

Abstract
There is a tremendous need for novel strategies aimed at directly assessing activities of histone modifiers to probe epigenetic determinants associated with disease progression. Here, we developed a high-throughput peptide microarray assay to identify altered histone lysine (de)acetylation activity in prostate cancer (PCa). This microarray-based activity assay revealed up-regulated histone acetyltransferase (HAT) activity against specific histone H3 sites in a castrate-resistant (CR) PCa cell line compared to its hormone-sensitive (HS) isogenic counterpart. NAD+-dependent deacetylation assays revealed down-regulated sirtuin activity in validated CR lines. Levels of acetyltransferases GCN5, PCAF, CBP, and p300 were unchanged between matched HS and CR cell lines. However, autoacetylation of p300 at K1499, a modification known to enhance HAT activity and a target of deacetylation by SIRT2, was highly elevated in CR cells, while SIRT2 protein level was reduced in CR cells. Interrogation of HS and matched CR xenograft lines reveals that H3K18 hyperacetylation, increased p300 activity, and decreased SIRT2 expression are associated with progression to CR in 8/12 (66%). Tissue microarray analysis revealed that hyperacetylation of H3K18 is a feature of CRPC. Inhibition of p300 results in lower H3K18ac levels and increased expression of androgen receptors. Thus, a novel histone array identifies altered enzyme activities during the progression to CRPC and may be utilized in a personalized medicine approach. Reduced SIRT2 expression and increased p300 activity lead to a concerted mechanism of hyperacetylation at specific histone lysine sites (H3K9, H3K14, and H3K18) in CRPC.
Graphical Abstract
Histone post-translational modifications (PTMs) exist in highly specific and combinatorial fashion, impinging upon other epigenetic marks and influencing specific transcriptional regulation.1–3 Dysregulated histone modifications alter intrinsic chromatin structure as well as the surface architecture, leading to erroneous recruitments of gene regulatory machineries at specific genomic loci.4 Therefore, altered cellular abundance and activity of histone modifiers plays a key role in cancer progression. Many PTMs are reversible and in dynamic interaction with antagonistic enzyme activities. A systematic interrogation of aberrant enzyme activities leading to distinctive histone PTM patterns would be crucial to understanding the molecular mechanism by which particular disease states are established. Such knowledge is essential for developing therapeutics to target these enzymes.
Recent advances in genomic and proteomic data mining tools together with chromatin immunoprecipitation (ChIP)-based profiling have been instrumental in unraveling aberrant expression of histone-modifying complexes in specific disease progression.5–8 However, post-transcriptional and -translational regulation and longevity of gene products poorly correlates with genome-wide transcript levels and their protein amounts.9 Furthermore, protein PTM, subcellular targeting, and the level of small molecule activators or inhibitors make it difficult to directly link protein levels with their biological activities.10 Hence, there is tremendous need for novel strategies aimed at directly assessing activities of histone modifiers as an alternative and complementary avenue for probing epigenetic determinants associated with disease progression.
Prostate cancer is the most commonly diagnosed disease in American men, resulting in 28 000 deaths annually.11 After an initial response to hormone ablation therapy, metastatic HS cancers relapse and progress to CRPC.12 Targeting the molecular determinants of this event would be pivotal for developing new prognostic and therapeutic strategies. Previous studies report that global levels of several histone modifications including H3K18ac and H3K4me2 are associated with PCa tumor recurrence, suggesting a role for histone modifications in cancer progression.13,14 Isolated histone-modifying enzymes have been found to have impaired activity in various cancers;15,16 however, a systematic evaluation of aberrant epigenetic enzyme activity in the progression to CRPC is lacking.
In this study, we examine dysregulation of global (genome-wide) lysine (de)acetylation activities during PCa progression by utilizing novel peptide microarray technology in cell extracts from relevant cancer models and human tissues. Previously, we had demonstrated the utility of peptide microarrays for identifying targets of chromatin “reader” proteins against combinatorial histone PTMs in a high-throughput protein binding assay.17 Herein, we prepared histone peptide libraries consisting of 932 unique peptides covering human histone H3, H4, H2A, and H2B sequences of different histone variants. This library includes combinatorial histone PTMs likely to be found in human chromatin,18 allowing interrogation of HAT and histone deacetylase (HDAC) activities that we applied to investigate the development of CRPC.
RESULTS AND DISCUSSION
Histone Peptide Microarray for HAT and Sirtuin Assays.
The primary goal was to establish a microarray enzyme assay to detect changes in the acetylation status of hundreds of immobilized histone peptides that are either synthetically acetylated or unmodified. The peptides were prepared as cellulose conjugates by the SPOT synthesis19 and printed on nitrocellulose-coated microscopic slides. Changes in acetylated lysine (Kac) states were quantified by utilizing a “pan-specific” anti-Kac antibody which is routinely used in mass spectrometry for enrichment of acetylated peptides.20 Adopted from immunodetection methods, antibody-bound peptides are identified using a red AlexaFluor647-tagged secondary antibody (Figure 1A). We incorporated a second green Cy3 tracer dye (532 nm) in all peptide stock solutions, effectively creating a dual-channel detection system for quality control (Figure 1B, Supporting Information (SI) Figure S1). The green signal facilitates peptide spot recognition for quantification and also serves to identify any printing errors introduced during the arraying of large peptide libraries (SI Figure S1).
Figure 1.
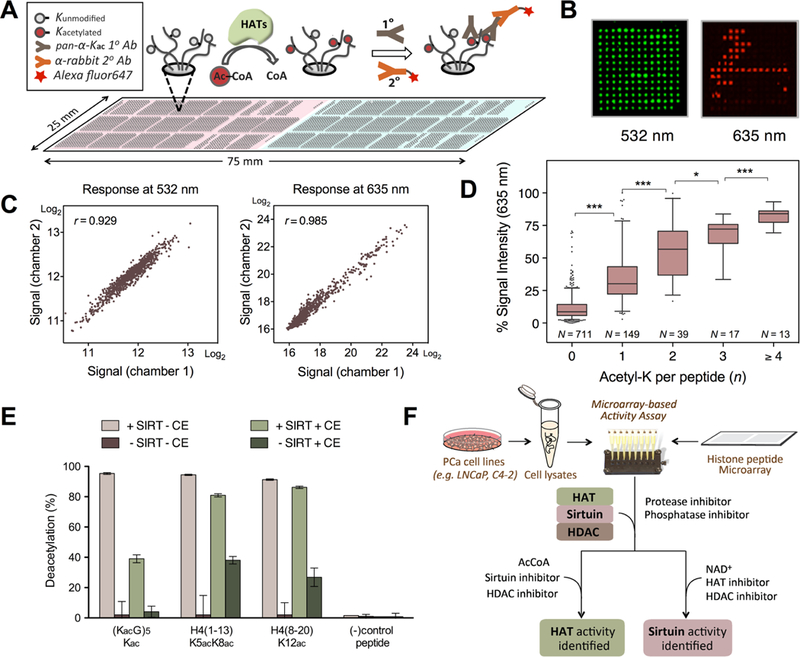
Histone (de)acetylation activity assay on a peptide microarray platform. (A) The assay scheme represents HAT-dependent lysine acetylation monitored by an immunoassay using the anti-pan-Kac antibody. A duplicate set (represented by pink and light turquoise) of 932-peptide libraries is each probed with a different cell lysate, separated by a duplex chamber. (B) Cy3-cellulose dye in peptide stock solution is used as a spot tracer and fluoresces at 532 nm (green), while acetylated lys (Kac) that binds with Alexa Fluor647-conjugated antibody fluoresces at 635 nm (red). Each peptide is printed in triplicate for statistical analysis. (C) A scatter plot of signals from peptides in two separately incubated subarrays at 532 nm and at 635 nm each respectively exhibits high correlation and intra-array concordance. (D) A box-and-whisker plot of % signal intensity at 635 nm across the peptide library at varying numbers of Kac (n). The whiskers represent 95% confidence intervals, while the outliers are shown as dots. The lower and upper lines of the boxes show the quartile range with a center cross line representing a median at n = 0 (median, M = 8.6, population, N = 711), 1 (M = 30.2, N = 149), 2 (M = 56.7, N = 39), 3 (M = 72.2, N = 17), and ≥4 (M = 84.0, N = 13). Statistical significance of signal intensity between each group is shown (*0.01 < P ≤ 0.05; **0.001 < P ≤ 0.01; ***P ≤ 0.001). (E) Deacetylation activity is measurable in a small pilot microarray. In the absence of recombinant sirtuin cocktail (Sirt1–3) and HEK293T cell extracts (CE), deacetylation activity is almost negligible (brown), while addition of sirtuins removed most of the acetyl mark (pink, pistachio). Addition of CE with the sirtuin cocktail led to some inhibition of deacetylation activity (pistachio), while CE in the absence of sirtuin supplementation revealed deacetylation up to 40% (dark olive). (F) Microarray-based assay scheme used in this study for identification of HAT and sirtuin activity.
