

Abstract
Nowadays, chemically defined cell culture media (CCM) have replaced serum‐ and hydrolysate‐based media that rely on complex ingredients, such as yeast extracts or peptones. Benefits include a significantly lower lot‐to‐lot variability, more efficient manufacturing by reduction to essential components, and the ability to exclude components that may negatively influence growth, viability, or productivity. Even though current chemically defined CCMs provide an excellent basis for various mammalian biotechnological processes, vitamin instabilities are known to be a key factor contributing to the variabilities still present in liquid CCM as well as to short storage times. In this review, the chemical degradation pathways and products for the most relevant vitamins for CCM will be discussed, with a focus on the effects of light, oxygen, heat, and other CCM compounds. Different approaches to stabilize vitamins in solution, such as replacement with analogs, encapsulation, or the addition of stabilizing compounds will also be reviewed. While these vitamins and vitamin stabilization approaches are presented here as particular for CCM, the application of these concepts can also be considered relevant for pharmaceutical, medical, and food supplement purposes. More precise knowledge regarding vitamin instabilities will contribute to stabilize future formulations and thus decrease residual lot‐to‐lot variability.
Keywords: cell culture media, solubility, stability, stabilization strategies, vitamins
1. INTRODUCTION
Cell culture media (CCM) are designed to provide an environment that supports the growth and maintenance of cells in vitro as well as the production of therapeutically relevant proteins like monoclonal antibodies. Whereas older formulations require the use of undefined components like serum or hydrolysates, newly developed formulations are completely chemically defined. These media regulate both the pH and the osmotic pressure of the culture and consist of several energy sources (e.g., glucose and pyruvate), salts, trace elements, buffers, shear stress protectants, large amounts of amino acids (AAs), and small quantities of vitamins (Landauer, 2014).
Vitamins form a group of essential organic compounds that are required in small amounts for highly specific intracellular functions (Bender, 2003a). They are unable to be synthesized in vivo in adequate amounts to meet cellular needs, and so need to be obtained from the environment (i.e., diet or CCM). If absent in the cell, specific deficient syndromes will occur, such as scurvy or beriberi (Combs, 2012). The classes of vitamins themselves have little in common, either chemically or in their metabolic function. Vitamins can act as enzyme cofactors (e.g., vitamin K and most B vitamins), as biological antioxidants (e.g., vitamins C and E), or even as hormones (e.g., vitamins A and D) (Bühler, 1988; Combs, 2012). Vitamins can be classified as being either fat‐soluble (vitamins A, D, E, and K) or water‐soluble (vitamins C and B) (Combs, 2012). The water‐soluble vitamins are either charged or possess highly polar functional groups, such as carboxyl, hydroxyl, or phosphoryl moieties, whereas the fat‐soluble vitamins are predominantly large, hydrocarbon structures, often with a high degree of unsaturation (Combs, 2012).
This review will focus on the B vitamins that are considered essential for CCM, covering literature that deals with vitamin degradation as a result of light, heat, oxygen, or reactive oxygen species (ROS) and methods to protect against these effects. Furthermore, essential positive and negative interactions between CCM components that are known or likely to occur in liquid CCM will be summarized. Being aware of the tremendous power of serum proteins to stabilize vitamins, the review will also focus on strategies for developing stable vitamin formulations in serum‐free, chemically defined CCM. These aspects of degradation and stabilization have been summarized in Table 1.
Table 1.
Vitamin reactivity and stability in CCM: An overview of the conditions and CCM components that cause vitamin degradation and viable stabilization strategies
| Vitamins | Conditions that degrade the vitamin | Compounds in CCM that enhance degradation of the vitamin | CCM components destroyed by the vitamin | Viable stabilization strategies |
|---|---|---|---|---|
| Riboflavin (B2) | Light, oxygen, strong alkali, divalent anions | Metal cations, thiamine HCl | AAs with heteroatoms in the side chain (e.g., Cys, Trp, Tyr), folic acid, cyanocobalamin, thiamine HCl | Avoid light or filtering out most damaging wavelengths, complexing agents, encapsulation |
| Folic acid (B9) | Light + oxygen, acid | Riboflavin, ascorbic acid, thiamine, reducing sugars | – | Additives to enhance solubility at low pH, avoiding light, antioxidants |
| Cyanocobalamin (B12) | Light, oxygen, strong acid or alkali | Ascorbic acid, riboflavin, nicotinamide, Cys, GSH, thiamine (following degradation) | – | Ferric salts, phosphate buffer, potassium ferrocyanide, filtering out most damaging wavelengths in the light source |
| Thiamine (B1) | Strong acid or alkali, light, oxygen, other oxidants | Sulfites, cystine, ketoacids, aldehydes (e.g., reducing sugars), metal cations, nicotinamide | Folic acid, riboflavin | Formation of/replacement with stable disulfide, additives to chelate metals, thiol additives, antioxidants, replacement with analog (nitrate) |
| Pyridoxine (B6) | Light, high temperatures | Any primary amine‐containing compounds (all AA, especially lysine) – pyridoxal only | Any primary amine‐containing compounds (all AA, especially lysine) – pyridoxal only | Choice of vitamer – avoid pyridoxal |
| Biotin (B7) | UV light, strong acid or alkali | – | – | Vitamin is stable |
| Pantothenate (B5) | Strong acid or alkali | Phosphate buffer, nicotinamide | – | Vitamin is stable |
| Nicotinamide (B3) | Acid/alkali conversion to vitamer (nicotinic acid) | – | Cyanocobalamin, thiamine, pantothenate | Vitamin is stable |
CCM: cell culture media; GSH: glutathione.
