unlike plasma lipoproteins. Serendipity led Shu et al. [28] to discover that apoA5 associates with intracellular LDs in rat hepatoma McA-RH7777–18A cells following transient transfection. The original question probed by Shu et al. was whether apoA5 associates with nascent apoB-containing VLDL in the ER lumen prior to secretion. Transient expression of the APOA5 gene in McA-RH7777 cells demonstrated unequivocally that apoA5 does not associate with apoB within cells but, unexpectedly, co-localized with LD. Subsequent studies revealed that LD number and size are affected by the concentration of apoA5, wherein high expression levels lead to increased numbers and larger sized LD [29]. Subsequent molecular evolutionary genetics analysis revealed structural homology between apoA5 and perilipins, providing a molecular basis for the binding of apoA5 to LDs [30]. The finding that transient expression of APOA1 did not result in apoA1 binding to LD [28] suggests a unique feature of apoA5 structure, possibly its strong hydropobicity, may be important for its LD homing ability.
Biogenesis of LD occurs in the ER, where TG accumulation creates a lipid lens between leaflets of the bilayer that grows in size, budding toward the cytosol and pinching off to form a discrete organelle [31]. In hepatocytes, TG lens budding toward the ER lumen supports the process of TG-rich apoB-containing lipoprotein assembly and secretion. Recognizing the unique property of liver to store TG in LD or to secrete it as a component of lipoproteins, Gao et al. [29] speculated that apoA5 plays a role in regulating the directionality of intracellular TG flux. A model was proposed wherein apoA5 in the ER lumen interferes with LD budding toward the lumen while promoting cytosolic LD formation. It was further suggested that interaction of lumenal apoA5 with ER membrane defects leads to its translocation to the cytosol and stable association with the surface of newly formed LD [review, 32].
As mentioned above, it is conceivable that apoA5 association with LD is initiated from the ER lumen. This is consistent with the fact that nascent apoA5 possesses a 23 amino acid N-terminal signal sequence that is recognized by the signal recognition particle (SRP). The SRP in turn, interacts with the ER membrane translocon machinery for co-translational passage of newly synthesized apoA5 into the ER lumen. Within the lumen, signal peptidase cleaves the signal sequence yielding mature apoA5 (Fig. 1A). Mature apoA5 has two possible fates: 1) interaction with membrane defects caused by nascent LD formation, leading to its association with nascent LDs or 2) passage from the ER to the Golgi and secretion from the cell. Recognizing that apoA5 association with cytosolic LD may occur in the absence of co-translational translocation to the ER, an alternate post-translational route is depicted in Fig. (1B). In this scheme, protein translation is completed in the cytosol followed by direct binding of nascent apoA5 to pre-existing LD, thereby avoiding the ER secretory pathway altogether.
Fig. (1). Alternate pathways for production and trafficking of nascent apoA5.
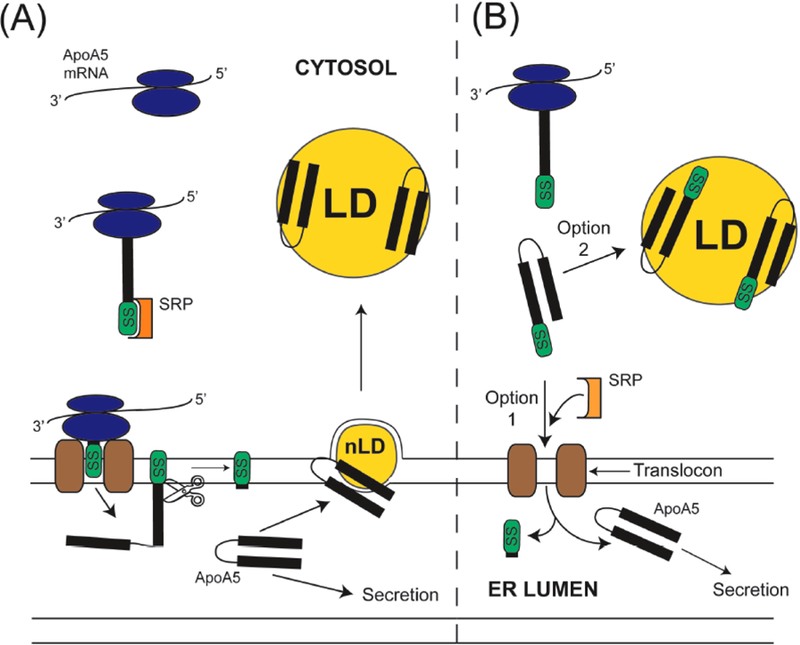