We examined the reproducibility of signal responses to antibody detection between peptides in each subarray after treating them separately using a duplex assay chamber. The signal intensity of each peptide between subarrays exhibited high intersubarray concordance as represented by scatter plots with strong positive correlation (Figure 1C). Coefficients of variation (CV) as a function of signal intensity (F635) across three technical replicates of 997 peptides (including controls) were low (mean = 4.2%, median = 2.6%), and >98% of detectable peptides showed CV not greater than 20%. Together with the high correlation between subarrays, the low technical variability was a strong indication that this microarray assay platform can be used with high confidence. The dynamic range of anti-pan-Kac antibodies reflected a linear increase of signal intensity in response to the increased number of acetylated lysine residues per peptide (n), as represented in a box-and-whisker plot (Figure 1D). A low percentage of false positives (<5% of 711 peptides) and false negatives (<3% of 218) was observed. The difference in signal intensity among the same n group is in part caused by variable affinities of the anti-pan-Kac antibody toward each peptide’s distinctive epitope (with different sequences and PTMs flanking a Kac site) as well as by slight concentration differences in peptide stock solutions. This assay is, therefore, best utilized to determine changes in the HAT and HDAC activity levels between samples.
To investigate HAT-dependent acetylation on the peptide microarray platform, we utilized recombinantly expressed and purified HATs with unique substrate specificity. In this pilot assay, 16 peptides were probed in a multiplex assay chamber, which allows multiple assay conditions to be tested simultaneously on a single array (SI Figure S2). Acetylation was detected by phosphor imaging of the [14C]-acetyl moiety transferred from [14C]-acetyl-CoA to peptide lysine. In this radio-isotopic detection, NuA4 displayed HAT activity toward peptides corresponding to the N-terminal H4 sequence, while showing no detectable activity toward N-terminal H3 peptide or globular regions of H4. Similarly, fluorescence immunodetection of acetyl lysine using pan-Kac antibody revealed that lysine acetylation was HAT-dependent, consistent with the substrate specificity of both NuA4 and PCAF reported previously.21,22 The two independent detection methods yielded consistent results. Peptides containing synthetically installed Kac (positive controls) produced positive signals while peptides lacking lysine residue (negative controls) did not exhibit a measurable fluorescent signal.
To examine deacetylation activity in the microarray format, we performed pilot assays with NAD+-dependent class III HDACs, known as sirtuins.23 Here, we employed a small custom-built library for the sirtuin microarray assays that contained physiologically relevant acetyl-lysine peptide substrates, including diacetylated H4K5 and K8, H4K12acK20me, H3R2me2-sK4ac, and H3K4acT6ph. The library also included a peptide of acetyl-lysine and glycine repeats and a negative control peptide containing the same sequence with unmodified lysine groups. Using recombinantly expressed sirtuins 1–3 with the NAD+ cosubstrate, sirtuin-dependent deacetylation was prominent, while nonenzymatic deacetylation was negligible (Figure 1E). All acetylated peptides present in the library were enzymatically deacetylated; however deacetylation was more complete on the synthetic acetyl-lysine and glycine repeat sequence and on peptides from H4. A lower overall signal from the H3K4ac peptides indicates a sequence dependent differential binding of the pan-Kac antibody and not substrate selectivity of the sirtuin deacetylases.
To evaluate whether endogenous enzymatic activity can be measured in these peptide arrays, we repeated the experiments with whole-cell extracts from HEK293T cells. Inhibitors of phosphatase, protease, HDAC (I, II), and HAT activity were included to prevent any complicating modifications (other than deacetylation by sirtuins) that can result in Kac signal change. In HEK293T cell extracts without added recombinant sirtuins, the Kac signal was reduced by up to 40%, suggesting endogenous sirtuin activity can be captured by this microarray assay. When cell extracts were supplemented to the recombinant sirtuins, the deacetylation activity was slightly reduced, suggesting the presence of proteins or small molecules that may occupy and compete for the specific deacetylation sites. Overall, the favorable results of the pilot HAT and sirtuin assays indicate that this novel microarray platform is compatible for enzyme activity assays using recombinant enzymes and cell lysates.
Differential Histone (De)acetylation in Prostate Cancer Cells.
Next, we investigated dysregulated HAT or sirtuin activities, during progression of the HS cell line, LNCaP, to its CR daughter cell line, C4–2.24 The LNCaP and C4–2 PCa model system follows phenotypic behavior similar to clinical models providing a unique opportunity for studying PCa progression in cell lines.24,25 Using cell extracts, we adopted a similar assay scheme used in the pilot enzyme study and compared the resulting Kac signals in one subarray treated with the LNCaP cell lysate to that in the second subarray treated with C4–2 (Figure 1F). In the HAT specific assay, acetyl-CoA, TSA (HDAC inhibitor), and nicotinamide (sirtuin inhibitor) were supplemented in addition to protease and phosphatase inhibitors. In the sirtuin-specific assay, NAD+, TSA, and anarcardic acid (HAT inhibitor) were added while acetyl-CoA was excluded. After each experiment, the peptide array was processed for data analysis. The Kac signals were determined and presented as log2 of the C4–2 signal over the LNCaP signal. For each biological replicate, cells were grown separately and lysed individually.
Figure 2 summarizes the representative histone peptides having the highest Kac signal difference between LNCaP and C4–2. In the HAT activity assays, C4–2 displayed increased acetylation on the N-terminal (amino acids 1–20) lysines of H3 peptides in the order of K18, K4, K9, and K14, while there were reduced relative Kac signals in H3K79 and H2A K13/K15 in C4– 2 compared to LNCaP (Figure 2A). For the sirtuin activity assay, we observed higher sirtuin activity on the H3K122ac and H3K56ac in C4–2, while there was lower activity on the H3K9ac, H3K14ac, and to a lesser extent on the N-terminal lysines of H4 (Figure 2B). Most up-regulation (positive log2) in HAT activity or down-regulation (negative log2) of sirtuin activity in C4–2 took place on the same histone lysine residues, leading to marked combined effects on Kac status. For instance, up-regulation in HAT activity against H3K9 was observed while sirtuin assays revealed decreased deacetylation activity on H3K9ac, leading to a potential net increase in H3K9ac signal. Increased Kac signals are shown as blue bars, while red bars represent a decreased Kac signal. Overall, the net effects on the Kac signal changes in peptides containing H3K9, H3K14, and H3K18 were most prominent, and these PTMs were investigated in the following experiments.