2. RIBOFLAVIN
2.1. Function
Riboflavin (vitamin B2) is essential for the metabolism of carbohydrates, AAs, and lipids, where it assists in enzymatically controlled one‐ or two‐electron redox reactions (Cardoso, Libardi, & Skibsted, 2012; Choe, Huang, & Min, 2005; Combs, 2012). In the cell, riboflavin is converted to either flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD) (Cardoso et al., 2012; Choe et al., 2005; Combs, 2012; Sheraz, Kazi, Ahmed, Anwar, & Ahmad, 2014). All three species function as prosthetic groups, covalently linked between an electronegative heteroatom in AA side chains of the enzyme (e.g., cysteine, tyrosine, and histidine) and one of the methyl groups on the dimethyl‐isoalloxazine core structure (Figure 1) (Bender, 2003b). The redox processes in which flavins are utilized typically involve the addition or removal of hydrogen, even in energetically high oxidation processes, such as the oxidation of a saturated hydrocarbon to a C–C double bond. Following reduction of the flavin moiety, the catalyst is regenerated by O2 or NADP+, producing either superoxide or NADPH. More than 100 enzymes are known to utilize riboflavin as a cofactor for redox processes (Combs, 2012).
Figure 1.
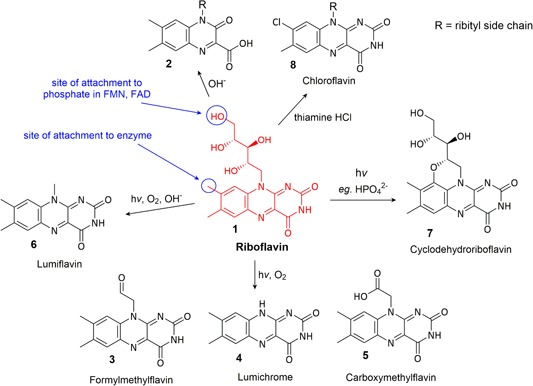
Major degradation products of riboflavin [Color figure can be viewed at wileyonlinelibrary.com]
2.2. Stability, reactivity, degradation products
2.2.1. Impact of external factors
Riboflavin (1) is quite stable to heat and atmospheric oxygen (Ottaway, 1993). It is also stable in acidic solutions but decomposes in alkaline environments to urea and 1,2‐dihydro‐6,7‐dimethyl‐2‐keto‐1‐d‐ribityl‐3‐quinoxalinecarboxylic acid (2) (Figure 1) (Kearsley & Rodriguez, 2007; Surrey & Nachod, 1951).
The most well‐characterized aspect of riboflavin reactivity is its sensitivity to light in an aerobic environment. Riboflavin becomes highly reactive upon exposure to UV or visible light in the presence of oxygen, and subsequent degradation processes are varied, dependent upon the conditions such as pH and the presence of other compounds in solution (Sheraz et al., 2014). Formylmethylflavin (3) and lumichrome (4) are major degradation products in all conditions and carboxymethylflavin (5) is a minor product (Ahmad, Anwar, Ahmed, Sheraz, & Khattak, 2017; Cairns & Metzler, 1971; Sheraz et al., 2014) (Figure 1). In alkaline solutions, lumiflavin (6) is also generated (Pan et al., 2001). In the presence of divalent anions, such as HPO4 2− or SO4 2−, cyclodehydroriboflavin (7) is formed (Ahmad, Ahmed, Sheraz, Vaid, & Ansari, 2010).
The light sensitivity of riboflavin is a result of its redox‐active aromatic isoalloxazine moiety, which leads to riboflavin functioning as a photosensitizer, wherein it catalyzes the oxidation of substrates as a consequence of absorption of UV light. Upon exposure to light, riboflavin is excited to a short‐lived singlet excited state (1Riboflavin*), which then readily undergoes intersystem crossing to the longer‐lived excited triplet state (3Riboflavin*) (9) (Figure 2b). This excited triplet state has a considerably higher standard reduction potential compared with the ground state of riboflavin (1.7 V vs. −0.3 V) (Cardoso et al., 2012; Pan et al., 2001), and so oxidizes other species (including other riboflavin molecules) via pathways that are classified as either Type I or Type II mechanisms (Figure 2a).
Figure 2.
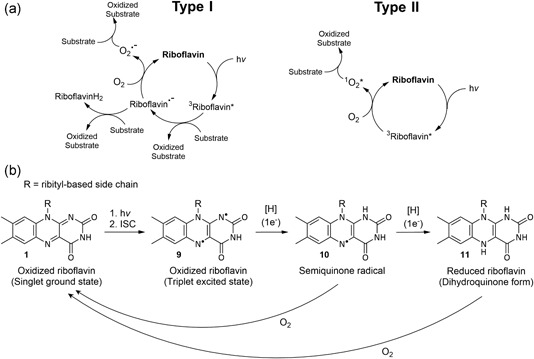
(a) Type I and Type II photooxidation mechanisms for riboflavin and subsequent mechanisms for substrate oxidation (modified from Cardoso et al. (2012)); (b) key forms of riboflavin in electronic excitation and redox processes
These photooxidation mechanisms induced by the light absorption of riboflavin have three central components: (a) the generation of ROS, such as singlet oxygen, the superoxide anion radical, and the hydroperoxide radical; (b) the decomposition of other compounds by reaction with the generated ROS or the intermediate semiquinone radical riboflavin species (10) (Figure 2b); (c) the catalytic regeneration of riboflavin, thus affecting large amounts of decomposition even in small amounts (Cardoso et al., 2012; Choe et al., 2005). The fully reduced form of riboflavin (11) is easily reoxidized to riboflavin in the presence of air (Carr, 1960) (Figure 2b).
