

Abstract
Vascular malformations are part of overgrowth syndromes characterized by somatic mosaic mutations or rarely by germline mutations. Due to their similarities and diversity, clinicopathological classification can be challenging. A comprehensive targeted Next Generation Sequencing screen using Unique Molecular Identifiers with a technical sensitivity of 1% mutant alleles was performed for frequently mutated positions in ≥21 genes on 319 formalin‐fixed paraffin‐embedded samples. In 132 out of 319 cases pathogenic mosaic mutations were detected affecting genes previously linked to vascular malformations e.g. PIK3CA (n=80), TEK (TIE2) (n=11), AKT1 (n=1), GNAQ (n=7), GNA11 (n=4), IDH1 (n=3), KRAS (n=9), and NRAS (n=1). Six cases harbored a combination of mutations in PIK3CA and in GNA11 (n=2), GNAQ (n=2), or IDH1 (n=2). Aberrations in PTEN and RASA1 with a variant allele frequency approaching 50% suggestive of germline origin were identified in six out of 102 cases tested; four contained a potential second hit at a lower allele frequency. Ninety‐one of the total 142 pathogenic mutations were present at a variant allele frequency <10% illustrating the importance of sensitive molecular analysis. Clinicopathological characteristics showed a broad spectrum and overlap when correlated with molecular data. Sensitive screening of recurrently mutated genes in vascular malformations may help to confirm the diagnosis and reveals potential therapeutic options with a significant contribution of PIK3CA/mTOR and RAS‐MAPK pathway mutations. The co‐existence of two activating pathogenic mutations in parallel pathways illustrates potential treatment challenges and underlines the importance of multigene testing. Detected germline mutations have major clinical impact.
Keywords: molecular genetics, mosaic mutations, overgrowth syndromes, vascular malformations
1. INTRODUCTION
Somatic overgrowth syndromes encompass entities with predominant vascular malformations on the one hand and conditions with more pronounced overgrowth of tissue with only a limited vascular component on the other hand.1, 2, 3, 4 This spectrum of lesions is characterized by pathogenic somatic mosaic mutations resulting in a variable clinicopathological presentation.5 Most of these mutations are activating in genes related to cell growth, proliferation, survival, and cell cycle progression.6 In comparison with rare familial vascular malformation syndromes that occur with typical patterns of inheritance,7 sporadic cases are more common.8
Recurrently mutated genes were identified in several vascular anomalies including overgrowth/vascular malformation syndromes like PIK3CA‐related overgrowth syndrome (PROS),2, 8, 9, 10, 11 Proteus syndrome (activating mutations in AKT1),12 Sturge Weber syndrome (activating mutations in GNAQ)13 and Maffucci syndrome (transforming mutations in IDH1 and IDH2).14
Other somatically mutated genes comprise TEK/TIE2 (activating mutations, mainly described in venous malformations),8 KRAS, NRAS, and HRAS (activating mutations, mainly described in arteriovenous malformations),15, 16 GLMN (disrupting mutations in glomuvenous malformations)17 and GNA11 and GNA14 (activating mutations in vascular lesions described as vascular tumors including congenital tufted angiomas and kaposiform hemangioendotheliomas).18
Germline RASA1‐mutations have been described in patients with Parkes Weber Syndrome and Capillary Malformation‐ArterioVenous Malformations (CM‐AVM).19, 20 The PTEN Hamartoma Tumor Syndrome includes Bannayan‐Riley‐Ruvalcaba syndrome (BRRS) and Cowden syndrome (CS) harboring disrupting (germline) mutations in PTEN. 21, 22 Hereditary hemorrhagic telangiectasia bears disrupting germline mutations in ENG and ACVRL1.19 Other known syndromes and disorders that may include vascular anomalies are Beckwith‐Wiedemann syndrome, Noonan syndrome, Turner's syndrome, Klinefelters syndrome, and Neurofibromatosis.1
To gain better insight into the biology of vascular malformations and overgrowth syndromes, their mutational spectrum and potential therapeutic options, we performed Next Generation Sequencing (NGS) analysis with a sensitivity of 1% variant allele frequency to allow detection of low abundant mosaic mutations in formalin‐fixed paraffin‐embedded (FFPE) tissue from 319 clinical cases.
2. MATERIALS AND METHODS
The 319 included cases are combined from a prospective and a retrospective cohort. The prospective samples (n = 217) were from routine diagnostics. The retrospective samples, (n = 102) retrieved from our pathology files, were all histologically reviewed and if possible, clinically and radiologically confirmed as vascular malformation according to the INTERNATIONAL SOCIETY FOR THE STUDY OF VASCULAR ANOMALIES (ISSVA) classification. In a few cases a somatic overgrowth syndrome (SOGS) without a prominent vascular component was diagnosed. The study was conducted in accordance with the Code of Conduct of the Federation of Medical Scientific Societies in the Netherlands after consent of the internal ethical review board (2016‐2310).
All samples were fixed in 4% buffered formalin, routinely processed and embedded in paraffin. Four micrometer thick sections were stained with hematoxylin and eosin.
DNA isolation and library preparation were performed as previously described.23, 24 Briefly, gDNA was isolated from FFPE tissue sections (generally 3 × 20 μm) using 5% Chelex‐100 and 400 μg proteinase K and DNA concentrations were measured using the Qubit Broad Range kit (Q32853; ThermoFisher). When available, 150 ng of gDNA representing an equivalent of ~25 000 cells was used per library preparation. In the remaining cases, the available amount of DNA was used with a minimum of 5 ng representing gDNA from approximately 800 cells.
Libraries for sequencing on the NextSeq 500 (Illumina) were generated using Single Molecule Molecular Inversion Probes (smMIPs).24 This method uses unique molecule identifiers (UMI's) to allow consensus‐based error correction and the deduction of the actual number of sequenced gDNA molecules. Variant calling required detection of ≥3 mutant gDNA molecules representing a minimum of 1% of the total unique coverage. The strand‐specific nature allows the distinction between deamination artifacts from genuine C:G > T:A mutations.24 This method therefore allows both sensitive detection of variants down to 1% variant allele frequency and specification of the sensitivity of sequencing on a case‐by‐case basis.