Figure 2.
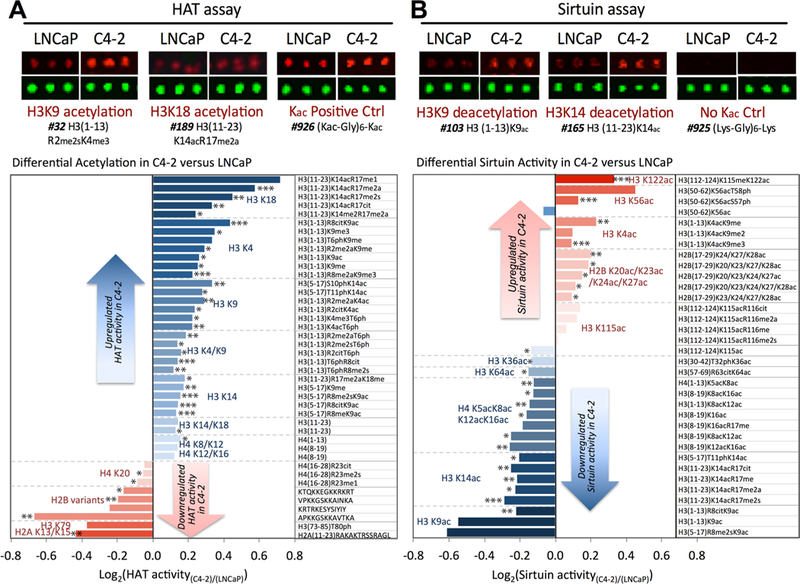
Differential activity profiles of HAT and sirtuin against histone peptides in C4–2 versus LNCaP. (A) In the global HAT activity assay, cell lysate of LNCaP or C4–2 was added to each subarray chamber in the presence of acetyl-coA, TSA, and nicotinamide. Shown here is a snapshot of a triplicate peptide spots after a HAT assay. The Alexa Fluor647 signal (red) became more intensive when peptides with either H3K9 or H3K18 residue were incubated with C4–2 in a HAT activity assay compared to with LNCaP. Peptides with preacetylated lysine (#926) did not show changes in the signal. The peptides with most differential HAT activity are shown in the table in the decreasing order of Kac signal. Increased acetylation in C4–2 was observed at several lysine sites, most prominently at H3K18, H3K4, H3K9, and H3K14. Statistical significance was calculated for each microarray slide, and experiments were performed in triplicate. Each peptide with p < 0.05 was labeled with *, **, or *** to denote significance of p < 0.05 in one, two, or three microarray experiments. (B) Sirtuin activity was monitored after incubation with NAD+, anarcardic acid, TSA, and cell lysates from either LNCaP or C4–2. The loss of Kac fluorescent signal due to deacetylation on the peptides H3K9ac (#103) or H3K14ac (#165) was lower (i.e., higher Kac signal) with C4–2 as shown in the snapshot. Peptides with the highest differential deacetylation activity were summarized in the table with higher differences in peptides harboring acetylation at H3K9, H3K14, and H4K5/K8/K12/K16. Lower Kac signals were detected at peptides with acetylation at H3K122, H3K56, and H3K4. In both acetylation and deacetylation profiles, a blue bar indicates higher Kac in C4–2 (CR) compared to LNCaP (HS), while a red bar indicates decreased Kac signal in C4–2 versus LNCaP. The column on the right represents the position of the peptide sequence within histone and the PTM mark introduced. Statistical significance was calculated as in A.
To validate the microarray results, we performed a highly sensitive in-solution HAT assay and demonstrated that the differential histone acetylation at specific lysine sites could be confirmed by independent methods (Figure 3). An H3 histone peptide (aa 1–20) bearing lysine sites with the highest differential activity (K4, K9, K14, and K18 of histone H3) was coincubated with 3H-acetyl CoA and extracts from either C4–2 or LNCaP cells, and incorporation of the radioactive acetyl group on the histone peptide was quantified by scintillation counting. The results revealed that both LNCaP and C4–2 exhibited increased acetylation over time with 55% higher HAT activity in the C4–2 cells over LNCaP after 2 h of incubation at 30 °C (Figure 3A). All the negative controls that lacked either cell extracts or a substrate peptide showed no signs of acetylation.
Figure 3.
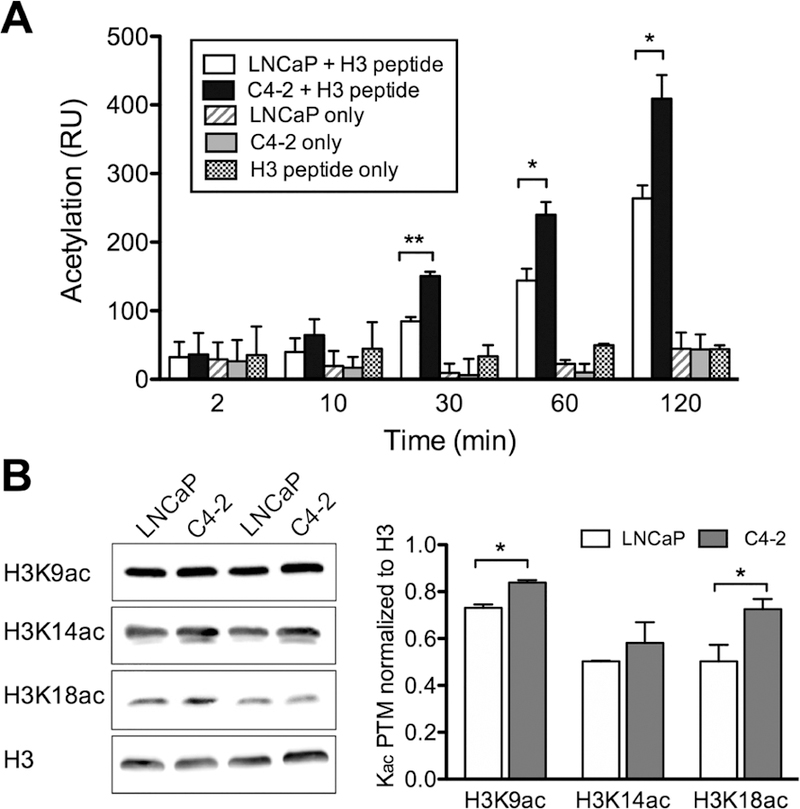
Upregulation of lysine acetylation of histone H3 N-terminal tail in castrate-resistant C4–2. (A) Lysine acetylation of histone H31–20 peptide by endogenous HAT activity in C4–2 vs LNCaP as monitored by isotopic incorporation of a tritium-labeled acetyl group from 3H-acetyl-CoA shows higher acetylation on the H3 peptide when incubated with C4–2 compared with LNCaP. (B) Quantification of Western blot analyses showing endogenous acetylation level at H3K9, H3K14, and H3K18 sites in LNCaP compared to C4–2. Each histone PTM was normalized against the histone H3 band (n = 3 for each PTM). All data represent the mean ± s.d. *P ≤ 0.05 and **P ≤ 0.01.