Generally being stable in the absence of light, other external factors combined with light exposure will enhance the reactivity and, hence, degradation of riboflavin further. The rate of photolysis can be promoted by complexation with some metal cations (e.g., Cu2+ and Zn2+) at the isoalloxazine moiety, whereas other metal cations will directly reduce riboflavin (e.g., Fe2+) (Ahmad et al., 2017). Degradation is also enhanced by increasing temperature (Ahmad et al., 2017; Bühler, 1988; Cardoso et al., 2012; Choe et al., 2005; Combs, 2012; Sato, Yokoo, Takahashi, & Takahashi, 1982). Photodegradation of riboflavin is also known to be enhanced by the presence of sodium bicarbonate (Combs, 2012) and increased ionic strength of the solution (Ahmad et al., 2016). Sunlight degrades riboflavin faster than UV light and degradation is fastest with irradiation at 450 nm, the absorbance maxima of riboflavin (Choe et al., 2005).
2.2.2. Interaction with other CCM components
Riboflavin is not usually affected by other CCM components but riboflavin will induce the degradation of many other CCM components via photosensitization. The only known alteration of riboflavin by other CCM components is the reaction with thiamine hydrochloride (HCl), in which a high concentration of thiamine HCl (relative to riboflavin) results in the oxidation of thiamine by riboflavin, followed by the precipitation of chloroflavin as the riboflavin degradation product (8) (Gambier & Rahn, 1957). This formation of chloroflavin from reaction with thiamine HCl is enhanced in the presence of ascorbic acid (Gambier & Rahn, 1957).
The type I photooxidation processes are particularly damaging to some of the AAs in CCM, particularly those with heteroatoms and aromatic groups in the side‐chains, most notably methionine, cysteine, tryptophan, and tyrosine. The resultant degradation processes and products will depend on what types of ROS are generated/present in the system, that is, whether it involves type I or type II photooxidation. For further details on the photooxidative riboflavin‐induced degradation processes and products of these particular AAs, we recommend the following resources: methionine (Tzeng, Lee, Chung, & DeVay, 1990; Yang, Ku, & Pratt, 1967); cysteine (Huvaere et al., 2006; Obata & Tanaka, 2014; Sarkar, Das, Bhattacharyya, & Bose, 1997); tryptophan (Kanner & Fennema, 1987; Yoshimura & Ohno, 1988); and tyrosine (Choe et al., 2005; Silva & Godoy, 1994).
2.3. Stabilization
Riboflavin photodegradation could be avoided by storing CCM in the complete absence of light, or laboratories that store and use CCM could utilize a light source that filters out the wavelengths of light known to cause the most riboflavin degradation, that is, wavelengths below 520 nm (Sheraz et al., 2014). Inhibition of light‐induced excitation processes might also be possible via complexing agents or encapsulation. Otherwise, extended degradation catalyzed by initial light‐initiated oxidative processes could hypothetically be mitigated by the scavenging of formed radicals.
The use of complexing agents has been suggested to inhibit riboflavin photodegradation. The monomeric interaction and complex formation of caffeine with riboflavin has been linked to the inhibition of riboflavin photolysis (Ahmad, Ahmed, Sheraz, Aminuddin, & Vaid, 2009) and the photochemical stability of riboflavin was observed to increase at pH 7 with the addition of disodium ethylenediaminetetraacetic acid (EDTA), possibly through forming a complex that protects the ribityl side chain from degradation (Asker & Habib, 2008; Fife & Moore, 1979).
Encapsulation is also a successful strategy to prevent riboflavin degradation. A range of cyclodextrins (α‐, β‐, and γ‐cyclodextrins) have been shown to stabilize riboflavin via encapsulation (Terekhova, Koźbiał, Kumeev, & Alper, 2011; Terekhova, Tikhova, Volkova, Kumeev, & Perlovich, 2010; X.‐M. Wang & Chen, 1996). However, the use of hydroxypropylated cyclodextrins did not show a positive effect on riboflavin stabilization (Terekhova et al., 2011).
Regarding the use of riboflavin derivatives, most of the studied molecules like FMN or FAD are quite susceptible to light‐induced degradation and minor changes in the structure (e.g., modification of the ribityl side chain or the NH and methyl groups of the isoalloxazine core structure) of riboflavin result in a loss of vitamin activity (Bühler, 1988; Casirola, Kasai, Gastaldi, Ferrari, & Matsui, 1994; Insińska‐Rak & Sikorski, 2014). Riboflavin tetraacetate is photochemically more stable than riboflavin but also enhances the photooxidized degradation of some compounds better than riboflavin, and is thus not beneficial for CCM (Larson, Stackhouse, & Crowley, 1992).
3. FOLIC ACID
3.1. Function
Folic acid (vitamin B9), also known as pteroylmonoglutamic acid, functions as a coenzyme in many metabolic reactions, such as AA metabolism, purine and pyrimidine synthesis, and S‐adenosylmethionine (SAM) formation, via acting as a transport vessel for single‐carbon units like methyl and formyl groups (Bailey & Gregory, 1999; Combs, 2012). Various redox forms and substituted forms of folic acid exist and these intracellularly interconvertible species are collectively known as folates. Folates can enter the cell in the form of folic acid, 5‐methyl‐tetrahydrofolic acid, and 5‐formyl‐tetrahydrofolic acid either by diffusion or by Na+‐coupled protein transporters (Combs, 2012; Matherly & Hou, 2008). Once inside the cell, monoglutamyl folates are converted into polyglutamyl forms to prevent outward transport (Combs, 2012).