The prospective cohort was analyzed using a panel targeting the following “cancer” hotspots and surrounding sequences (the exact positions analyzed for variants are shown in Eijkelenboom et al24): AKT1 (NM_005163.2): codon 17; BRAF (NM_004333.4): codon 582‐615; CTNNB1 (NM_001904.3): codon 19‐48; EGFR (NM_005228.3): codon 465‐499, 688‐823, 849‐875; ERBB2 (NM_004448.3): codon 770‐785; GNA11 (NM_002067.4): codon 183 and 209; GNAQ (NM_002072.3): codon 183 and 209; GNAS (NM_000516.4): codon 201 and 227; H3F3A (NM_002107.4): codon 28 and 35; H3F3B (NM_005324.4): codon 37; HRAS (NM_005343.2): codon 12, 13, 59 and 61; IDH1 (NM_005896.3): codon 132; IDH2 (NM_002168.3): codon 140 and 172; JAK2 (NM_004972.3): codon 617; KIT (NM_000222.2): codon 412‐513, 550‐591, 628‐713, 799‐828; KRAS (NM_004985.4): codon 12, 13, 59, 61, 117 and 146; MPL (NM_005373.2): codon 515; MYD88 (NM_002468.4): codon 265; NRAS (NM_002524.4): codon 12, 13, 59, 61, 117 and 146; PIK3CA (NM_006218.2): codon 520‐554, 1020‐1069; PDGFRA (NM_006206.4): codon 552‐596, 632‐667, 814‐848.24
For the retrospective cohort our panel was supplemented with GNA14 (NM_004297.3) codon 205, 206; PIK3CA (NM_006218.2): codon 420 and TEK (NM_000459.4) codon 897, 915‐920, 925, 1100; exons 3 and 13 of GLMN (NM_053274); PTEN (NM_000314), RASA1 (NM_002890), ACVRL1 (NM_000020) and ENG (NM_001114753) (smMIP sequences of these genes are shown in Supporting Information Table S1.
We excluded cases without identified (likely) pathogenic mutations if less than 125 individual genomic DNA (gDNA) molecules were analyzed at the frequently mutated positions. For NGS analyses above this threshold, the presence of mutations with an allele frequency > 5% could be excluded with 95% confidence.24 A total of 286 cases (89.6%) fulfilled these requirements for reliable analysis.
No informed consent for the possibility of detecting germline mutations (PTEN, RASA, and TEK) was obtained for the retrospective cohort. Data could therefore only be analyzed anonymously at the group level. Based on very low variant allele frequencies in our TEK positive cases (all 12% or lower [see Table]) none of the identified variants were likely germline mutations. As a consequence, we repeated the analyses for GNA11, GNAQ, KRAS, and TEK mutated cases to correlate phenotype and genotype. To increase the size of the anonymous group we also excluded the 27 PIK3CA‐mutated cases because clinical (and histopathological) characteristics are well‐studied.25, 26
Genetic data from our entire prospective cohort were correlated with clinical and histopathological features.
3. RESULTS
3.1. Molecular results
The identified pathogenic and likely pathogenic variants are listed in Table 1 and Supporting Information Table S2 (all PIK3CA mutated cases).
Table 1.
Clinical information and all (likely) pathogenic mutations and mutant allele frequencies identified in the prospective and retrospective cohorts are listed (with the exception of the 80 PIK3CA mutations, see Supporting Information Table S1)
| Gene | Identified variant(s) | Variant allele frequency (%) | Age/sex | Site | Clinical features | Histology |
|---|---|---|---|---|---|---|
| Prospective panel | ||||||
| GNA11 | c.627G > C (p.Gln209His) | 9 | 64/m | Hand | Unknown | Veno‐capillary malformation |
| GNA11 | c.547C > T (p.Arg183Cys) | 5 | 61/m | Upper eyelid | Congenital port wine stain; progressive hypertrophy | Veno‐capillary malformation |
| GNA11 | c.626A > T (p.Gln209Leu) | 3 | 72/m | Acetabulum | Unknown | Veno‐capillary malformation |
| GNAQ | c.548G > A (p.Arg183Gln) | 8 | 67/f | Skin neck | Residue after venous malformation | Veno‐capillary malformation |
| GNAQ | c.627A > C (p.Gln209His) | 4 | 57/f | Arm | Atypical lesion; spectrum non‐involuted congenital hemangioma (NICH)/ venous malformation | Veno‐capillary malformation |
| GNAQ | c.548G > A (p.Arg183Gln) | 8 | 81/f | Lower lip | Port wine stain in Sturge Weber Syndrome | Combined vascular malformation |
| GNAQ | c.627A > C (p.Gln209His) | 18 | 64/f | Epidural | Epidural mass | Veno‐capillary malformation |
| GNAQ | c.548G > A (p.Arg183Gln) | 8 | 43/f | Upper lip | Port wine stain with firm hypertrophy; KTS spectrum | Venous malformation |
| GNAQ | c.626A > T (p.Gln209Leu) | 21 | 15/m | Skin clavicle | Non congenital vascular high flow lesion | Veno‐capillary malformation |
| AKT‐1 | c.49G > A (p.Glu17Lys) | 8 | 5/m | Saphenous vein | Congenital port wine stain of one leg with firm overgrowth; mild phenotype of Proteus. | Venous malformation |
| KRAS | c.35_38delinsCTCA (p.Gly12_Gly13delinsAlaHis) | 32 | 56/f | Flank | Vascularized tumor | Veno‐capillary malformation |
| KRAS | c.64C > A (p.Gln22Lys) | 22 | 32/m | Cheek | KTS‐spectrum with lymphedema and port‐wine stain leg and multiple vascular lesions left side face | Veno‐capillary malformation |
| KRAS | c.35G > C (p.Gly12Ala) | 18 | 32/m | Cheek | Lipoma with vascular lesion | Venous malformation |
| KRAS | c.436G > A (p.Ala146Thr) | 17 | 45/f | Arm | KTS spectrum with venolymphatic lesion and port wine stain | Venous malformation |
| KRAS | c.37G > T (p.Gly13Cys) | 2 | 8/f | Lower jaw | Unknown | Veno‐capillary malformation |
| KRAS | c.35G > A (p.Gly12Asp) | 9 | 70/f | Subcutis buttock | Unknown | Veno‐capillary malformation |