The enzyme assays clearly indicate that C4–2 cells display higher HAT activity and lower sirtuin activity on several overlapping lysine residues. To investigate whether these dysregulated activities represent true cellular dysfunction between C4–2 and LNCaP cells, we investigated the site-specific acetylation levels (immunoblots) on histone extracted directly from each cell line. We predicted that differential (de)acetylation activity observed in the microarray assay should be reflected in the acetylation status of endogenous histones. The top three “hits” in the microarray data were examined by an in-solution assay, which revealed that the combined levels of endogenous acetylation at H3K9, H3K14, and H3K18 in C4–2 cells were elevated compared to LNCaP (Figure 3B). Increases in the level of H3K9ac and H3K18ac at the global level reached statistical significance. Together, these results confirm the utility of our microarray analysis in detecting a specific difference in the chromatin modifying activities found among highly related cancers.
Endogenous Protein Levels of HATs and Sirtuins.
An important feature of the microarray assay is the ability to detect differences in enzymatic activity even when comparisons of protein levels in Western blots (or comparable ELISA) reveal no significant changes. To identify putative enzymes responsible for the altered (de)acetylation activities, we first investigated the endogenous protein levels of particular HATs that affect H3 acetylation. These HATs include p300/CBP that regulates gene expression by acetylation of histone and other transcription factors and are reported to be more promiscuous with respect to substrate sequence recognition.26 Other putative HATs include the GNAT family, GCN5, and PCAF. No significant changes were detected in the level of GCN5, PCAF, and p300 by immunoblotting (Figure 4A). The possibility that a PTM-regulated mechanism is involved therefore merited investigation. p300 undergoes autoacetylation at the amino acid residue K1499, which results in enhanced HAT catalytic activity,27 so we performed Western analysis to detect the endogenous amount of acetyl-p300 (K1499ac), which revealed a marked increase in C4–2 compared to LNCaP (Figure 4A). Collectively, these observations suggest that increased HAT activity in C4–2 can be attributed to activated, acetylated p300.
Figure 4.
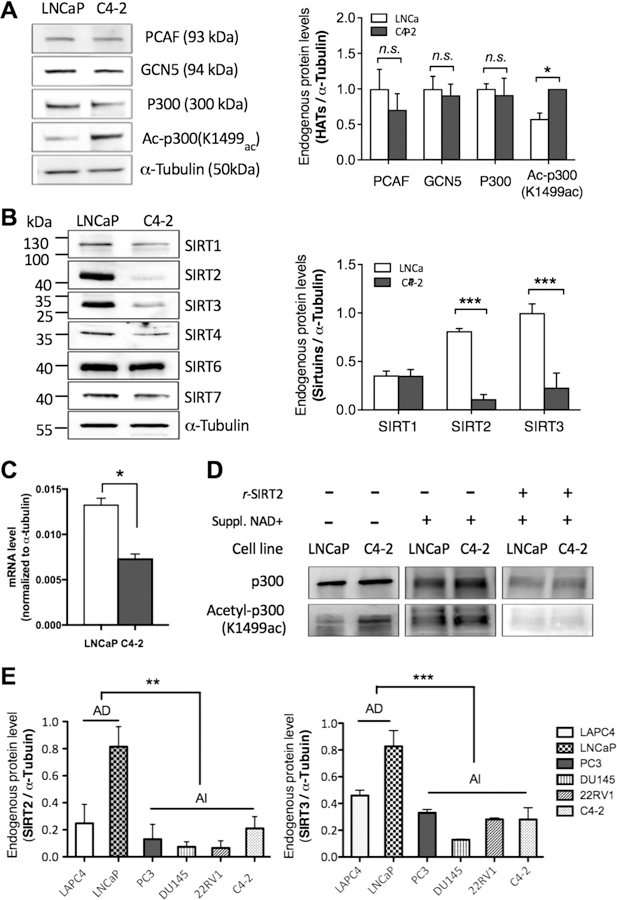
Endogenous protein and gene expression levels against HATs and sirtuins in HS vs CR PCa cell lines. (Full images of the blots can be found in SI Figure S4.) For all the data shown in this figure, n > 3; n.s., P > 0.5; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. (A) Representative immunoblots showing the levels of HATs, PCAF, Gcn5, p300, and acetyl-p300(K1499ac), normalized to the α-tubulin level. (B) Sirtuin levels and quantification for Sirt1–4, 6, and 7 in total cell extract of LNCaP versus C4–2 as normalized to α-tubulin level. The cellular level of Sirt2 and Sirt3 was greatly reduced in C4–2, while the cellular level of other sirutins between the two cell lines was not significant. (Sirt5 was not measured as it displays undetectable deacetylase activity.) (C) Quantitative analysis of SIRT2 mRNA level in LNCaP versus C4–2. The reduction in SIRT2 level in the castrate-resistant (CR) C4–2 cell is consistent with other CR cell lines (DU145) as shown in the SI (Figure S3). (D) SIRT2-dependent deacetylation of acetyl-p300(K1499ac) was shown in both LNCaP and C4–2 after 1 h of incubation with recombinant SIRT2 (r-SIRT2) and NAD+. When LNCaP and C4–2 were not treated with NAD+ and SIRT2, the ac-p300(K1499ac) level was higher in C4–2, while the p300 level remained constant. (E) Sirt2 and Sirt3 protein levels in the whole cell extracts of PCa cell lines (two HS cells, LAPC4 and LNCaP; and four CR cell lines, PC3, DU145, 22RV1, and C4–2) were determined by immunoblots.
Next, we investigated the protein levels of the six sirtuins (SIRT1–4, 6, and 7) in C4–2 and LNCaP cells to assess whether altered protein expression of a specific sirtuin enzyme was responsible for the overall lowered NAD+-dependent deacetylation observed in the C4–2 cells. Quantitative immunoblot analysis revealed that among the six sirtuins, only SIRT2 and SIRT3 displayed a significant loss of protein expression during the transition from LNCaP to C4–2 (Figure 4B). SIRT3 is a mitochondrial enzyme, while SIRT2 is known to function in the nucleus and the cytoplasm. Quantitative analysis of SIRT2 mRNA level in LNCaP versus C4–2 (Figure 4C) shows a reduction in SIRT2 level in castrate-resistant (CR) C4–2 cells, consistent with other CR cell lines (DU145) as shown in the SI (Figure S3). Most importantly, SIRT2 was shown to regulate the acetylation level of the activated form of acetyl-p300 (K1499ac).28 The combined observations of acetylated, hyperactive p300 and loss of SIRT2 expression in the C4–2 cells suggest that the dramatically decreased protein levels of SIRT2 lead to the inability to down-regulate the hyper-acetylated form of p300.
SIRT2-Dependent Deacetylation of p300 and SIRT2 Expression in Other PCa Cell Lines and Human Tissue.
To further establish the link between hyperacetylated activated p300 and decreased sirtuin activity in the CR cell line C4–2, we performed an in vitro NAD+-dependent deacetylation assay using cell extracts from both LNCaP and C4–2 cells with or without recombinant SIRT2 (Figure 4D). The addition of NAD+ to C4–2 cell extracts was not sufficient to completely remove acetyl modification from p300, while the addition of both SIRT2 and NAD+ led to a loss of immunoreactivity against acetyl-p300 (K1499ac). Consistent with the protein abundance of acetyl-p300 (K1499ac) observed from immunoblot (Figure 4A), LNCaP cells displayed lower acetylation at K1499 whether or not NAD+ was added (Figure 4D). Next we examined whether SIRT2 and SIRT3, the only sirtuins significantly lower in C4–2 compared to LNCaP were altered in extracts from other HS cells (LAPC4 and LNCaP) and CR cells (PC3, DU145, 22RV1, and C4–2). Lower levels of both proteins in CR cells relative to the two HS cell lines were seen (Figure 4E). Consistent with this observation, analysis of gene expression data obtained from the NCBI Gene Expression Omnibus (GEO) data depository for prostate cancer cell lines indicate that there is a statistically significant downregulation of SIRT2 and SIRT3 expression in the CR cell line DU145 when compared to the HS LNCaP cells while p300 gene expression data showed the levels of p300 remains unaltered between LNCaP and DU145 (SI Figure S3A–C).