For the functional utilization of the folates, the pteridine system is reduced enzymatically to afford tetrahydrofolic acid (12) (Figure 3). Enzymes subsequently substitute the N5 and N10 positions with a range of single‐carbon units (Figure 3), which direct the folate derivatives to particular metabolic pathways, where they are used for the catabolism and metabolism of other intracellular components (Combs, 2012). As a food additive and in CCM, vitamin B9 is exclusively administered in the form of folic acid.
Figure 3.
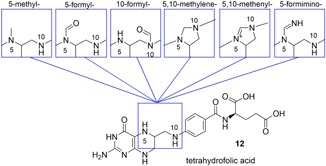
Overview of the single‐carbon units carried by tetrahydrofolic acid between N5 and N10 (modified from Combs (2012)) [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Stability, reactivity, degradation products
3.2.1. Impact of external factors
Folic acid (13) (Figure 4) can be inactivated in physiological conditions by heat, acid, light, and reducing agents (Akhtar, Khan, & Ahmad, 1999, 2000; De Brouwer, Zhang, Storozhenko, Straeten, & Lambert, 2007; Koft & Sevag, 1949; Off et al., 2005; Steindal, Porojnicu, & Moan, 2007). It has limited solubility in acidic and neutral conditions (Wu, Li, Hou, & Qian, 2010), so to achieve higher concentrations in solution, folic acid is often dissolved in alkaline solutions, which are then acidified to the desired pH, achieving a supersaturated solution (a solution with a higher concentration of dissolved substance than could normally be achieved by dissolution of the solid directly in the same solvent). These supersaturated solutions are stabilized to a point (determined by the thermodynamics of the compound and the solvent environment) but upon reaching the boundaries of saturation for the compound at a given pH, the compound will precipitate out of solution. Acidifying below about pH 6 results in folic acid undergoing either degradation and/or precipitation (Biamonte & Schneller, 1951; Taub & Lieberman, 1953).
Figure 4.
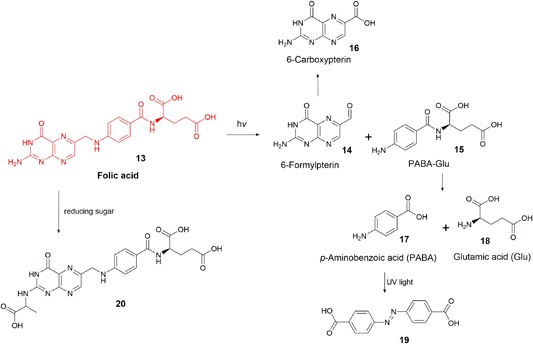
Overview of known folic acid degradation products [Color figure can be viewed at wileyonlinelibrary.com]
In aqueous aerobic solutions, folic acid is sensitive to light. In the absence of oxygen, folic acid is photostable. The pterin moiety of folic acid can absorb UV–visible light and transition to a reactive excited state in much the same way as a photosensitizer, however, the low quantum yields of subsequent energy transfers means that folic acid is not classified as a photosensitizer (e.g., for 1O2 production, Φ[folic acid] < 0.02 [Thomas et al., 2003]); Φ(riboflavin) = 0.54 (Baier et al., 2006)). Although rare, the absorption of UV light and subsequent intermolecular reaction catalyzes the degradation of folic acid (Figure 4) forming 6‐formylpterin (14) and para‐aminobenzoic acid (PABA)‐Glu (15) (Dántola et al., 2010; Lowry, Bessey, & Crawford, 1949; Stokstad, Fordham, & De, 1947). 6‐Formylpterin is a photosensitizer (Φ(1O2 generation) = 0.45 [Thomas et al., 2003]), and promotes extensive folic acid degradation, affording the same degradation products as pure folic acid (Off et al., 2005). 6‐Formylpterin under the same conditions may also be further oxidized to 6‐carboxypterin (16) (Akhtar et al., 1999; Dántola et al., 2010). While some studies show that the PABA‐Glu residue available following folic acid oxidative cleavage can undergo further UV‐induced oxidative degradation, there appear to be no primary studies confirming any products beyond PABA‐Glu, though some have hypothesized that the products are simply PABA (17) and glutamic acid (18) (Dántola et al., 2010; Gazzali et al., 2016; Lowry et al., 1949).
PABA (formerly classified as vitamin B10) has been used as an additive in CCM as an antioxidant and as a protective agent against UV radiation, as it absorbs light in the UVB range (Akberova, 2002; Hu, Chen, Chen, & Sano, 1995). UV radiation degrades PABA to the potentially toxic 4,4′‐azodibenzoic acid (19) (Gasparro, 1985).
3.2.2. Interaction with other CCM components
Nicotinamide was shown to enhance the long‐term stability of folic acid in neutral and mildly acidic solutions by inhibiting precipitation of folic acid that normally occurs at lower pH values (Taub & Lieberman, 1953).
The degradation of folic acid is accelerated by riboflavin in the presence of UV light (Akhtar et al., 2000; Scheindlin, Lee, & Griffith, 1952; Scurachio, Skibsted, Metzker, & Cardoso, 2011), by ascorbic acid even in absence of light (Scheindlin & Griffith, 1951), and by the degradation products of thiamine (Biamonte & Schneller, 1951; DeRitter, 1982). It is possible that ascorbic acid may actually protect folic acid against degradation in the presence of light but the authors did not provide details regarding the illumination of the sample during the study (Liang, Zhao, & Hao, 2013).