In pathway (A) the ribosomal translation machinery generates a nascent apoA-V signal sequence (SS) that binds to the Signal Recognition Particle (SRP) with subsequent docking to the SRP receptor (not shown) with continued translation through the translocon and into the endoplasmic reticulum (ER) lumen. Translated preprotein in the ER lumen is a substrate for signal peptidase (depicted as scissors), which cleaves the signal sequence, releasing mature apoA5. In Pathway A, mature apoA5 either associates with membrane defects at the site of nascent lipid droplet (nLD) assembly or transits the ER for secretion from the cell. In pathway (B) apoA5 translation is completed within the cytoplasm. Once formed, the apoA5 preprotein may undergo post-translational translocation to the ER for secretion from the cell (option 1) or it may escape this fate by interaction, and stable binding to, cytosolic LD (option 2).
Signal sequences are variable in amino acid composition and peptide length, features that can affect the efficiency of protein secretion [33]. It is conceivable that apoA5’s signal sequence is an inefficient SRP ligand such that LD association occurs as a default event. Overexpression of apoA5 with a subsequent over-abundance of newly synthesized protein that overwhelms the translocation machinery, results in trafficking of apoA5 to LD as suggested by the study of Shu et al. [18]. Once associated with the surface of LD, the protein will be protected from ER translocation / secretion as well as proteasomal degradation. Although this could explain the appearance of over-expressed apoA5 on LD in transfected cell culture models, it does not explain the finding that apoA5 is also found on LD isolated from mouse livers where apoA5 is extremely low [14]. The latter, however, supports the premise that apoA5 LD association is a physiologically relevant and natural process.
A recent report [34] described a child with severe HTG and no detectable apoA5 in plasma who was found to be homozygous for a partial signal sequence deletion (c.16– 39del; p.Ala6-Ala13). The parents, who were heterozygotes, had low plasma apoA5 levels and elevated TG. When primary hepatocytes from apoa5 (−/−) mice were transfected with wild type (WT) or p.Ala6-Ala13 mutant APOA5, WT apoA5 was secreted into the medium while mutant apoA5 remained intracellular, in association with LD [34]. Thus, expression of APOA5 with a defective signal sequence increased its association with LD. Deletion of 8 residues from the core region of the signal sequence likely prevents SRP binding and ER translocation, resulting in direct interaction of nascent apoA5 with LD (Fig. 1B), explaining the lack of apoA5 in plasma and severe HTG in this subject.
It is noteworthy that a coding SNP within the signal sequence, c.56C>G (S19W apoA5), has a relatively high incidence in the population [19] and is correlated with low plasma levels of apoA5 and corresponding high plasma TG levels. Furthermore, this SNP is associated with increased risk of myocardial infarction [35]. If this amino acid substitution affects SRP binding or signal peptide cleavage efficiency, then it is anticipated that nascent S19W apoA5 may be attracted to the surface of LD. On the one hand, it is possible that poor signal sequence cleavage efficiency affects the membrane association properties of apoA5 in the ER, thereby enhancing its interaction with nascent LD from the ER lumen. The Trp substitution is 4 residues from to the signal sequence cleavage site and early studies, using a secreted alkaline phosphatase reporter construct to evaluate the effect of the S19W SNP on signal sequence function, revealed a correlation with low secretion efficiency [21]. Alternatively, it is possible that S19W apoA5 is not efficiently recognized by SRP, allowing apoA5 to escape ER translocation such that it associates with LD directly.