| NRAS | c.182A > G (p.Gln61Arg) | 14 | 12/m | Thoracal skin | Multiple eruptive pyogenic granuloma | Capillary malformation |
| IDH1 | c.394C > T (p.Arg132Cys) | 2 | 12/f | Upper arm muscle | Venous malformation or non‐involuted congenital hemangioma | Veno‐capillary malformation |
| IDH1 | c.394C > T (p.Arg132Cys) | 14 | 38/f | Foot | Unknown | SCH |
| IDH1 | c.394C > T (p.Arg132Cys) | 10 | 15/m | Soft tissue foot | Unknown | SCH |
| PIK3CA; GNAQ | c.3140A > G (p.His1047Arg); c.627A > C (p.Gln209His) | 26; 26 | 53/m | Skull/dura | Vascular proliferation with Masson‐like features | Veno‐capillary malformation |
| PIK3CA; GNA11 | c.3140A > G (p.His1047Arg); c.627G > C (p.Gln209His) | 17; 17 | 57/m | Skin pericumbilical | Unknown | Veno‐capillary malformation |
| PIK3CA; GNA11 | c.1633G > A (p.Glu545Lys); c.627G > T (p.Gln209His) | 7; 6 | 45/m | Skin nose | Venous malformation | Veno‐capillary malformation |
| PIK3CA; IDH1 | c.3140A > G (p.His1047Arg); c.394C > T (p.Arg132Cys) | 12; 9 | 5/f | Scapula | Venolymphatic malformation, atypical by ultrasound | SCH |
| Retrospective panel | ||||||
| TEK | c.2690A > G (p.Tyr897Cys) | 3 | 11/f | Jaw | Venous malformation | Venous malformation |
| TEK | c.2740C > T (p.Leu914Phe) | 5 | 20/f | Labia minora | Venous malformation | Venous malformation |
| TEK | c.2740C > T (p.Leu914Phe) | 11 | 15/m | Soft tissue leg | Venous malformation | Veno‐capillary malformation |
| TEK | c.2740C > T (p.Leu914Phe) | 9 | 67/f | Upper lip | Venous lake | Combined malformation (extensive lymphatic component) |
| TEK | c.2690A > G (p.Tyr897Cys) | 4 | 80/f | Subcutis | Unknown | Combined malformation (extensive lymphatic component) |
| TEK | c.2690A > G (p.Tyr897Cys) | 4 | 9/f | Groin | Venous malformation, also anemia. d.d. Blue rubber bleb nevus. | Combined malformation (extensive lymphatic component) |
| TEK | c.2740C > T (p.Leu914Phe) | 8 | 33/m | Lower arm | Venous malformation | Venous malformation |
| TEK | c.2740C > T (p.Leu914Phe) | 5 | 17/f | Leg | Unknown | Veno‐capillary malformation |
| TEK | c.2740C > T (p.Leu914Phe) | 5 | 10/m | Lip | Venous malformation | Veno‐capillary malformation |
| TEK | c.2740C > T (p.Leu914Phe) | 9 | 15/m | Soft tissue arm | Unknown | Veno‐capillary malformation |
| TEK | c.2689 T > C (p.Tyr897His) | 12 | 2/m | Arm | Localized atypical tumor by ultra sound. d.d. vascular malformation | Veno‐capillary malformation |
| GNAQ | c.548G > A (p.Arg183Gln) | 7 | 56/f | Face | Port wine stain with hypertrophy | Venous malformation |
| KRAS | c.34G > T (p.Gly12Cys) | 6 | 76/m | Lower leg | Arteriovenous malformation (AVM) with ulceration | AVM |
| KRAS | c.34G > T (p.Gly12Cys) | 6 | 68/f | Finger | AVM | AVM |
| KRAS | c.35G > T (p.Gly12Val) | 3 | 17/f | Brain | Unknown | Venous malformation |
| GNA11 | c.626A > T (p.Gln209Leu) | 9 | 45/m | Skin ear canal | Unknown | Veno‐capillary malformation |
| PIK3CA; GNAQ | c.1624G > A (p.Gln542Lys); c.627A > C (p.Gln209His) | 15; 13 | 27/m | Skin | High flow lesion, d.d. AVM | Veno‐capillary malformation |
| PIK3CA; IDH1 | c.1624G > A (p.Gln542Lys); c.394C > T (p.Arg132Cys) | 24; 20 | 21/m | Subcutis leg | Atypical vascular lesion | SCH |
Abbreviations: KTS, Klippel Trenaunay syndrome; AVM, arteriovenous malformation; SCH, spindel cell hemangioma.
3.1.1. The spectrum of gain of function (hotspot mutations)
Prospective cohort
Nineteen out of 217 prospective cases obtained insufficient coverage. Of the remaining 198 cases, 77 had mutations: 53 PIK3CA, 6 GNAQ, 6 KRAS, 3 IDH1, 3 GNA11, 1 NRAS, and 1 AKT1 pathogenic mutations. Four cases harbored two mutations: PIK3CA and GNA11 (n = 2), PIK3CA and GNAQ, and PIK3CA (n = 1) and IDH1 (n = 1).
Retrospective cohort
In the retrospective cohort (n = 102), 14 samples were excluded due to insufficient coverage. Among the remaining 88 cases, 45 cases had mutations: 27 PIK3CA, 11 TEK (one with two mutations in cis, TEK: c.[2690A > G; c.2752A > G], p.([Tyr897Cys; Arg918Cys]), three KRAS, one GNAQ and one GNA11 hotspot mutation. There was one case with double‐mutations in PIK3CA and GNAQ and one affecting PIK3CA and IDH1. Mutations and histological types of both cohorts are shown in Figure 1.
Figure 1.
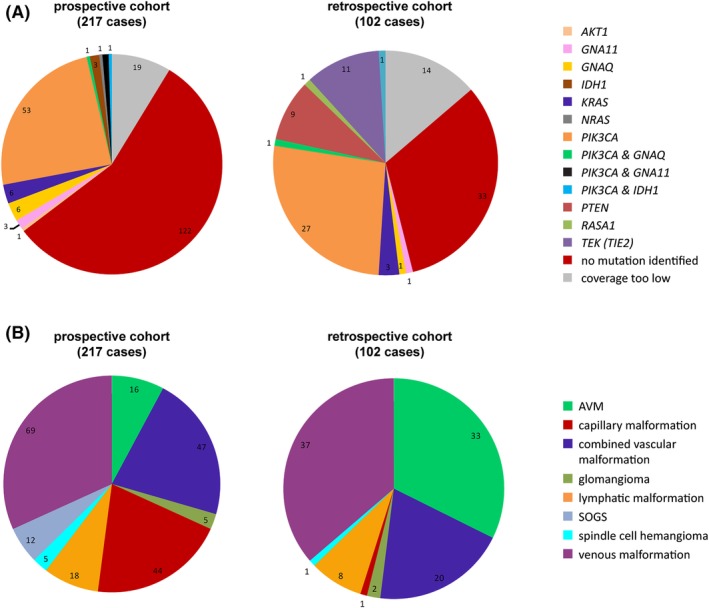
Overview of prospective and retrospective vascular malformation cohort. NGS‐based sequence analyses of a total of 319 cases of vascular malformations was performed in a routine diagnostic (prospective) and retrospective cohort consisting of histologically confirmed vascular anomalies. Molecular results grouped per mutated gene (top) and the clinicopathological entities in both cohorts (bottom). Abbreviations: AVM, arteriovenous malformation; SOGS, somatic overgrowth syndrome
Overall, we discovered gain of function (hotspot) mutations in a total of 122 cases (43%), with allele frequencies ranging from 2% to 24%. The mutant allele frequencies of 72% of cases (n = 88) were below the presumed detection limit of 10% for Sanger and first generation NGS approaches (Figure 2). Six of all mutated cases (5%) harbored a combination of known pathogenic mutations in PIK3CA with mutations in GNA11, GNAQ, or IDH1 with comparable mutant allele frequencies (Figure 3).