Using a well-described human xenograft model system,29 we interrogated the SIRT2 mRNA expression in matched human tumors before and after the development of CRPC. We observed that in the majority of CR samples (7 out of 12 sets; 58%), SIRT2 expression was downregulated when compared to its HS paired precursor (Figure 5A). Several paired lines were analyzed for relative Sirt2 protein levels, showing good agreement between mRNA and protein for samples 35, 77, and 70 (Figure 5B). Collectively, these data indicate that reduced expression of SIRT2 is associated with the development of CR in the majority of human tumors directly assessed during progression.
Figure 5.
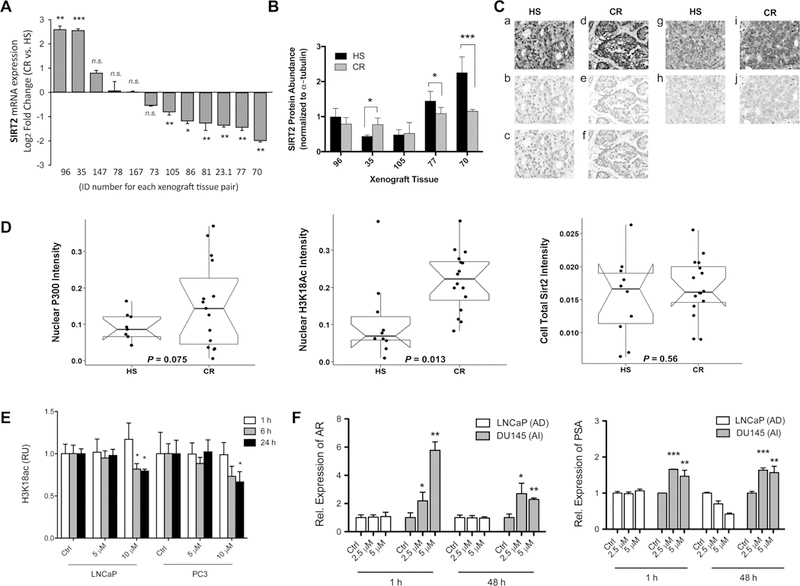
Changes in the abundance of histone modifiers in HS vs CR human PCa. (A) Sirt2 gene expression is represented by a log2 fold change of CR over HS cells. All of the compared pairs are generated from the same individuals as the tissue progressed from AS to CR by serial xenografts. The bar graph is the average of three technical replicates with standard error represented by s.d. The x axis represents each patient number with at least seven xenografts exhibiting Sirt2 down-regulation in CR. (B) Sirt2 relative protein levels in CR and HS select cells from A. The bar graph is the average of three technical replicates with standard error represented by s.d. (C) Representative staining of HS (a, b, c, g, and h) and CR (d, e, f, i, and j) PCa. Composite images of multiplexed staining are shown in the top panel (a, d, g, and i). Spectral libraries of individual stains were used to segment composite images for quantitative analysis. The selected images demonstrate segmented stains for Sirt2 (b, e), H3K18ac (c, f), and P300 (h, j). These images are representative of the mean intensity of HS and CR PCa for each marker shown (±10% optical density). (D) Box plots of the optical density (OD) of p300, H3K18ac, and Sirt2 immunohistochemistry in HS and CR PCa tissue microarray. Each box represents the 25% to 75% percentile range. Solid horizontal lines represent medians, and notches represent 95% confidence intervals of the medians. Dots represent individual patient tumors in each group. Tissue and cellular segmentation of IHC staining revealed significantly increased nuclear H3K18ac in the epithelium of CR PCa samples (p = 0.013), while epithelial total Sirt2 and nuclear P300 expression did not significantly change in CR vs HS samples. (E) Reduction of the H3K18ac level in C646-treated LNCaP and PC3 cells. At 10 μM C646, both cells showed a decrease in the level of H3K18ac over time as detected by Western blot analysis. Triplicate data were averaged and represented with s.d. The significance of the values was shown using standard convention of p > 0.5, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. (F) Changes in the mRNA expression of AR and PSA in PCa cell lines after treatment with C646 up to 48 h are shown. Overall, both AR and PSA showed higher expression after C646 treatment in Du145 cell lines, while LNCaP did not show significant changes compared to the control experiment.
Having shown that a loss of SIRT2 expression, increased p300 activity, and histone H3 hyperacetylation are associated with the transition to CR, we next examined a unique tissue microarray to determine whether any of these observations were evident among a series of HS and CR samples of human PCa tissue.30 The arrays were analyzed and quantified for protein abundance of p300, H3K18ac, and SIRT2 using VECTRA automated analysis (Figure 5C). Acetyl-p300 (K1499ac) protein levels were not examined due to antibody limitations. P300 levels trended lower in HS compared to CRPC tissues with broad expression patterns noted in the CRPC group (p = 0.075; Figure 5D). However, CRPC showed a marked increase in H3K18ac (p = 0.013) compared to the HS tumors. SIRT2 expression was highly variable in both groups (p = 0.56), with 8/15 (45%) of CPRC specimens demonstrating lower expression than the HS mean (Figure 5D). These specimens were not able to be matched with regard to progression in individual patients.
Inhibition of p300 activity has previously been demonstrated to decrease the proliferation of PC3 and other CR prostate cancer cell lines when exposed to the inhibitor C646,31 a response we confirmed (data not shown). To confirm the dependency of H3K18ac on p300 activity in these lines, exposure to C646 resulted in a consistent decrease in H3K18ac levels (Figure 5E). Finally, to further investigate this phenotype after p300 inhibition, LNCaP and PC3 cell lines were exposed to C646 and an increase in androgen receptor (AR) and its target PSA were seen in the AR negative DU145 cell line after treatment (Figure 5F). No increase was seen in the LNCaP line already robustly expressing these genes. Taken as a whole, evaluation of cell lines, xenografts, and tissue microarrays implicates the SIRT2 and p300/CBP in dictating the CR PCa transition in chromatin dynamics and transcription.
High-throughput approaches to discover critical histone modifying enzymes during cancer progression offer a unique approach to improving cancer therapy strategies. For the first time, histone peptide microarrays were used to quantify differential lysine acetylation and deacetylation activities during the progression to castrate resistance in PCa cells. Covering the entire human histone sequences with prevailing combinatorial PTMs, we show that the site-specific acetylation and deacetylation activities can be resolved reliably with high sensitivity when comparing two conditions. Indeed, our screening of two cells lines (C4–2 vs LNCaP) of the same genetic origin uncovered dysregulated acetyl-p300 and SIRT2. Thus, such microarray-based enzymatic assays can be successfully employed as a discovery tool to reveal novel mechanisms by which histone-modifying activities might drive the epigenetic landscapes to pathological states.