Expression of apoA5 without a signal sequence in McA-RH7777 cells resulted in cytosolic accumulation of apoA5 and no secretion [18]. Thus, the signal sequence plays an integral role in trafficking of this protein for secretion. Shu et al. [14] examined the relationship between apoA5 concentration and liver TG content in vivo in APOA5 transgenic mice and apoa5 (−/−) mice and noted that the TG content of livers of transgenic mice was elevated compared to apoa5 (−/−) mice. These data suggest that intracellular apoA5 levels influence intrahepatic TG accumulation. A major question that remains relates to the function of apoA5 on LD. Does it represent an inert storage pool of apoA5 or does it function in a manner akin to perilipins [review, 36], influencing TG accumulation in liver? In the latter case, apoA5 may play a role in the pathophysiology of non-alcoholic fatty liver disease (NALFD).
NON-ALCOHOLIC FATTY LIVER DISEASE (NAFLD) AND APOA5
NAFLD is frequently referred to as the “silent liver disease” and is a pathological disorder that is rapidly increasing in developed countries, paralleling increases in obesity and diabetes. The latter are major public health concerns. The prevalence of NAFLD is estimated at 80 – 90 % in obese adults and up to 40 – 70 % in obese children [37]. NAFLD is closely linked with metabolic syndrome, a condition manifest by obesity, impaired glucose tolerance, dyslipidemia, hypertension and insulin resistance. NAFLD is characterized by increased hepatic TG synthesis with ectopic accumulation of LD [38–40]. In its early stages, NAFLD is considered relatively benign and reversible. Inflammation in liver, along with continued lipid accumulation, leads to non-alcoholic steatohepatitis (NASH). This condition is associated with fibrosis, a pathological event that can progress to cirrhosis [41]. Ultimately, NASH can progress to end-stage liver disease and development of hepatocellular carcinoma. It is not yet known which specific intracellular factors contribute to unregulated lipid accumulation in hepatocytes. Proposed candidates include insulin resistance, oxidative stress, ER stress, proinflammatory stimuli, impaired ß-oxidation, increased availability of fatty acids, increased de novo lipogenesis, and dysregulation of lipoprotein assembly and secretion [review, 42]. In addition, a number of studies implicate a genetic component wherein a familial predisposition to NAFLD exists [43, 44]. The appearance of apoA5 on hepatic LD has led to speculation that this protein may have an active role in NALFD [45].
Ress et al. [45] carried out seminal studies with morbidly obese patients who underwent bariatric surgery. Following surgery, there was significant weight loss as well as a decrease in insulin levels and insulin resistance. Importantly, there was also a profound reduction in histological scores for steatosis. This observation was paralleled by a significant reduction in apoA5 mRNA, suggesting that apoA5 contributes to NAFLD. To further characterize apoA5’s role in NAFLD, the authors employed HepG2 hepatocarcinoma cells in which apoA5 mRNA was silenced. Transfection with apoA5 siRNA resulted in almost complete loss of intracellular apoA5 and a significant reduction in intracellular TG, compared to cells transfected with a control siRNA. These results corroborate results obtained with patient biopsies. A question arising from these studies is whether apoA5 knockdown in this cell culture model affected enzymes and/or transcription factors implicated in lipid metabolism. Upon analysis, the authors found no apparent effect on mRNAs encoding DGAT-2, CPT-1, FAS, GPAT-1, SREBP-1c or PPARα [45]. PPARγ, is a member of the nuclear hormone receptor family found in low levels in liver. In obese subjects with fatty liver disease there was no correlation between apoA5 mRNA and PPARγ mRNA. In fact, there was no change in PPARγ expression following bariatric surgery. Thus, understanding exactly how apoA5 levels influence LD accumulation in NAFLD remains elusive.
Pediatric obese subjects have recently been shown to manifest the same disease characteristics as adults wherein patients exhibited elevated mRNA apoA5 levels in liver, as compared to a pediatric control population [46]. Furthermore, apoA5 mRNA levels in pediatric NAFLD subjects were positively correlated with hepatic TG accumulation. However, there was no correlation with plasma TG levels. As might be expected, insulin resistance was elevated. When a high fat diet rat model was examined [46], the results obtained recapitulated NAFLD characteristics found in humans, lending support to the proposal that apoA5-induced hepatic TG storage contributes to NAFLD.