Folic acid reacts with reducing sugars via nonenzymatic glycation (aka. Maillard‐type reactions), predominantly forming N2‐[1‐(carboxyethyl)] folic acid (20). The degradation occurs quickly at temperatures above 70°C but the same products are observed in reactions at 37°C after 12 weeks. It was hypothesized that 20 is formed by the condensation of folic acid and the known sugar degradation product, dihydroxyacetone (M. Schneider et al., 2002).
3.2.3. Stabilization
Folic acid degradation is primarily light‐induced, so the easiest recommendation for inhibiting this process remains to store solutions containing folic acid protected from light. As oxygen effects the degradation of folic acid, scavenging oxygen with antioxidants has been found to stabilize folic acid solutions. Such antioxidants include phenolic compounds like butylated hydroxyanisole, nordihydroguaiaretic acid, and ethyl hydrocaffeate (Tansey & Schneller, 1955), salts of EDTA (Weidenheimer & Carstensen, 1954) and ascorbic acid (Liang et al., 2013).
Many stability studies also focus on adding chemicals to solutions to enhance the solubility of folic acid at low pH values. The protein lactoferrin was patented for its ability to complex folic acid, and thus stabilize it in acidic media for application in food, drinks, or medicines (Uchida, Suguri, & Harjinder, 2002). It was found to result in solutions with much higher concentrations of folic acid at low pHs (one protein molecule was able to complex up to 200 folic acid molecules) and also minimized the photodegradation of folic acid. Stabilization of folic acid in fruit juices was achieved via encapsulation with polyamine‐gated mesoporous silica particles (Ruiz‐Rico et al., 2017). This encapsulation inhibited folic acid precipitation at low pH values but the folic acid was released into solution at neutral pH values, making this method unfeasible for application in neutral CCM. In addition, the folic acid‐containing particles were >0.8 µm in size, so this method would also be unfeasible for cell culture applications due to the folic acid‐containing particles being filtered out of solution during the sterilization step (CCM is typically sterilized via filtration through 0.2 µm sterilization filters before use in culture). Small molecules shown to provide higher folic acid concentrations at lower pH values include nicotinamide (Taub & Lieberman, 1953) and higher viscosity liquids, such as syrups and propylene glycol, though the percentage of these liquid additives (usually >20%), while fine in pharmaceutical preparations, are generally too high to be considered applicable for CCM (Biamonte & Schneller, 1951).
Replacement of folic acid with the reduced folate form is not as beneficial as the fully oxidized folic acid is considered the most stable form. Photostability of folates might be achieved by chemical modifications on the glutamate residue. It was shown, for example, that the monosubstituted derivative 6‐deoxy‐6‐[(1‐(2‐amino)ethylamino)folate]‐β‐cyclodextrin (CDEnFA) is more photostable than free folic acid (Blakley, 1969; Gregory, 1997; Tofzikovskaya, O’Connor, & McNamara, 2012; X. Wang & Fenech, 2003).
4. CYANOCOBALAMIN
4.1. Function
Cyanocobalamin (vitamin B12) (21) (Figure 5) is a coenzyme, playing a key role in propionate, AA, and single‐carbon metabolism (Allen & Prentice, 2012; Combs, 2012). Like most vitamins, cyanocobalamin is converted between multiple active forms intracellularly. It does not naturally (i.e., in cells) exist in the cyano form but rather gains the cyano group only during the process of isolating the vitamin from cells (Reddy, 1999). The interconvertible forms can generically be classified as cobalamins, i.e. cobalt‐centered coordination complexes, with a single 5‐coordinate ligand consisting of a corrin ring with a 5,6‐dimethylbenzimidazole ribonucleotide moiety tethered to the corrin via an aminopropyl group. In humans, the two functional coenzyme forms of cobalamin are methylcobalamin (22), used to methylate substrates (e.g., the conversion of homocysteine to methionine) and 5′‐deoxy‐5′‐adenosylcobalamin (23), used to isomerize chiral substrates (e.g., methylmalonyl CoA to succinyl‐CoA) (Allen & Prentice, 2012). A vitamin B12 analog, hydroxocobalamin (24), is often described as a degradation product of cyanocobalamin but is actually still biologically active as the hydroxy ligand can be substituted for the biologically functional ligands in vivo (Ahmad, Hafeez, Akther, Vaid, & Qadeer, 2012; Ansari, Vaid, & Ahmad, 2004; Jägerstad & Arkbåge, 2003). In some enzymes, the cobalamin is bound to the protein by replacement of the α axial ligand with a histidine residue in the enzyme (25) (Figure 5). The benzimidazole tether assists this binding (Banerjee & Ragsdale, 2003).
Figure 5.