Figure 2.
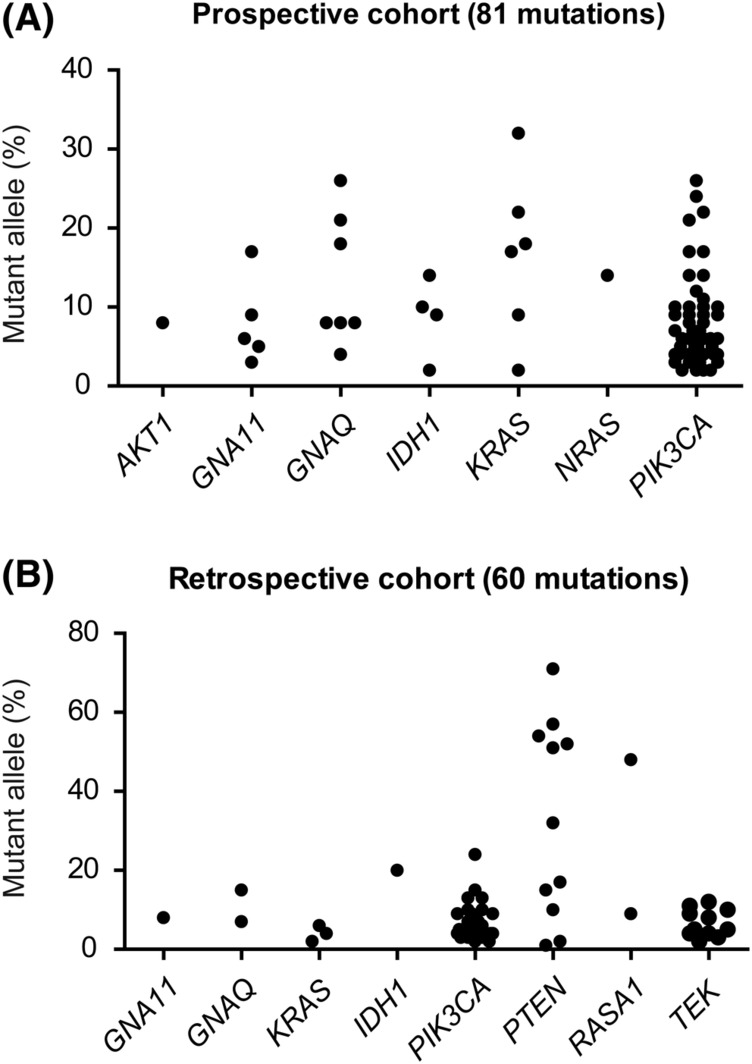
Mutant allele frequencies of identified mutations. The mutant allele frequencies of all (likely) pathogenic mutations in both, the prospective (top) and retrospective (bottom) cohort, grouped per gene
Figure 3.
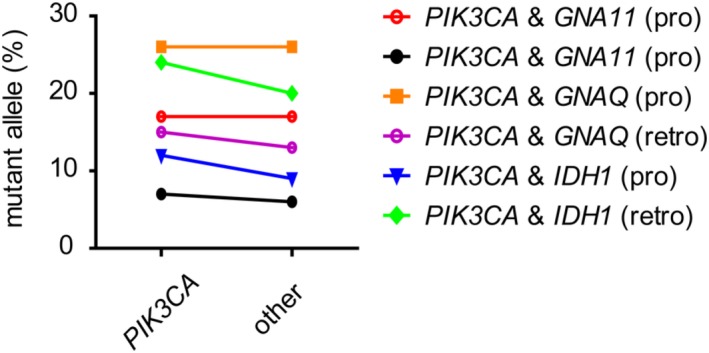
Mutant allele frequencies in cases with two pathogenic hotspot mutations. In 6 cases, a combination of one PIK3CA with an additional pathogenic activating (GNA11 and GNAQ) or transforming (IDH1) hotspot mutation was identified. The mutant allele frequencies of both mutations are depicted and connected per case
3.1.2. Loss of function mutations
In addition to the mutations investigated earlier, the retrospective cohort (n = 88/102) was analyzed for loss of function mutations in PTEN, RASA1, ACVRL1, ENG, and relevant exons of GLMN. We identified pathogenic loss of function mutations in PTEN (9 cases) and RASA1 (1 case) (Figure 4), mutually exclusive with the earlier described hotspot mutations. Six cases had mutant allele frequencies >40%. Because the tissue selection for DNA isolation in these cases was identical to the other cases in the cohort and mutant allele frequencies >40% were not identified for any of the other genes, this was interpreted as being germline variants in one RASA1 positive case and at least 4 out of 9 PTEN positive cases. In 3 of these 6 cases with a variant allele frequency >40% a potential second hit was observed at a lower allele frequency (≤10%), indicative of a combination of a germline pathogenic mutation with a mosaic second hit. In the fifth possible PTEN germline case, the variant allele frequency of 71% suggested loss of the wild type allele and thus complete loss of a functional PTEN copy in a subset of cells. We did not find any pathogenic inactivating variants in GLMN, ENG, and ACVRL1, making the presence of mutations less likely, although horizontal and vertical coverage may have been insufficient to exclude low mosaic mutations (unique coverage of both activating and inactivating genes is shown in Supporting Information Tables S3 and S4). Also unique coverage of RASA1 was insufficient to exclude the presence of mutations in other cases.
Figure 4.
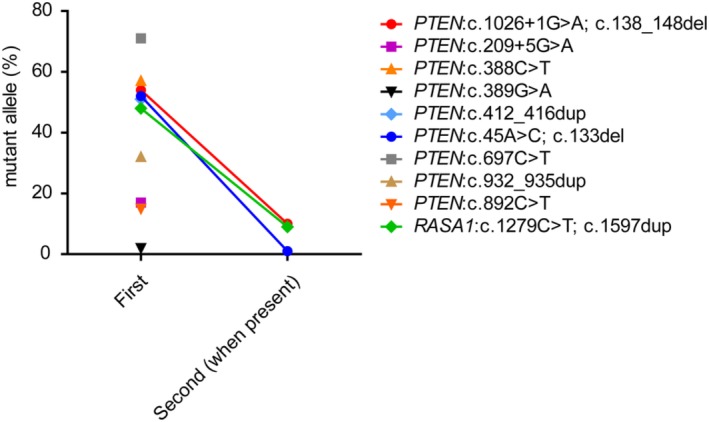
Mutant allele frequencies in cases with disrupting mutations. In 10 cases of the retrospective cohort, disrupting mutations were identified in PTEN (9 cases) and RASA1 (1 case). In a subset of cases, an additional mutation was found. The mutant allele frequencies are depicted per case, with connected data points in case two mutations were identified
3.2. Clinical data
The PIK3CA mutated lesions were mainly low flow (lymphatic and/or venous) malformations, but comprised heterogeneous clinical aspects as reported in the literature.2, 25, 26 Three patients had clinical characteristics of Klippel Trenaunay syndrome (Table 1).