A recent study by Camporez et al. [47] examined the relationship between apoA5 expression and lipid deposition in liver tissue of C57Bl/6 mice. The mice were maintained on either a regular chow diet or a high fat diet and were treated with apoa5 antisense oligonucleotide (ASO) for 8 weeks to reduce apoA5 expression up to 70%. The reduction in apoA5 expression was paralleled by a 3-fold increase in plasma TG concentration. The latter extracellular process was associated with a reduction in TG clearance. Similar to the observation of Ress et al. [45] in apoA5 knockdown HepG2 cells, ASO-induced reduction of apoA5 expression in this mouse model was commensurate with a reduction of liver TG accumulation. Furthermore, the reduction of intracellular TG correlated with improved hepatic and peripheral insulin responsiveness. Thus, human and mouse studies support the premise that apoA5 has a direct effect on ectopic TG accumulation in liver. In obesity and its associated insulin resistance, it is possible that apoA5 acts as a sensor for increased fatty acid production and increased accumulation of fatty acids derived from adipocytes (Fig. 2B) thereby resulting in increased numbers and size of hepatic LD. Unlike a normal insulin response, where plasma triglyceride-rich lipoproteins (TRLs) are low (Fig. 2A), in subjects with insulin resistance plasma TRL levels along with apoA5 levels are elevated reflecting a dysfunction in TG hydrolysis and clearance (Fig. 2B). Indeed, it has been shown in obese and diabetic subjects that plasma apoA5 and TG are elevated [48, 49]. Other factors, such as increased apoC3 or reduced apoC2, may be contributing factors.
Fig. (2). Effect of obesity and insulin resistance on hepatocyte apoA5 levels and LD production.
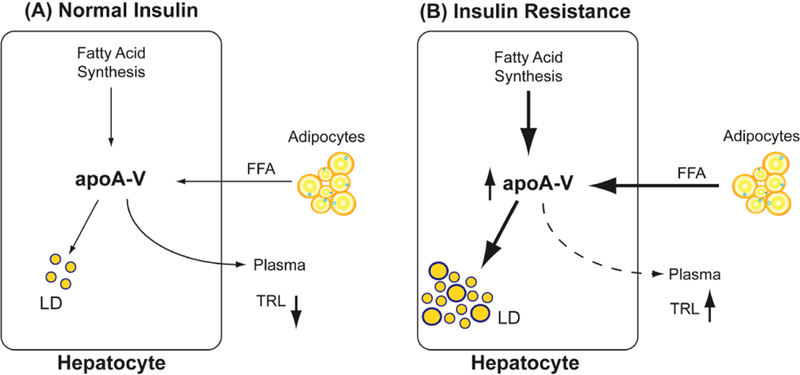
(A) Under conditions of normal apoA5 expression, de novo synthesis of free fatty acid (FFA) in hepatocytes is low; so also is the delivery of FFA from adipocytes. In this case, there are few cytosolic apoA5-containing LDs and apoA5 secreted into the plasma efficiently hydrolyzes plasma TRLs and stimulates uptake of remnants, thus accounting for low plasma TG levels. (B) In the case of insulin resistance, de novo synthesis of fatty acid and influx of FFA from adipocytes are elevated. These processes stimulate apoA5 synthesis and accumulation of LDs (a hallmark of NAFLD). The flux of apoA5 to the plasma compartment is unknown as indicated by the dashed arrow; however, plasma TRLs and apoA5 remain elevated, most likely due to a secondary dysfunction in normal TG hydrolysis and remnant clearance linked to the insulin resistance phenotype.
Not only does apoA5 play a central role in lipid metabolism, but it also has a role in energy balance. Examining more closely the role of apoA5 in energy balance, van den Berg et al. [50] showed that apoa5 (−/−) mice fed a high fat diet became more obese and manifest greater hepatic steatosis and insulin resistance, compared to control mice. In this study aberrant phenotypes were reversed by adenovirus-mediated overexpression of APOA5 in liver. These findings suggest that, in this model, apoA5 overexpression prevents ectopic lipid accumulation rather than increasing it. These results are opposite those reported for obese humans and rats and mice fed a high fat diet [45–47] where apoA5 appears to be necessary for ectopic lipid accumulation in liver. The difference, in part, may reflect the use of the apoa5 (−/−) mouse where lipid and apolipoprotein metabolism are not necessarily similar to that of humans and/or the fact that apoa5 antisense oligonucleotide treated mice have residual apoA5 synthesis capability.