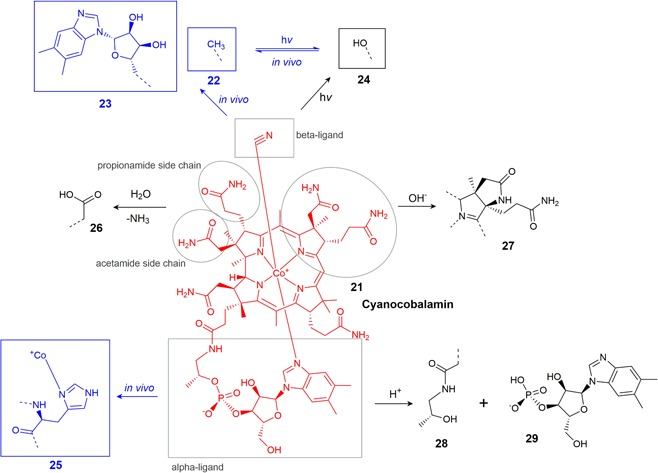
Overview of known cyanocobalamin degradation products and some in vivo alterations [Color figure can be viewed at wileyonlinelibrary.com]
4.2. Stability, reactivity, degradation products
4.2.1. Impact of external factors
Taking into account all degradation mechanisms (oxidative, photolytic, thermal, etc.) cyanocobalamin is the most stable of the cobalamin derivatives. Any cobalamin with a carbon‐linked β‐ligand (e.g., methyl and cyano) will be quickly photolyzed, in which the β‐ligand is cleaved, reducing Co(III) to Co(II), forming vitamin B12r. In the presence of oxygen, the cobalt center is reoxidized and the β‐ligand becomes a hydroxy group, yielding hydroxocobalamin (24) (Hogenkamp, Barker, & Mason, 1963; Kirschbaum, 1981; Reddy, 1999). UV light will degrade hydroxocobalamin further, but this subsequent degradation proceeds much more slowly than the initial conversion of cyanocobalamin or methylcobalamin to hydroxocobalamin. Additionally, this conversion to the hydroxo form is much faster for methylcobalamin or adenosylcobalamin than cyanocobalamin (Juzeniene & Nizauskaite, 2013).
The cobalt center in the cobalamins is very tightly bound by the ligand and thus cannot be chemically substituted for other metals (Bieganowski & Friedrich, 1981; Z. Schneider & Stroiński, 1987; Toohey, 1965).
Hydrolysis of the propionamide and acetamide side chains to carboxylic acid groups (26) (Figure 5) can occur under acidic or alkaline conditions, yielding biologically inactive products (Bonnett, 1963; Bonnett et al., 1957). At basic pH and elevated temperatures, lactam formation by cyclization of the acetamide side chain of the B ring produces biologically inactive dehydrovitamin B12 (27) (Bonnett et al., 1957; Z. Schneider & Stroiński, 1987); (Figure 5). In highly acidic conditions, hydrolysis of the phosphate bond yields cobinamide 28 and the benzimidazole nucleotide moiety 29, which can break down further to multiple products (Bonnett, 1963; Z. Schneider & Stroiński, 1987).
4.2.2. Interaction with other CCM components
The most well‐characterized incompatibility of cyanocobalamin is with ascorbic acid. A one‐electron reduction by ascorbate produces vitamin B12r, which then yields hydroxocobalamin via oxidation by air (Gakenheimer & Feller, 1949; Macek, 1960; Reddy, 1999; Trenner, Buhs, Bacher, & Gakenheimer, 1950). Degradation by ascorbate proceeds further, and it was shown that ascorbate‐mediated degradation of hydroxocobalamin occurs at a rate about 10x that of cyanocobalamin (Ahmad et al., 2014). This ascorbate‐mediated degradation is enhanced in the presence of Cu2+ salts, and cyanocobalamin can also be degraded by the reduced form of ascorbic acid and dehydroascorbic acid (Bartilucci & Poss, 1954; Rosenberg, 1956). Other reducing agents, such as sodium bisulfite, reducing sugars, or ferrous salts may have similar destructive effects on cyanocobalamin (Macek, 1960). The photolytic degradation of other cobalamins to hydroxocobalamin is mildly enhanced by nicotinamide (Ahmad, Ansari, & Ismail, 2003) and riboflavin (Iqbal Ahmad et al., 2012), and riboflavin also further enhances the photolytic degradation of hydroxocobalamin (Juzeniene & Nizauskaite, 2013). The hydroxy β‐ligand of hydroxocobalamin may be readily substituted by thiolates (e.g., Cys and glutathione [GSH]) and imidazoles (e.g., His) (Hannibal et al., 2007; Pezacka, Green, & Jacobsen, 1990; Pratt, 1964).
Cyanocobalamin/thiamine solutions are stable at low pH values (~3–4) but in conditions that degrade thiamine (higher pH, temperature, Section 4.2.1) the degradation products of thiamine (mostly thiazole products) induce the degradation of cyanocobalamin (Feller & Macek, 1955). This effect is mostly significant with solutions at high concentrations or high thiamine‐cyanocobalamin ratios (Macek, 1960) but may also be enhanced in the presence of nicotinamide (Blitz, Eigen, & Gunsberg, 1956) or pyridoxine (Monajjemzadeh, Ebrahimi, Zakeri‐Milani, & Valizadeh, 2014). These effects are only really observed at higher temperatures or upon exposure to light. In prolonged storage at room temperature, mixtures of B vitamins do not cause any significant degradation of cyanocobalamin (Feller & Macek, 1955).