The clinical data of the 42 cases harboring hotspot mutations other than PIK3CA are shown in Table 1. Briefly, 21 patients were female and 21 were male aged between 2 and 81 years (median 35 years and mean 38 years). Patients had diverse clinical presentations with lesions arising at various anatomical sites with most of them localized in the subcutis/skin (n = 30). Other lesions were located in soft tissue (n = 6), bone (n = 3), (epi)dural (n = 2), and intracerebral (n = 1). Two patients had a vascular malformation associated with other anomalies in the context of Proteus (n = 1), and Sturge Weber syndromes (n = 1). The case with a double mutation in TEK clinically fitted well with a Blue Rubber Bleb Nevus (BRBN) syndrome.
Of the cases with double mutations, the first case with a PIK3CA and GNAQ mutation was an intraosseous/dural vascular lesion and the second case was a small relatively well‐circumscribed high flow skin lesion. One of the two cases with both a PIK3CA and a GNA11 mutation concerned a low flow lesion in the nose with bleeding. The clinical information of the other case was unknown. Finally, the two cases with both a PIK3CA and an IDH1 mutation were both clinically atypical vascular skin lesions localized on the site of the scapula and the leg, respectively. Overall, these 6 cases had no special clinical signs indicating their double mutations.
Reasons for obtaining tissue were also diverse. In 13 patients a diagnostic biopsy was taken for genetic analysis. In 17 cases tissue was surgically removed for various reasons: for correction (n = 7), to reduce pain (n = 3), and because of progression (n = 5) involving one case with paraplegia due to an epidural mass and twice an amputation of a lower leg and a finger, respectively. Other reasons were treatment of varicosity, phlebitis, and a debulking resection because of a massive lesion in the upper lip. Only sparse clinical information was available for 12 patients.
3.3. Correlation of histology and molecular findings
The PIK3CA mutated cases depicted variable histology ranging from simple to combined capillary, lymphatic, and venous malformations and AVMs. All 11 cases with activating mutations in TEK were noncircumscribed mostly combined venous, capillary, and/or lymphatic lesions without an arterial component. Eight of the eleven cases with RAS (9 KRAS, 1 NRAS, and 1 RASA1) mutations were well‐circumscribed capillary and/or venous/cavernous malformations (Figure 5). The remaining 2 cases were noncircumscribed AVMs (Figure 6). Two of three cases with an IDH1 mutation, were typical spindle cell hemangiomas (SCH) and the third sample showed an intramuscular veno‐capillary malformation. All 4 GNA11 mutated cases had a veno‐capillary morphology. Of the 7 cases with a GNAQ mutation, 4 were veno‐capillary malformations, 2 VMs, and 1 had combined features including an arterial component. The AKT1 mutated case consisted of veins only. The 2 cases harboring an IDH1 and PIK3CA mutation were SCHs. The other 4 cases with a PIK3CA mutation combined with either a GNA11 (n = 2) or GNAQ (n = 2) mutation were all veno‐capillary malformations. Histology of the RASA1 and PTEN mutated cases could not be determined due to anonymization.
Figure 5.
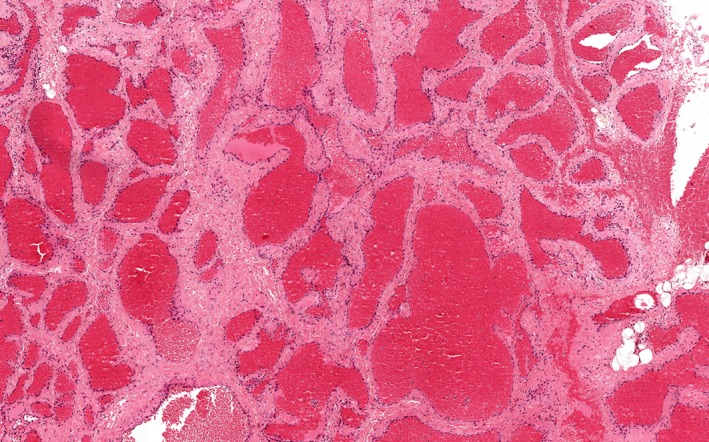
Venous/cavernous malformation. HE, ×100 [Color figure can be viewed at wileyonlinelibrary.com]
Figure 6.
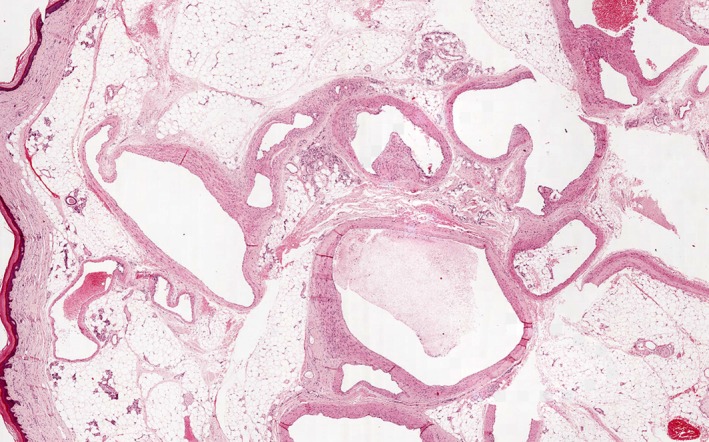
Arteriovenous malformation showing variation in wall thickness. HE, ×20 [Color figure can be viewed at wileyonlinelibrary.com]
In the prospective cohort (n = 198/217), mutations and the percentages of mutated cells within the histologically determined groups are shown in Supporting Information Table S5.
The spectrum of mutations per group was, based on clinical and histological features, relatively heterogeneous. The capillary malformations, for example, showed 3 GNAQ and 3 KRAS mutations, 1 NRAS and 1 PIK3CA mutation, and also a combined PIK3CA with GNA11 mutation. In AVMs (4 out of 17) and LMs (8 out of 14), without exception however, PIK3CA mutations were identified.
4. DISCUSSION
Vascular malformations belonging to the spectrum of overgrowth syndromes are, in most cases, genetically characterized by somatic mosaic pathogenic hotspot mutations or, more rarely, by germline mutations.1 We analyzed frequently mutated positions in more than 20 genes in clinically/radiologically and/or histopathologically diagnosed vascular malformations/overgrowth syndromes to gain more insight into their molecular biology linked to clinicopathological characteristics.