The key factor(s) that regulates apoA5 expression in the liver, and hence intracellular TG accumulation, is not well understood. However, a recent study of Nur77 may offer some clues. Nur77 is a member of the NR4A1 orphan nuclear receptor family and is known to modulate apoptosis, gluconeogenesis and lipogenesis in liver. Nur77 binds the promoter region of APOA5 and recent studies suggest Nur77 regulates apoA5 expression [51] in HepG2 cells. It is not evident, however, whether Nur77 affects LD formation / TG accumulation in NAFLD. Regulation of apoA5 mRNA and protein in liver cells is incompletely understood. Furthermore, if apoA5 is causal for LD formation and accumulation, does it accomplish this by regulating gene(s) involved in lipid metabolic pathways? Thus far, such a linkage has not been demonstrated. Another possibility is that apoA5 contributes indirectly to LD accumulation by stabilizing preformed LD.
In summary, it is acknowledged that, based on the limited data available on apoA5 in steatosis in humans, the mechanism whereby apoA5 contributes to the pathophysiology of hepatic LD accumulation has yet to be elucidated. ApoA5 is unique in that it plays a critical role in regulation of plasma TG homeostasis as well as TG accumulation in liver. Whereas there have been substantial breakthroughs in understanding of the function of apoA5 in plasma, its role in hepatic LD biology and NAFLD are less well understood.
ACKNOWLEDGEMENTS
The authors thank Jennifer Beckstead for her assistance in generating the figures used in this review. Research in the authors’ laboratory was supported by NIH grants HL-64159 and HL-073061.
ABBREVIATIONS
- apoA5
Apolipoprotein A-V
- ASO
Antisense oligonucleotide
- CPT-1
Carnitin-palmitoyl-transferase-1
- DGAT
Diacyl-glycerol-acyl-transferase-2
- EC
Endothelial cells
- ER
Endoplasmic reticulum
- FAS
Fatty acid synthase
- GPAT-1
Glycerol-3-phosphatase 1
- GPIHBP1
Glycosylphosphatidylinositol-anchored HDL binding protein 1
- HTG
Hypertriglyceridemia
- LD
Lipid droplet
- LPL
Lipoprotein lipase
- NAFLD
Non-alcoholic fatty liver disease
- PLIN
Perilipin
- PPARα
Peroxisome proliferator-activated receptor alpha
- PPARγ
Peroxisome proliferator-activated receptor gamma
- SNP
Single nucleotide polymorphism
- SREBP-1c
Sterol regulatory element-binding protein-1c
- SRP
Signal recognition protein
- TG
Triacylglycerol
- TRL
Triglyceride rich lipoprotein
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- [1].Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, Krauss RM Rubin EM. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science 2001; 294: 169–73. [DOI] [PubMed] [Google Scholar]
- [2].van der Vliet HN, Sammels MG, Leegwater AC, Levels JH, Reitsma PH, Boers W, Chamuleau RA. Apolipoprotein A-V: a novel apolipoprotein associated with an early phase of liver regeneration. J Biol Chem 2001; 276: 44512–20. [DOI] [PubMed] [Google Scholar]
- [3].O’Brien PJ, Alborn WE, Sloan JH, et al. The novel apolipoprotein A5 is present in human serum, is associated with VLDL, HDL, and chylomicrons, and circulates at very low concentrations compared with other apolipoproteins. Clin Chem 2005; 51: 351–9. [DOI] [PubMed] [Google Scholar]
- [4].Priore Oliva C, Pisciotta L, Li Volti G, Sambataro MP, Cantafora A, Bellocchio A, Catapano A, Tarugi P, Bertolini S, Calandra S. Inherited apolipoprotein A-V deficiency in severe hypertriglyceridemia. Arterio Throm Vasc Biol 2005; 25: 411–7. [DOI] [PubMed] [Google Scholar]