P300 is autoacetylated on multiple lysine sites by an intermolecular mechanism, and one of these sites, K1499, is critical for enhanced HAT activity.27,32 It was previously reported that deacetylation of p300 at K1499ac is SIRT2-dependent.28 Our unbiased histone array assays detected increased HAT and decreased NAD+-dependent deacetylase activities against specific lysine sites in histones. Strong activation of H3K18 acetylation occurred in CRPC tissues compared to HS primary and metastatic tissues in the unique series of tissue arrays tested (Figure 5). SIRT2 expression was not uniformly decreased in all CRPC samples tested, but SIRT2 functional mutations have been recognized,33 and activity may also be altered through other post-translational mechanisms.34 Follow-up experiments strongly suggested that these activities could be assigned to activated p300 and reduced SIRT2/3. Taken together, the results suggest that the loss of SIRT2 expression (activity) in some CR cells leads to uncontrolled activation of autoacetylated p300, which has been previously linked to CR gene expression.35
While this mechanism is likely, it is important to point out that we observed a decreased ability of C4–2 cell extracts to catalyze the NAD+-dependent deacetylation of H3K9, H3K14, and H3K18, which are targets of p300/CBP and SIRT2. Thus, while K1499ac is a target of SIRT2, the observed endogenous hyperacetylation of H3K9, H3K14, and H3K18 could be the result of SIRT2 loss or increased p300/CBP activity or both. Adding to the p300/CBP-SIRT2 connection, it has been reported that p300 acetylates SIRT2 to attenuate its deacetylation activity.36 SIRT2 and p300 have multiple layers of antagonizing activities that would be entirely consistent with the majority of trends observed in the cell lines, xenografts, and tissue microarray. Previous studies have shown a close link between SIRT2 expression and suppression of tumorigenesis.37,38 To our knowledge, no direct evidence has been reported to date whether SIRT2 affects the progression to CRPC. Expression of SIRT2 in PCa cells has not been examined, although deacetylation of α-tubulin, which is SIRT2-dependent, was shown to be up-regulated in CR PCa cell PC3.39 Furthermore, recent data suggest SIRT2 plays a role as a mitotic checkpoint, maintaining genomic integrity40 and impairing cell motility through perturbation of microtubule dynamics.41,42
In this novel, unbiased analysis of histone modifying enzymes, we have uncovered a distinct molecular pathway in progression to CRPC in which SIRT2 appears to play an important role. This has potential to be utilized in a personalized medicine approach to direct therapy against altered enzymes with further development. A variety of new inhibitors targeting histone modifying enzymes are being developed offering new options for cancer treatment.43 Other enzyme alterations strongly supported by our study include SIRT3, which deserves further investigation. In addition, other dysregulated activities identified in our micro-array study include deacetylation at H3K56ac. The enzyme responsible for this modification is SIRT6, of which the cellular protein abundance remained unchanged in our study; however, there remains the possibility that SIRT6 catalytic efficiency is affected in a similar fashion as in the SIRT2-p300 cross-talk mechanism. While this speculation lends further support, together these results provide a proof of concept for the unbiased use of our histone array assays to uncover aberrant histone modifying activities in cell extracts derived from different disease states.
MATERIALS AND METHODS
Preparation of Histone Peptide Microarray.
Peptides (13 amino acids) were synthesized on modified cellulose discs (celluspots) with a ResPepSL automated synthesizer as previously described in Su et al.17 Each synthesis included a peptide with an acid cleavable linker for quality assessment by HPLC and mass spectrometry. The cellulose peptide conjugates were extracted, diluted, and printed onto nitrocellulose slides (Intuitive Biosciences) with a Gene Machines OmniGrid Arrayer (Genomic Solutions) with TeleChem SMP3 quill-like pins. Each array housed duplicate sets of the peptide library (“subarrays”), allowing two reaction conditions to be probed on the same slide. A total of 932 histone peptides which make up our histone library were printed in triplicate. Positive control spots consisted of acetyllysine (Kac)-containing peptides, and negative control spots were cellulose without any peptide conjugation or peptides without any Lys residues. Fluorescent Cy3 dye (Lumiprobe Life Science Solutions) was conjugated to cellulose and mixed to each peptide stock solution to serve as a spot tracer (green, 532 nm), while peptides carrying Kac PTM fluoresced at 635 nm (red).
Microarray Enzyme Activity Assay and Data Analysis.
All the microarray assays were performed with a multichamber simplex gasket, which allows multiple reaction conditions for each array. Each array was blocked with 2% bovine serum albumin (BSA) to eliminate nonspecific protein binding to the nitrocellulose surface. Following blocking, each chamber was filled with each reaction buffer containing cofactors and cell lysate. For HAT-specific assays, 50 μM acetyl-CoA and 5 mM nicotinamide were supplemented, while for sirtuin deacetylation assays, 50 μM NAD+ and 2 mM anarcardic acid (HAT inhibitor) were added. In each chamber was added the total cell extract from each cell type at a concentration of 0.5–3 mg mL−1. The detailed method of the preparation of cell extracts and procedures to a detailed microarray assay have been included in the Supporting Information. The reaction was stopped by removing the assay components by aspiration. The array was washed with TBST before being incubated with the rabbit-derived pan-Kac antibody. Subsequently, the slide was incubated with anti-rabbit IgG conjugated to Alexa647. The slide was washed with TBST before being scanned on an Axon GenePix 4000B microarray fluorescence scanner (Molecular Devices) at a 5-μm pixel resolution and 33% laser power. Slide images were collected at a 532 nm/635 nm dual channel and analyzed using GenePix Pro 6.1 software (Molecular Devices). For alternative detection of HAT assays, isotopic incorporation of the acetyl group from a trace amount of radiolabeled 14C-acetyl-CoA (2.5 μCi, Moravek Biochemicals) was performed. Once the reaction was complete, the array was rinsed, and radiolabeled peptide was exposed to a BAS-IP TR2015 tritium screen for phorphorimaging. For GenePix data analysis of pan-Kac antibody detection, total signal intensities of both Alexa647 (635 nm) and Cy3 (532 nm) were utilized. The spot quality was evaluated, and intensity was analyzed and quantified as described in the SI (Figure S1 and SI Materials and Methods). The microarray design and the list of peptides in the histone library are provided in SI (XLSX).
Pilot microarray deacetylation assays were performed with a multichamber simplex gasket. The microarray contained a 20-member peptide library with controls and printed eight times per microarray slide in a 4 by 2 layout. Each array was blocked with 2% bovine serum albumin (BSA) to eliminate nonspecific protein binding to the nitrocellulose surface. Following blocking, each chamber was filled with each reaction buffer containing a combination of a final concentration of 1 mM NAD+; 1 μM purified recombinant of Sirt1, Sirt2, and Sirt3; 1 mg of HEK293T cell lysate in 50 mM Tris-HCl pH 8.0 buffer; and 1 mM DTT. The reaction was incubated for 1 h on the slide with gentle rocking and stopped by removing the assay components by gentle aspiration. The array was washed with TBST before being incubated with rabbit-derived pan-Kac antibody. Subsequently, the slide was incubated with anti-rabbit IgG conjugated to Alexa647. The slide was washed with TBST before being scanned on an Axon GenePix 4000B microarray fluorescence scanner (Molecular Devices) at 5-μm pixel resolution and 33% laser power. Slide images were collected at 532 nm/635 nm dual channel and analyzed using GenePix Pro 6.1 software (Molecular Devices). Data analysis was performed by utilizing the average total intensity at 635 nm of each peptide triplicate spot for a 200 μm diameter circle. The percent deacetylation was calculated by the determining the loss of signal in comparison to the well containing no enzyme. Each condition was run in duplicate.
Studies with Human Cell Lines and Mouse Xenograft.