4.1. Technical challenges for molecular diagnostics
The range of identified mutant allele frequencies shows that it is highly valuable to analyze vascular malformations with NGS, especially with the inclusion of UMIs allowing an analytical sensitivity of 1% variant allele frequency in sequence analyses of FFPE tissue. At least 65% of mutations would have been missed using Sanger sequencing, based on a detection level of at least 10% mutant alleles. More sensitive techniques such as digital droplet PCR would potentially result in identification of more pathogenic mutations.27 However, the variety of mutations in combination with limited amounts of affected cells and available tissue require an approach in which a larger number of positions can be tested simultaneously. The identified low frequency pathogenic variants are in line with the mosaicism.
The detection of mosaic mutations with variant allele frequencies <10% in gDNA from FFPE tissue depends on quality and quantity of isolated gDNA and the presence of cytosine deamination resulting in C:G > T:A mutations. False‐negative calls can result from the overestimation of the actual number of analyzed DNA molecules and potential false‐positive calls can arise from deamination artifacts.
4.2. Vascular malformations span a mutational spectrum affecting multiple signaling pathways
4.2.1. PI3K/AKT/mTOR pathway
Activating mutations in PIK3CA, AKT1, and TEK, and alternatively disrupting mutations in PTEN, activate the mTOR pathway, regulating cell growth, proliferation, survival and cell cycle progression.2, 6, 28, 29 PIK3CA mutations are the most ubiquitous ones in vascular malformations. This was reflected in our cohorts with ~30% (n = 86) of the cases being affected. They show a variable phenotype both histologically (LMs, VMs, and AVMs) and clinically (low flow malformations), which is in accordance with the literature.26, 30 AKT1 was mutated in only 1 case (~0.3%) of the retrospective cohort, showing a p.Glu17Lys mutation, the same mutation that has been most frequently described in the literature.12 Mutations in TEK/TIE2 with about 85% of cases harboring a p.[Leu914Phe] missense mutation results nearly exclusively in venous malformations.31, 32, 33 In our retrospective cohort, 11 cases (12.5%) were mutated. A c.2740C > T (p.(Leu914Phe)) mutation was found in 7 of them. Other mutations we identified were c.2690A > G (p.[Tyr897Cys]) (n = 3) and c.2689 T > C (p.[Tyr897His]) (n = 1) indicating that only screening of p.(Leu914Phe) is insufficient. Clinically, all cases had a venous phenotype including 1 case with a BRBN syndrome. Both TEK mutations found in the BRBN case were present in cis. As all mutated reads contained both mutations, the same variant allele frequency was suggested.34 In contrast to the literature,31, 32, 33 our TEK mutated cases were histologically more heterogeneous showing also combined lesions with a venous, capillary, and/or a lympathic component.
The retrospective cohort contained 9 PTEN mutated cases (~10%) (see later), adding up to a total of 107 cases containing mutations indicative of mTOR activation in the PI3K/AKT/mTOR pathway.
4.2.2. RAS–RAF–MEK–ERK signaling pathway
RAS signaling can be stimulated by both activating (somatic) mutations in KRAS, HRAS, NRAS as well as inactivating (germline) mutations in RASA1, which is a RAS‐GAP (Guanine Activating Protein) and negative regulator of the RAS–RAF–MEK–ERK signaling pathway. Subsequently, diverse cellular signaling networks including the mTOR and ERK pathways are activated.35 Histologically, in contrast with the heterogeneity seen for the other mutations, 8 out of 11 RAS mutated vascular malformations were well‐circumscribed (capillary and/or venous), while lesions with other mutations were almost all noncircumscribed.
GNAQ is a gene encoding guanine nucleotide binding protein G(q)alpha, a subunit within a complex that hydrolyses the intracellular messenger GTP to GDP.36 GNA11 and GNA14 are analogous binding proteins with nearly the same protein sequence as GNAQ.18, 36 When mutated, these genes induce changes in cellular morphology and cell growth by upregulation of mainly the RAS‐RAF‐MEK‐ERK pathway.18
Combining the two cohorts, we detected 9 KRAS, 1 NRAS, 7 GNAQ, and 4 GNA11 hotspot mutations; GNAQ and GNA11 were in 2 cases each combined with a PIK3CA mutation. These, and one inactivating mutation in RASA1, suggest a more significant role (about 8% of all the included cases) of the RAS‐RAF‐MEK‐ERK pathway in vascular malformations than may be expected from the literature.15, 16
We did not detect GNA14 mutations. This is in accordance with the literature reporting that such mutations are restricted to tufted hemangioma and kaposiform hemangioendothelioma, which were not included in our cohorts.22
4.3. IDH 1/2 mutated vascular lesions are malformations rather than tumors
Five cases showed an IDH1 mutation, in two cases in combination with a PIK3CA mutation (one of these cases was included in a previously published cohort37). Isocitrate dehydrogenases (IDH) are metabolic enzymes which, as a result of a somatic mutation (in both IDH1 and IDH2), mostly inhibit the catalyzation of isocitrate to α‐ketoglutarate (α‐KG). This results in aberrant DNA methylation and histone modification leading to stabilization of hypoxia inducible factor‐1α subsequently inducing angiogenesis.38 Four of our cases were spindle cell hemangiomas (SCH), in line with previous findings.37 Interestingly, the fifth lesion was a small biopsy of an intramuscular veno‐capillary malformation without a spindle cell component. Vice versa, we also found a SCH with only a PIK3CA mutation (17% variant allele frequency) confirming that SCHs belong to the heterogeneous spectrum of vascular malformations rather than to vascular tumors.4, 37
4.4. Combined mutations
In 6 cases we identified a combination of an activating mutation in PIK3CA with an additional pathogenic mutation in GNAQ, GNA11, or IDH1. As these cases had a heterogeneous/combined phenotype without specific clinical symptoms, no conclusion can be drawn regarding the significance of the dominant mutation.
To our knowledge, such a combination of mutations in two individual genes in vascular malformations/overgrowth syndromes has not been reported before. It is likely that this can be explained by the presence of two related events in the same cells, because in all cases the mutant allele frequencies of both mutations were very similar. However, an in situ approach would be required to formally prove this concept. From a signaling perspective, the pathogenic mutations in GNAQ, GNA11, and IDH1 are expected to provide additional growth advantage in parallel with PIK3CA‐driven mTOR activation. One could argue that different pathways in parallel are involved in aberrant development of vascular structures and other affected tissue. This may be relevant information if application of targeted treatments is considered.