- [5].Marcais C, Verges B, Charriere S, Pruntea V, Merlin M, Billon S, Perrot L, Drai J, Sassolas A, Pennacchio LA, Fruchart-Najib J, Fruchart J-C, Durlach V, Moulin P. Apoa5 Q139X truncation pre-disposes to late onset hyperchylomicronemia due to lipoprotein lipase impairment. J Clin Invest 2005; 115: 2862–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schaap FG, Rensen PC, Voshol PJ, Vrins C, van der Vliet HN, Chamuleau RA, Havekes LM, Groen AK, van Dijk KW. ApoAV reduces plasma triglycerides by inhibiting very low density lipoprotein-triglyceride (VLDL-TG) production and stimulating lipoprotein lipase-mediated VLDL-TG hydrolysis. J Biol Chem 2004; 279: 27941–7. [DOI] [PubMed] [Google Scholar]
- [7].Lookene A, Beckstead JA, Nilsson S, Olivecrona G, Ryan, RO. Apolipoprotein A-V-heparin interactions: implications for plasma lipoprotein metabolism. J Biol Chem 2005; 280: 25383–87 [DOI] [PubMed] [Google Scholar]
- [8].Beignaux AP, Davies BS, Gin P, Weinstein MM, Farger E, Qiao X, Peale F, Bunting S, Walzem RL, Wong JS, Blaner WS, Ding ZM, Melford K, Wongsirioj N, Shu X, de Sauvage F, Ryan RO, Fong LG, Bensadoun A, Joung SG. Glycosylphosphatidylinositol-anchored high density lipoprotien-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab 2007; 5: 279–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shu X, Nelbach L, Weinstein MM, Burgess BL, Beckstead JA, Young SG, Ryan RO, Forte TM. Intravenous injection of apolipoprotein A-V reconstituted high-density lipoprotein decreases hypertriglyceridemia in apoav−/− mice and requires glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1. Arterioscler Thromb Vasc Biol 2010; 30: 2504–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nilsson SK, Lookene A, Beckstead JA, Gliemann J, Ryan RO, Olivecrona G. Apolipoprotein A-V interaction with members of the low density lipoprotein receptor gene family. Biochem 2007; 46: 3896–904. [DOI] [PubMed] [Google Scholar]
- [11].Merkel M, Loeffler B, Kluger M, Fabig N, Geppert G, Pennacchio LA, Latsch A, Heeren J. Apolipoprotein A-V accelerates plasma hydrolysis of triglyceride-rich lipoproteins by interaction with proteoglycan-bound lipoprotein lipase. J Biol Chem 2005; 280: 21553–60. [DOI] [PubMed] [Google Scholar]
- [12].Gonzales JC, Gordts LSM, Foley EM, Esko JD. Apolipoproteins E and A-V mediate lipoprotein clearance by hepatic proteoglycans. J Clin Invest 2013; 123: 2742–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Merkel M, Heeren J. Give me A5 for lipoprotein hydrolysis! J Clin Invest 2005; 115: 2694–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shu X, Nelbach L, Ryan RO, Forte TM. Apolipoprotein A-V associates with intrahepatic lipid droplets and influences triglyceride accumulation. Biochim Biophys Acta 2010; 1801: 605–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Blade AM, Fabritius MA, Hou L, Weinberg RB, Shelness GS. Biogenesis of apolipoprotein A-V and its impact on VLDL triglyceride secretion. J Lipid Res 2011; 52: 237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wong K, Ryan RO. Characterization of apolipoprotein A-V structure and mode of plasma triacylglycerol regulation. Curr Opin Lipidol 2007; 18: 319–24. [DOI] [PubMed] [Google Scholar]
- [17].Sharma V, Ryan RO, Forte TM. Apolipoprotein A-V dependent modulation of plasma triacylglycerol: a puzzlement. Biochim Biophys Acta 2012; 1821: 795–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shu X, Ryan RO, Forte TM. Intracellular lipid droplet targeting by apolipoprotein A-V requires the carboxyl-terminal segment. J Lipid Res 2008; 49: 1670–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pennacchio LA, Olivier M, Hubacek JA, Krauss RM, Rubin EM, Cohen JC. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Human Molec Gen 2002; 11: 3031–8. [DOI] [PubMed] [Google Scholar]