Before each assay, cell pellets from PCa cell lines (LAPC4, LNCaP, PC3, DU145, 22RV1, and C4–2) were thawed on ice and resuspended in 300 μL of lysis buffer; 50 mM Tris-HCl at pH 7.5; 150 mM NaCl; 1% Triton X-100; DNase I (Roche); protease inhibitor cocktail (Promega); and NaF and incubated at RT for 10 min with gentle rotation. The cell suspension was passed through a 27-gauge needle 10 times, and the lysate was centrifuged at 14000g for 5 min at 4 °C. For the mouse xenograft study, 12 pairs of each HS and CR human prostate tissue were prepared. LuCaP patient-derived xenografts were grown subcutaneously in intact (HS lines) and castrated (CR lines) SCID male mice.29 RNA was isolated from the tissues using a Perfect Pure RNA Tissue Kit (5-Prim) in the presence of DNase I. cDNA was synthesized with qScript cDNA superMix (Quanta Biosciences) using 1 μg of total RNA with both Oligo(dT)’s and Random Haxamer Primer present. To further eliminate gDNA contamination, the amplification of cDNA is achieved by designing exon-primed intron crossing primers, in which the forward and reverse primers flank at different exons. qPCR was performed to measure gene expression, and GAPDH was used as a housekeeping gene.
Histone PTM Assays.
Recombinant full-length histone (Xenopus laevis), H2A, and H3, used in the enzyme assay were expressed in BL21(DE3)pLysS and purified according to Luger et al.44 Histone peptides that were not cellulose-conjugated were synthesized by Fmoc-based peptide synthesis (University of Wisconsin Peptide Synthesis Facility) and purified on a C18 column by Beckman BioSys 510 HPLC. N-terminally His6-tagged PCAF (human), piccolo NuA4 (yeast), and SIRT2 (human) were overexpressed in BL21(DE3) and purified on a Ni-charged HiTrap HP affinity column (GE Healthcare). All the recombinant enzymes used in the pilot experiments were purified to near homogeneity and were tested for their specific activities against histone protein substrates. For measuring the HAT activity of purified recombinant HATs as well as that in nuclear extracts, a filter-binding assay was used as previously reported.45
Immunohistochemical Staining of Tissue Microarrays.
A tissue microarray containing 18 HS and 18 CR cancer tissues in quadruplicate was obtained from the Prostate Cancer Biorepository Network. Slide preparation and image analysis were conducted as previously described.46 Briefly, 5 μm sections were taken through routine deparaffinization and rehydration. One triple stain (H3K18ac, SIRT2, and E-cadherin) and one double stain (p300, E-cadherin) were performed from two TMA sections. E-cadherin antibody was used to define the epithelial compartment for automated tissue segmentation. Stained slides were scanned using VECTRA as previously described.46 Cores with <5% epithelial component or tissue loss were excluded from the analysis. InForm 1.2 software was used to segment tissue subcellular compartments and tissue compartments. Epithelial staining intensity automatically quantified using inForm software was used for analysis of protein expression. Quadruplicate cores were averaged for a more precise estimate of protein expression. Tissues were required to have at least two cores with sufficient epithelium and tissue to be included in the analysis.
C646 p300 Inhibition.
mRNA expression of AR and PSA in prostate cancer cell lines LNCaP (HS) and DU145 (CR) were measured after treatment with C646 (p300 inhibitor). The cell lines were treated with C646 at a concentration of 2.5 and 5 μM. For a control experiment, DMSO was substituted for C646. After 1 h and 48 h, cells were harvested, and RNA was isolated from the cells using a Perfect Pure RNA Tissue Kit (5-Prism) following the manufacturer’s protocol. The same mRNA measurement procedure as described in the SIRT2 mRNA measurement (previous section) was used. The expression was normalized by GAPDH, and the data are shown as relative expression mean ± s.d. The list and sequence of primers used in this study is provided in Supporting Information.
Statistical Analysis.
Data are shown as a mean ± s.d., with a minimum N of three replicates unless otherwise noted. All the graphs were generated using Prism (GraphPad software) except for the tissue microarray immunostaining, which was generated using the R package (Bioconductor; ggplot2). Student’s t test was used to determine the significance of the differences between two groups. p < 0.05 was considered significant.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge members of the J.M.D. and D.F.J. research groups for technical advice and support, and particularly S. Oliver for the preparation of peptide microarrays. This study was supported by the National Institutes of Health (R01 GM065386) and Department of Defense (DOD) through PCa Research Program (W81XWH-15–1-0628). J.-H.L. is a recipient of an American Heart Association Postdoctoral Fellowship (15POST25680060). The tissue array was contributed by the Prostate Cancer Biorepository Network supported by the DOD PCa Research Program (W81XWH-14–2-0182, −0183, −0185, −0186, and −15–2-0062). We acknowledge the University of Wisconsin Translational Research Initiatives in Pathology laboratory for use of its facilities and services.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.6b01035.
Histone peptide list and their PTMs in the library, microarray design, summary of HAT and sirtuin activity from the microarray assay, and processed data files of the large microarray assay (XLSX)
Additional controls and results, and a list of the primers used for the mRNA measurements. (PDF)
The authors declare the following competing financial interest(s): J.M.D. consults for Bio-Techne and FORGE Life Science and is a co-founder of Galilei BioSciences.
REFERENCES
- (1).Oliver S, Musselman C, Srinivasan R, Svaren J, Kutateladze T, and Denu J (2012) Multivalent Recognition of Histone Tails by the PHD Fingers of CHD5. Biochemistry 51, 6534–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, and Allis CD (2000) Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell 5, 905–915. [DOI] [PubMed] [Google Scholar]
- (3).Jenuwein T, and Allis C (2001) Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- (4).Kouzarides T (2007) Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- (5).Esteller M (2007) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet 8, 286–298. [DOI] [PubMed] [Google Scholar]
- (6).Gygi SP, Rochon Y, Franza BR, and Aebersold R (1999) Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol 19, 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).LeRoy G, DiMaggio P, Chan E, Zee B, Blanco M, Bryant B, Flaniken I, Liu S, Kang Y, Trojer P, and Garcia B (2013) A quantitative atlas of histone modification signatures from human cancer cells. Epigenet. Chromatin 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Huang H, Lin S, Garcia B, and Zhao Y (2015) Quantitative Proteomic Analysis of Histone Modifications. Chem. Rev 115, 2376–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Vogel C, and Marcotte EM (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Stitt M, and Gibon Y (2014) Why measure enzyme activities in the era of systems biology? Trends Plant Sci 19, 256–265. [DOI] [PubMed] [Google Scholar]
- (11).Stephenson AJ, Kattan MW, Eastham JA, Bianco FJ Jr., Yossepowitch O, Vickers AJ, Klein EA, Wood DP, and Scardino PT (2009) Prostate cancer-specific mortality after radical prostatectomy for patients treated in the prostate-specific antigen era. J. Clin. Oncol 27, 4300–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Eisenberger MA, and Carducci MA (2002) Chemotherapy for Hormone-Resistant Prostate Cancer, 8th ed., Vol. 4, Philadelphia. [Google Scholar]