4.5. Potential germline aberrations in cases of vascular anomalies
Pathogenic germline mutations in PTEN lead to PTEN hamartoma tumor syndrome (PHTS) with susceptibility to multiple tumors as well as vascular malformations. This process is initiated after loss or inactivation of the second (wild type) allele by somatic mutation resulting in increased levels of PIP3 and increased activation of AKT.29 We found 9 cases (approximately 10% of the cases) with a (likely) pathogenic PTEN mutation, of which at least 4 were a germline mutation, in the retrospective cohort. This unexpectedly high percentage of PTEN mutations suggests a much more important role in the etiology of sporadic vascular malformations than anticipated, as PTEN‐associated anomalies are only described in the context of PHTS.29, 39
We also identified 1 case with a potential germline RASA1 mutation. Because data from our retrospective cohort were analyzed anonymously, we could not associate the PTEN and RASA1 positive lesions to individual histological and clinical characteristics. However, a vascular malformation may well represent the first clinical presentation of an eventually more extensive syndromal phenotype, which is especially relevant in the context of de novo germline mutations or atypical phenotypic presentation. Our findings urge for clinical awareness of this potentially unrecognized patient group of vascular malformations at risk for germline‐associated tumors.
Complete analyses of all coding and splice site sequences is required to identify all disrupting mutations, in contrast to the aforementioned “hotspot” analyses; this is challenging when using relatively poor quality gDNA obtained from FFPE tissue. Due to limitations in horizontal and vertical coverage for ACVRL1, ENG, and GLMN, but also PTEN and RASA1, pathogenic disruptions in these genes may have been remained undetected.
4.6. Therapeutic opportunities for vascular malformations
There is increasing evidence28, 40 that sirolimus, an mTOR inhibitor, can be used to treat a subset of the vascular malformations with a more favorable balance between effect and side effects than conventional treatments like surgery, embolization, or laser therapy.41 The precise mechanism behind this treatment is still unknown, but at least 107 of 286 cases in our series harbored mutations predicted to activate mTOR signaling. These mTOR activating cases are not limited to PIK3CA, so a more comprehensive analysis including additional genes involved in this pathway could increase the diagnostic and therapeutic yield (in our set‐up by ~25%). Similarly, MEK inhibitors seem to be a promising therapeutic agent for lesions harboring mutations involved in the RAS‐RAF‐MEK‐ERK pathway16; such mutations were found in 25 of our cases. The mapping of causative and druggable mutations by sequencing appears to be a big promise for patients with hitherto hardly treatable conditions in the vascular anomalies spectrum. Clinical trials with sirolimus with favorable initial results exemplify the potency of this new medical approach.42, 43
5. CONCLUSION
In summary, mutational analysis of vascular malformations is of high diagnostic value and an indispensable addition to regular diagnostics, because clinical, radiological, and pathological investigations often offer insufficient clarity. Furthermore, it can provide clues for an underlying hereditary condition due to a germline mutation. In addition, increased knowledge of pathogenic mutations present in these lesions can stratify patients for targeted therapeutic options.15, 40, 44 A comprehensive and sensitive analysis is required because of the spectrum of genes involved and the mosaicism with low levels of mutated cells and also combined mutations.
CONFLICT OF INTEREST
Authors have no disclosures/conflict of interest.
Supporting information
Table S1 smMIP sequences of the genes added to the more extensive panel
Table S2 Overview of molecular results of PIK3CA mutated cases. All PIK3CA mutations and mutant allele frequencies identified in the prospective and retrospective cohorts are listed
Table S3 Overview of coverage of genes with pathogenic gain‐of‐function (hotspot) mutations. Using the unique molecule identifiers (UMIs) the number of analyzed gDNA molecules is calculated. Here, we show the number of cases for which >250, >125 or > 63 gDNA molecules are sequenced for all indicated hotspots. Combined with our variant calling requirements, the chance of false negative is <5% for the specified mutant allele frequency per threshold (indicated between brackets)
Table S4 Overview of coverage of genes with possibly pathogenic inactivating variants. The number of analyzed gDNA molecules is determined for all genes in which disrupting mutations are expected. The percentage of cases in which >95% of targeted sequences is sequenced above the indicated thresholds is shown
Table S5 Overview of the mutations and their percentages within the histologically distinct groups in the prospective cohort (n = 198)
ACKNOWLEDGMENTS
We thank all technicians of the Molecular Diagnostic Group, especially Annemiek Kastner‐van Raaij and Sandra Hendriks‐Cornelissen and other members of the Genome Technology Unit, Laboratory of Tumor Genetics, Departments of Pathology and Genetics, especially Arjen Mensenkamp.
Ten Broek RW, Eijkelenboom A, van der Vleuten CJM, et al. Comprehensive molecular and clinicopathological analysis of vascular malformations: A study of 319 cases. Genes Chromosomes Cancer. 2019;58:541–550. 10.1002/gcc.22739
REFERENCES
- 1. Blei F. Overgrowth syndromes with vascular anomalies. Curr Probl Pediatr Adolesc Health Care. 2015;45(4):118‐131. [DOI] [PubMed] [Google Scholar]
- 2. Keppler‐Noreuil KM, Rios JJ, Parker VE, et al. PIK3CA‐related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. 2015;167A(2):287‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keppler‐Noreuil KM, Sapp JC, Lindhurst MJ, et al. Clinical delineation and natural history of the PIK3CA‐related overgrowth spectrum. Am J Med Genet A. 2014;164A(7):1713‐1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wassef M, Blei F, Adams D, et al. Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics. 2015;136(1):e203‐e214. [DOI] [PubMed] [Google Scholar]
- 5. Happle R. Mosaicism in human skin. Understanding the patterns and mechanisms. Arch Dermatol. 1993;129(11):1460‐1470. [PubMed] [Google Scholar]
- 6. Freed D, Stevens EL, Pevsner J. Somatic mosaicism in the human genome. Genes (Basel). 2014;5(4):1064‐1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uller W, Fishman SJ, Alomari AI. Overgrowth syndromes with complex vascular anomalies. Semin Pediatr Surg. 2014;23(4):208‐215. [DOI] [PubMed] [Google Scholar]
- 8. Boon LM, Ballieux F, Vikkula M. Pathogenesis of vascular anomalies. Clin Plast Surg. 2011;38(1):7‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uebelhoer M, Boon LM, Vikkula M. Vascular anomalies: from genetics toward models for therapeutic trials. Cold Spring Harb Perspect Med. 2012;2(8):a009688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vahidnezhad H, Youssefian L, Uitto J. Klippel‐Trenaunay syndrome belongs to the PIK3CA‐related overgrowth spectrum (PROS). Exp Dermatol. 2016;25(1):17‐19. [DOI] [PubMed] [Google Scholar]
- 11. Kang HC, Baek ST, Song S, Gleeson JG. Clinical and Genetic Aspects of the Segmental Overgrowth Spectrum Due to Somatic Mutations in PIK3CA. J Pediatr. 2015;167(5):957‐962. [DOI] [PubMed] [Google Scholar]