- [20].Palmen J, Smith AJ, Dorfmeister B, Putt W, Humphries SE, Talmud PJ. The functional interaction on in vitro gene expression of APOA5 SNPs, defining haplotype APOA5, and their paradoxical association with plasma triglyceride but not plasma apoA-V levels. Biochim Biophys Acta 2008; 1782: 447–52. [DOI] [PubMed] [Google Scholar]
- [21].Talmud PJ, Palmen J, Putt W, Lins L, Humphries SE. Determination of the functionality of common APOA5 polymorphisms. J Biol Chem 2005; 280: 28215–20. [DOI] [PubMed] [Google Scholar]
- [22].Johansen CT, Kathiresan S, Hegele RA. Genetic determinants of plasma triglycerides. J. Lipid Res 2011; 52: 189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Do R, Stitziel NO, Won HH. et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2014; 518: 102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kao JT, Wen HC, Chien KL, Hsu HC, Lin SW. A novel genetic variant in the apolipoprotein A5 gene is associated with hypertriglyceridemia. Nat Genet 2003; 12: 2533–9. [DOI] [PubMed] [Google Scholar]
- [25].Tang Y, Sun P, Guo D, Ferro A, Ji Y, Chen Q, Fan L. A genetic variant c.553G>T in the apolipoprotein A5 gene is associated with an increased risk of coronary artery disease and altered triglyceride levels in a Chinese population. Atherosclerosis 2006; 185: 433–7. [DOI] [PubMed] [Google Scholar]
- [26].Pullinger CR, Aouizerat BE, Movsesyan I, Durlach V, Sijbrands EJ, Nakajima K, Poon A, Dallinga-Thie GM, Hattori H Green LL, Kwok PY, Havel RJ, Frost PH, Malloy MJ, Kane JP. An apolipoprotein A-V gene SNP is associated with marked hypertriglyceridemia among Asian-American patients. J Lipid Res 2008; 49: 1846–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sharma V, Witkowski A, Witskowaska E, Dykstra A, Simonson JB, Nelbach L, Beckstead JA, Pullinger CR, Kane JP, Malloy MJ, Watson G, Forte TM, Ryan RO. Hypertriglyceridemia associated with the c.553G>T APOA5 SNP results from aberrant hetero-disulfide bond formation. Arterioscler Thromb Vasc Biol 2014; 34: 2254–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shu X, Chan J, Ryan RO, Forte TM. ApoA5 association with intracellular lipid droplets. J Lipid Res 2007; 48: 1445–50. [DOI] [PubMed] [Google Scholar]
- [29].Gao X, Forte TM, Ryan RO. Inifluence of apolipoprotein A-V on hepatocyte droplet formation. Biochem Biophys Res Comm 2012; 427: 361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yang L, Ding Y, Chen Y, Zhang S, Chaoxing H, Yang W, Yu J, Zhang P, Na H, Zhang H, Ma Y, Liu P. The proteomics of lipid droplets: structure, dynamics and functions of the organelle conserved from bacteria to humans. J Lipid Res 2012; 53: 1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Martin S, Parton RG. Lipid droplets: a unified view of a dynamic organelle. Nature Rev 2006; 7: 373–8. [DOI] [PubMed] [Google Scholar]
- [32].Sharma V, Forte TM, Ryan RO. Influence of apolipoprotein A-V on the metabolic fate of triacylglycerol. Curr Opin Lipidol 2013; 24: 153–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kober L, Zehe C, Bode J. Optimized signal peptides for the development of high expressing CHO cell lines. Biotechnol Bioeng 2013; 110: 1164–73. [DOI] [PubMed] [Google Scholar]
- [34].Albers K, Schlein C, Wenner K, Lohse P, Bartelt A, Heeren J, Santer R, Merkel M. Homozygosity for a partial deletion of apo-protein A-V signal peptide results in intracellular missorting of the protein and chylomicronemia in a breast-fed infant. Atherosclerosis 2014; 233: 97–103. [DOI] [PubMed] [Google Scholar]