- (13).Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, and Kurdistani SK (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435, 1262–1266. [DOI] [PubMed] [Google Scholar]
- (14).Bianco-Miotto T, Chiam K, Buchanan G, Jindal S, Day TK, Thomas M, Pickering MA, O’Loughlin MA, Ryan NK, Raymond WA, Horvath LG, Kench JG, Stricker PD, Marshall VR, Sutherland RL, Henshall SM, Gerald WL, Scher HI, Risbridger GP, Clements JA, Butler LM, Tilley WD, Horsfall DJ, and Ricciardelli C (2010) Global Levels of Specific Histone Modifications and an Epigenetic Gene Signature Predict Prostate Cancer Progression and Development. Cancer Epidemiol., Biomarkers Prev 19, 2611–2622. [DOI] [PubMed] [Google Scholar]
- (15).Dawson M, Prinjha R, Dittmann A, Giotopoulos G, Bantscheff M, Chan W, Robson S, Chung C, Hopf C, Savitski M, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman T, Roberts E, Soden P, Auger K, Mirguet O, Doehner K, Delwel R, Burnett A, Jeffrey P, Drewes G, Lee K, Huntly B, and Kouzarides T (2011) Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Pereź-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, and Esteller M (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet 37, 391–400. [DOI] [PubMed] [Google Scholar]
- (17).Su Z, Boersma MD, Lee JH, Oliver SS, Liu S, Garcia BA, and Denu JM (2014) ChIP-less analysis of chromatin states. Epigenet. Chromatin 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Britton L, Gonzales-Cope M, Zee B, and Garcia B (2011) Breaking the histone code with quantitative mass spectrometry. Expert Rev. Proteomics 8, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Thiele A, Stangl GI, and Schutkowski M (2011) Deciphering Enzyme Function Using Peptide Arrays. Mol. Biotechnol 49, 283–305. [DOI] [PubMed] [Google Scholar]
- (20).Choudhary C, Weinert B, Nishida Y, Verdin E, and Mann M (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol 15, 536–550. [DOI] [PubMed] [Google Scholar]
- (21).Arnold KM, Lee S, and Denu JM (2011) Processing Mechanism and Substrate Selectivity of the Core NuA4 Histone Acetyltransferase Complex. Biochemistry 50, 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Love IM, Sekaric P, Shi D, Grossman SR, and Androphy EJ (2012) The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle 11, 2458–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Feldman J, Dittenhafer-Reed K, and Denu J (2012) Sirtuin Catalysis and Regulation. J. Biol. Chem 287, 42419–42427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Thalmann G, Sikes R, Wu T, Degeorges A, Chang S, Ozen M, Pathak S, and Chung L (2000) LNCaP progression model of human prostate cancer: Androgen-independence and osseous metastasis. Prostate 44, 91–103. [DOI] [PubMed] [Google Scholar]
- (25).Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, Pathak S, Voneschenbach AC, and Chung LWK (1994) Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res 54, 2577–2581. [PubMed] [Google Scholar]
- (26).Liu X, Wang L, Zhao KH, Thompson PR, Hwang Y, Marmorstein R, and Cole PA (2008) The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 451, 846–850. [DOI] [PubMed] [Google Scholar]
- (27).Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D, An W, Ge Q, Roeder RG, Wong J, Levrero M, Sartorelli V, Cotter RJ, and Cole PA (2004) Regulation of the p300 HAT domain via a novel activation loop. Nat. Struct. Mol. Biol 11, 308–315. [DOI] [PubMed] [Google Scholar]
- (28).Black JC, Mosley A, Kitada T, Washburn M, and Carey M (2008) The SIRT2 deacetylase regulates autoacetylation of p300. Mol. Cell 32, 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Corey E, Quinn JE, Buhler KR, Nelson PS, Macoska JA, True LD, and Vessella RL (2003) LuCaP 35: a new model of prostate cancer progression to androgen independence. Prostate 55, 239–246. [DOI] [PubMed] [Google Scholar]
- (30).Ha S, Iqbal NJ, Mita P, Ruoff R, Gerald WL, Lepor H, Taneja SS, Lee P, Melamed J, Garabedian MJ, and Logan SK (2013) Phosphorylation of the androgen receptor by PIM1 in hormone refractory prostate cancer. Oncogene 32, 3992–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Debes J, Sebo T, Lohse C, Murphy L, Haugen D, and Tindall D (2003) p300 in prostate cancer proliferation and progression. Cancer Res 63, 7638–7640. [PubMed] [Google Scholar]
- (32).Karanam B, Jiang L, Wang L, Kelleher NL, and Cole PA (2006) Kinetic and mass spectrometric analysis of p300 histone acetyltransferase domain autoacetylation. J. Biol. Chem 281, 40292–40301. [DOI] [PubMed] [Google Scholar]
- (33).Finnin MS, Donigian JR, and Pavletich NP (2001) Structure of the histone deacetylase SIRT2. Nat. Struct. Biol 8, 621–625. [DOI] [PubMed] [Google Scholar]
- (34).North BJ, and Verdin E (2007) Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J. Biol. Chem 282, 19546–19555. [DOI] [PubMed] [Google Scholar]
- (35).Debes JD, Schmidt LJ, Huang HJ, and Tindall DJ (2002) p300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res 62, 5632–5636. [PubMed] [Google Scholar]
- (36).Han Y, Jin Y-H, Kim Y-J, Kang B-Y, Choi H-J, Kim D-W, Yeo C-Y, and Lee K-Y (2008) Acetylation of Sirt2 by p300 attenuates its deacetylase activity. Biochem. Biophys. Res. Commun 375, 576–580. [DOI] [PubMed] [Google Scholar]
- (37).Dryden SC, Nahhas FA, Nowak JE, Goustin AS, and Tainsky MA (2003) Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol 23, 3173–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hiratsuka M, Inoue T, Toda T, Kimura N, Shirayoshi Y, Kamitani H, Watanabe T, Ohama E, Tahimic CGT, Kurimasa A, and Oshimura M (2003) Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun 309, 558–566. [DOI] [PubMed] [Google Scholar]
- (39).Soucek K, Kamaid A, Phung AD, Kubala L, Bulinski JC, Harper RW, and Eiserich JP (2006) Normal and prostate cancer cells display distinct molecular profiles of alpha-tubulin posttranslational modifications. Prostate 66, 954–965. [DOI] [PubMed] [Google Scholar]
- (40).Inoue T, Hiratsuka M, Osaki M, Yamada H, Kishimoto I, Yamaguchi S, Nakano S, Katoh M, Ito H, and Oshimura M (2007) SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 26, 945–957. [DOI] [PubMed] [Google Scholar]
- (41).Pandithage R, Lilischkis R, Harting K, Wolf A, Jedamzik B, Lüscher-Firzlaff J, Vervoorts J, Lasonder E, Kremmer E, Knöll B, and Lüscher B. (2008) The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol 180, 915–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Inoue T, Nakayama Y, Yamada H, Li YC, Yamaguchi S, Osaki M, Kurimasa A, Hiratsuka M, Katoh M, and Oshimura M (2009) SIRT2 downregulation confers resistance to microtubule inhibitors by prolonging chronic mitotic arrest. Cell Cycle 8, 1279–1291. [DOI] [PubMed] [Google Scholar]
- (43).Arrowsmith CH, Bountra C, Fish PV, Lee K, and Schapira M (2012) Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 11, 384–400. [DOI] [PubMed] [Google Scholar]
- (44).Luger K, Rechsteiner TJ, and Richmond TJ (1999) Preparation of nucleosome core particle from recombinant histones. Methods Enzymol 304, 3–19. [DOI] [PubMed] [Google Scholar]
- (45).Berndsen C, and Denu J (2005) Assays for mechanistic investigations of protein/histone acetyltransferases. Methods 36, 321–331. [DOI] [PubMed] [Google Scholar]
- (46).Huang W, Hennrick K, and Drew S (2013) A colorful future of quantitative pathology: validation of Vectra technology using chromogenic multiplexed immunohistochemistry and prostate tissue microarrays. Hum. Pathol 44, 29–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.