4.3. Stabilization
The optimum pH range for prolonged storage of cyanocobalamin in solution is 4–7 (Macek, 1960; Monajjemzadeh et al., 2014). Most stabilization methods focus on methods to reduce the formation of hydroxocobalamin, produced by both oxidizing agents and exposure to light. Although there are many suggested additives that can stabilize cyanocobalamin, (see Macek, 1960; Kirschbaum, 1981 for comprehensive lists), the most recognized are ferric salts (e.g., FeCl3 and saccharated iron oxide) (Macek, 1960; Newmark, 1958; Skeggs, 1952), phosphate buffer (Monajjemzadeh et al., 2014), and potassium ferrocyanide (Zuck & Conine, 1963). A considerably novel method for minimizing the cyano‐to‐hydroxocobalamin conversion is the application of red light filters in the working laboratory, to filter out the lower wavelengths of light (<600 nm) that contribute most to this reaction (Du et al., 2018). This was shown to be especially relevant to the biomanufacturing industry as it was demonstrated that the B12 cobalamins can be incorporated into therapeutic mAbs as a result of thiolates in the protein reacting with hydroxocobalamin, forming a sulfide‐cobalt linkage, which is analogous to in vivo ligand‐exchange processes. Du et al. showed that mAbs were tainted pink by this cobalamin incorporation and that the red light was able to minimize this incorporation to nearly the same extent as the complete absence of light. The authors suggested that installing red light filters in the laboratory for the entire biomanufacturing process (from storage through cell culture) would be able to minimize this product contamination by inhibiting light‐induced cyanocobalamin‐to‐hydroxocobalamin degradation.
5. THIAMINE
5.1. Function
Thiamine (vitamin B1) functions as a coenzyme in various decarboxylation reactions including processes in energy metabolism, such as the conversion of pyruvate to acetyl‐coenzyme A or α‐ketoglutarate to succinyl‐coenzyme A, and the metabolism of glucose to form pentoses (Begley, 1996; Bühler, 1988; Combs, 2012; Lonsdale, 2006). The vitamin consists of a pyrimidine ring and a thiazolium ring linked by a methylene bridge, and upon being transported into the cell, it is phosphorylated at the primary hydroxyl group to the functional coenzyme form, thiamine pyrophosphate (aka. cocarboxylase) (Lonsdale, 2006). The vitamin is a zwitterion in its phosphate form but as a food additive or CCM component exists as a cation accompanied by a counter anion, usually chloride because of its high solubility compared with other derivatives (Bühler, 1988), but nitrate is also common.
5.2. Stability, reactivity, degradation products
5.2.1. Impact of external factors
Thiamine (30) (Figure 6) is sensitive to high temperatures, oxidation, reduction, and light and the mechanisms and rates of degradation are highly dependent on pH (Bühler, 1988; Dwivedi & Arnold, 1973). Thiamine HCl is the most widely studied form, and a pure solution of the salt exists at a pH of ~3–4 (thiamine HCl is technically thiamine chloride hydrochloride, a protonated doubly charged thiamine with two chloride counterions) (Al‐Rashood, Al‐Shammary, & Mian, 1990). Most conditions that degrade thiamine often result in multiple products and descriptions of the degradation of aqueous thiamine solutions often describe a color change from clear to yellow and the formation of precipitates, though these precipitates are rarely characterized (Bray, 1948; Kuijvenhoven & Westerterp, 1952) with a clear exception of the reported precipitation of the oxidation product, thiochrome (31) (Gambier & Rahn, 1957). Thiamine is often described as being both most stable and most soluble in mildly acidic conditions, and while compound degradation is indeed slower in acidic solutions (K. T. Farrer, 1941; Mulley, Stumbo, & Hunting, 1975), there appears to be no concrete data on thiamine solubility in differing pH environments. However, as thiamine HCl has a solubility of ~1,000 g/L as a pure solution in water (Al‐Rashood et al., 1990), it would be reasonable to assume that changing pH will not result in precipitation of thiamine (especially at the much lower concentrations that are usually applied in vitamin preparations or CCM) but rather that any precipitation observed is that of degradation products.
Figure 6.
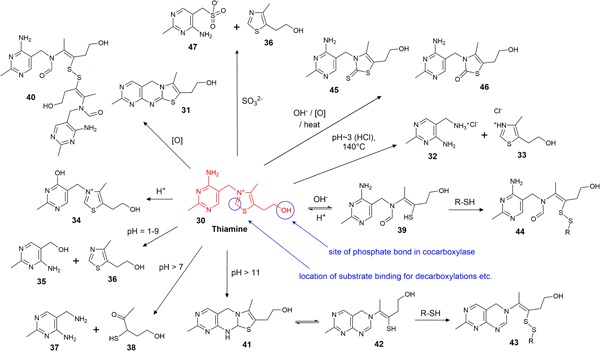
Overview of the major known thiamine degradation processes [Color figure can be viewed at wileyonlinelibrary.com]
Thiamine HCl solutions can be briefly boiled or heat‐sterilized at the pH generated in pure solutions (i.e., pH ~3–4) but heating at 140°C cleaves the methylene bridge to form pyrimidine 32 and thiazole 33 (Figure 6) (Watanabe, 1939). Thiamine HCl will degrade over time at these high temperatures and will degrade faster at higher pH levels (Beadle, Greenwood, & Kraybill, 1943). The rate of degradation is accelerated by buffers in the order: borate > unbuffered > acetate > phosphate (Beadle et al., 1943). In strongly acidic solutions (pH < 1) oxythiamine (34) is formed, however, in mildly acidic and neutral solutions the major degradation products are 5‐hydroxymethylpyrimidine 35 and sulfurol (36) (Windheuser & Higuchi, 1962), or 5‐aminomethylpyrimidine 37 and α‐aceto‐γ‐mercaptopropanol (38) (Matsukawa, Iwatsu, & Yuguri, 1951) (Figure 6). Upon raising the pH, an acid‐base reaction induces a reversible cleavage of the thiazolium ring, yielding thiol 39. Dimerization of this thiol via formation of a disulfide bond yields disulfide 40, which is still biologically active (Macek, 1960). In extreme alkaline conditions (pH > 11) deprotonation of the primary amine affords a yellow compound, identified as tricyclic structure 41, and subsequent ring opening of the dihydrothiazole produces thiol 42 (Dwivedi & Arnold, 1973; Maier & Metzler, 1957). Both thiol products 39 and 42 can react with other free thiols in solution (including themselves) under oxidative conditions, producing disulfide products 43 and 44 (Kawasaki, 1964; Matsukawa, Yurugi, & Oka, 1962). Combinations of destructive conditions (i.e., high pH, oxidation, and heat) can lead to further degradation products, such as thiothiamine (45) or oxothiamine (46) (Kawasaki, 1964; Matsukawa et al., 1962; Zima & Williams, 1940).