- [35].Hubacek JA, Skodova Z, Adamkova V, Lanska F, Poledne R. The influence of APOAV polymorphisms (T-1131>C and S19W) on plasma triglyceride levels and risk of myocardial infarction. Clin Genet 2004; 65: 126–30. [DOI] [PubMed] [Google Scholar]
- [36].Ducharme NA, Bickel PE. Minireview: lipid droplets in lipogenesis and lipolysis. Endocrinology 2008; 149: 942–9. [DOI] [PubMed] [Google Scholar]
- [37].Milic S, Stimac D. Nonalcoholic fatty liver disease/steatohepatitis: epidemiology, pathogenesis, clinical presentation and treatment. Digest Diseases 2012; 30: 158–62. [DOI] [PubMed] [Google Scholar]
- [38].Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2006; 114: 1147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Goldberg IJ, Ginsberg HN. Ins and outs modulating hepatic triglyceride and development of nonalcoholic fatty liver disease. Gastroenterology 2006; 130: 1343–6. [DOI] [PubMed] [Google Scholar]
- [40].Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008; 134: 424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sass DA, Chang P, Chopra KB. Nonalcoholic fatty liver Disease: A clinical review. Digest Dis Sci 2005; 50: 171–80. [DOI] [PubMed] [Google Scholar]
- [42].Erickson SK. Nonalcoholic fatty liver disease. J Lipid Res 2009; 50: S412–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Osterreicher CH, Brenner DA. The genetics of non-alcoholic fatty liver disease. Ann Hepatol 2007; 6: 83–8. [PubMed] [Google Scholar]
- [44].Cheung O, Sanyal AJ. Recent advances in nonalcoholic fatty liver disease. Curr Opin Gastro 2010; 26: 202–208. [DOI] [PubMed] [Google Scholar]
- [45].Ress C, Moschen AR, Sausgruber N, Tschoner A, Graziadei I, Weiss H, Schgoer W, Ebenbichler CF, Konrad RJ, Patsch JR, Tilg H, Kaser S. The role of apolipoprotein A5 in non-alcoholic fatty liver disease. Gut 2011; 60: 985–91. [DOI] [PubMed] [Google Scholar]
- [46].Feng Q, Baker SS, Liu W, Arbizu RA, Aljomah G, Khatib M, Nugent CA, Baker RD, Forte TM, Hu Y, Zhu L. Increased apolipoprotein A5 expression in human and rat non-alcoholic fatty livers. Pathology In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Camporez JPG, Kanda S, Petersen MC, Jornayvaz FR, Samuel VT, Bhanot S, Petersen KF, Jurczak MJ, Shulman GI. ApoA5 knockdown improves whole-body insulin sensitivity in high-fat fed mice by reducing ectopic lipid content. J Lipid Res 2014; 56: 526–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dallinga-Thie GM, van Tol A, Hattori H, van Vark-van der Zee LC, Jansen H, Sijbrands EJ. Plasma apolipoprotein A5 and triglycerides in type 2 diabetes. Diabetologia 2006; 49: 1505–11. [DOI] [PubMed] [Google Scholar]
- [49].Talmud PJ, Cooper JA, Hattori H, Miller IP, Miller GJ, Humphries SE. The apolipoprotein A-V genotype and plasma apolipoprotein A-V and triglyceride levels: prospective risk of type 2 diabetes. Results from the Northwick Park Heart Study II. Diabetologia 2006; 49: 2337–40. [DOI] [PubMed] [Google Scholar]
- [50].van den Berg SAA, Heemskerk MM, Geerling JJ, van Klinken JB, Schaap FG, Bijland S, Berbee JFP, van Harmelen VJA, Pronk ACM, Schreurs M, Havekes LM, Rensen PCN, van Dijk KW. Apolipoprotein A5 deficiency aggravates high-fat diet-induced obesity due to impaired central regulation of food intake. FASEB J 2013; 27: 3354–62. [DOI] [PubMed] [Google Scholar]
- [51].Song K-H. Orphan nuclear receptor Nur77 participates in human apolipoprotein A5 gene expression. Biochem Biophys Res Comm 2010; 392: 63–6. [DOI] [PubMed] [Google Scholar]