- 12. Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shirley MD, Tang H, Gallione CJ, et al. Sturge‐Weber syndrome and port‐wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971‐1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amary MF, Damato S, Halai D, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet. 2011;43(12):1262‐1265. [DOI] [PubMed] [Google Scholar]
- 15. Al‐Olabi L, Polubothu S, Dowsett K, et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J Clin Invest. 2018;128:5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nikolaev SI, Vetiska S, Bonilla X, et al. Somatic activating KRAS mutations in arteriovenous malformations of the brain. N Engl J Med. 2018;378(3):250‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brouillard P, Boon LM, Mulliken JB, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. 2002;70(4):866‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lim YH, Bacchiocchi A, Qiu J, et al. GNA14 somatic mutation causes congenital and sporadic vascular tumors by MAPK activation. Am J Hum Genet. 2016;99(2):443‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McDonald J, Wooderchak‐Donahue W, VanSant Webb C, Whitehead K, Stevenson DA, Bayrak‐Toydemir P. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet. 2015;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation‐arteriovenous malformation. Hum Mutat. 2013;34(12):1632‐1641. [DOI] [PubMed] [Google Scholar]
- 21. Nozaki T, Nosaka S, Miyazaki O, et al. Syndromes associated with vascular tumors and malformations: a pictorial review. Radiographics. 2013;33(1):175‐195. [DOI] [PubMed] [Google Scholar]
- 22. Tan WH, Baris HN, Burrows PE, et al. The spectrum of vascular anomalies in patients with PTEN mutations: implications for diagnosis and management. J Med Genet. 2007;44(9):594‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Leng WW, Gadellaa‐van Hooijdonk CG, Barendregt‐Smouter FA, et al. Targeted next generation sequencing as a reliable diagnostic assay for the detection of somatic mutations in tumours using minimal DNA amounts from formalin fixed paraffin embedded material. PloS one. 2016;11(2):e0149405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eijkelenboom A, Kamping EJ, Kastner‐van Raaij AW, et al. Reliable Next‐Generation sequencing of Formalin‐Fixed, Paraffin‐Embedded tissue using single molecule tags. J Mol Diagn. 2016;18(6):851‐863. [DOI] [PubMed] [Google Scholar]
- 25. Kurek KC, Luks VL, Ayturk UM, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90(6):1108‐1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luks VL, Kamitaki N, Vivero MP, et al. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. 2015;166(4):1048‐1054. e1041‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next‐generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet. 2018;19(5):269‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boscolo E, Limaye N, Huang L, et al. Rapamycin improves TIE2‐mutated venous malformation in murine model and human subjects. J Clin Invest. 2015;125(9):3491‐3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nathan N, Keppler‐Noreuil KM, Biesecker LG, Moss J, Darling TN. Mosaic disorders of the PI3K/PTEN/AKT/TSC/mTORC1 signaling pathway. Dermatol Clin. 2017;35(1):51‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Limaye N, Kangas J, Mendola A, et al. Somatic activating PIK3CA mutations cause venous malformation. Am J Hum Genet. 2015;97(6):914‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Du Z, Zheng J, Zhang Z, Wang Y. Review of the endothelial pathogenic mechanism of TIE2‐related venous malformation. J Vasc Surg Venous Lymphat Disord. 2017;5(5):740‐748. [DOI] [PubMed] [Google Scholar]
- 32. Soblet J, Limaye N, Uebelhoer M, Boon LM, Vikkula M. Variable Somatic TIE2 Mutations in Half of Sporadic Venous Malformations. Mol Syndromol. 2013;4(4):179‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Uebelhoer M, Natynki M, Kangas J, et al. Venous malformation‐causative TIE2 mutations mediate an AKT‐dependent decrease in PDGFB. Hum Mol Genet. 2013;22(17):3438‐3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Soblet J, Kangas J, Natynki M, et al. Blue rubber bleb nevus (BRBN) syndrome is caused by somatic tek (TIE2) mutations. J Invest Dermatol. 2017;137(1):207‐216. [DOI] [PubMed] [Google Scholar]
- 35. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170(1):17‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ayturk UM, Couto JA, Hann S, et al. Somatic activating mutations in GNAQ and GNA11 are associated with congenital hemangioma. Am J Hum Genet. 2016;98(4):789‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ten Broek RW, Bekers EM, de Leng WWJ, et al. Mutational analysis using Sanger and next generation sequencing in sporadic spindle cell hemangiomas: A study of 19 cases. Genes Chromosomes Cancer. 2017;56(12):855‐860. [DOI] [PubMed] [Google Scholar]
- 38. Schaap FG, French PJ, Bovee JV. Mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 in tumors. Adv Anat Pathol. 2013;20(1):32‐38. [DOI] [PubMed] [Google Scholar]
- 39. Mester J, Charis E. PTEN hamartoma tumor syndrome. Handb Clin Neurol. 2015;132:129‐137. [DOI] [PubMed] [Google Scholar]
- 40. Triana P, Dore M, Cerezo VN, et al. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg. 2017;27(1):86‐90. [DOI] [PubMed] [Google Scholar]
- 41. Hammill AM, Wentzel M, Gupta A, et al. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer. 2011;57(6):1018‐1024. [DOI] [PubMed] [Google Scholar]
- 42. Adams DM, Trenor CC 3rd, Hammill AM, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics. 2016;137(2):e20153257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hammer J, Seront E, Duez S, et al. Sirolimus is efficacious in treatment for extensive and/or complex slow‐flow vascular malformations: a monocentric prospective phase II study. Orphanet J Rare Dis. 2018;13(1):191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ranieri C, Di Tommaso S, Loconte DC, et al. In vitro efficacy of ARQ 092, an allosteric AKT inhibitor, on primary fibroblast cells derived from patients with PIK3CA‐related overgrowth spectrum (PROS). Neurogenetics. 2018;19:77‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 smMIP sequences of the genes added to the more extensive panel
Table S2 Overview of molecular results of PIK3CA mutated cases. All PIK3CA mutations and mutant allele frequencies identified in the prospective and retrospective cohorts are listed
Table S3 Overview of coverage of genes with pathogenic gain‐of‐function (hotspot) mutations. Using the unique molecule identifiers (UMIs) the number of analyzed gDNA molecules is calculated. Here, we show the number of cases for which >250, >125 or > 63 gDNA molecules are sequenced for all indicated hotspots. Combined with our variant calling requirements, the chance of false negative is <5% for the specified mutant allele frequency per threshold (indicated between brackets)
Table S4 Overview of coverage of genes with possibly pathogenic inactivating variants. The number of analyzed gDNA molecules is determined for all genes in which disrupting mutations are expected. The percentage of cases in which >95% of targeted sequences is sequenced above the indicated thresholds is shown
Table S5 Overview of the mutations and their percentages within the histologically distinct groups in the prospective cohort (n = 198)