The primary products of thiamine oxidation are thiochrome (31) and thiamine disulfide (40) (Kawasaki, 1964; Kinnersley, Brien, & Peters, 1935). The photolysis of thiamine in aqueous solution yields many of the same products that arise from hydrolysis or oxidation (40, 31, 37, 38) and the product distribution during photolysis is dependent on the light source (Vaid, 1997).
5.2.2. Interaction with other CCM components
Thiamine decomposition is enhanced in the presence of a multitude of bioactive compounds, and the reader is advised to refer to more comprehensive reviews for details on all of these interactions (Dwivedi & Arnold, 1973; K. T. H. Farrer, 1955; Macek, 1960; Vaid, 1997).
The most relevant interactions for CCM are the enhanced degradation by sulfites to pyrimidylmethane sulfonic acid 47 and thiazole 36 (Dwivedi & Arnold, 1973), oxidation by riboflavin to thiochrome (31) (Gambier & Rahn, 1957), and there is some suggestion of enhanced degradation through the reductive activity of ascorbic acid (Gambier & Rahn, 1957). Sulfur‐based oxidants, such as cystine, enhance the degradation of thiamine, whereas some sulfur‐based reductants, such as cysteine or other thiols, stabilize thiamine against degradation (Uprety, R., & Rao, 1961; Windheuser & Higuchi, 1963).
The precise effects of particular additives on thiamine stability seem to be heavily dependent on other conditions, such as temperature, pH, the presence of buffers, and access to light and oxygen. For example, although enhanced degradation by nicotinamide was observed in refluxing solutions at pH 6 (McIntire & Frost, 1944; Windheuser & Higuchi, 1962), no enhancement of degradation was seen in solutions with nicotinamide at pH 4 at room temperature (Feller & Macek, 1955). Also, cupric salts were claimed to enhance thermal degradation (Booth, 1943), but this was shown to only be reliable in phosphate buffers ‐ in other buffers cupric salts retarded the reaction (K. T. H. Farrer, 1947). Similarly, iron, zinc, and nickel salts had no effect in phosphate buffer but had variable effects on reaction rate in other buffers (Booth, 1943; K. T. K. Farrer, 1947). And yet another study demonstrated that over the course of almost a year, cupric, ferric, and ferrous salts all enhanced degradation at room temperature at pH 5 in unbuffered samples (Dutta, Mehta, & Narayanan, 1952).
Thiamine also reacts with ketoacids as in the enzymatic reactions, at the nucleophilic C2 in the thiazolium ring and the carbonyl carbon in the keto acid, in which it can effect decarboxylation, again, analogous to the enzymatic process. These reactions were carried out at pH 8 and at 40°C, forming adducts that were shown to retain the biological function of thiamine, perhaps suggesting this bond can be broken in vivo (Miller, Sprague, & Krampitz, 1962). Reducing sugars and other aldehydes can also react with thiamine, either as with the ketoacids at C2 or forming a Schiff base with the amine group on the pyrimidine moiety, but these reactions usually occur at higher temperatures and often degrade thiamine (Lee, 1988; Mizuhara & Handler, 1954).
5.3. Stabilization
In general, as thiamine degradation pathways are highly dependent on the storage conditions (i.e., temperature, pH, and buffer), a multitude of stabilization methods have been described in the literature.
Thiamine mononitrate was described as significantly more stable than thiamine hydrochloride in both solid and solution but is 10‐fold less water soluble, therefore, it is often used in a solid form for nutrition purposes and rarely in solution (Bühler, 1988; Coelho, 2002). In oral liquid preparations, the stability of thiamine is often enhanced by drastically altering the vehicle, that is, the solvent. Such vehicles successfully employed include propylene glycol, glycerol, propyl gallate, sucrose syrup, sorbitol, and combinations of these, but in these situations, such vehicles carry out this stabilizing function at concentrations that are not feasible for CCM (usually >20%) (Vaid, 1997).
Other small molecules are frequently added to solutions of thiamine to increase its stability. The most common are free thiols. Various disulfides, which are formed from the ring‐opened thiamine generated in alkaline conditions (Figure 6, the conversion of 39 to 44) were shown to be reduced in cell culture back to bioactive thiamine. This suggests that free thiols in thiamine solutions could be stabilizing thiamine by trapping it as a disulfide in this ring‐opened form. This already oxidized form appears inert to further oxidation and more stable to changes in pH than thiamine (Fujiwara & Watanabe, 1952; Mohberg & Johnson, 1963). The thiols could also be functioning as antioxidants, effectively mopping up dissolved oxygen. Thiamine could be substituted directly with a disulfide or similarly reducible thiol esters and thiol carbonates for this purpose. Some of these disulfides are described as passively absorbed by cells due to their increased lipophilicity (Le Huerou et al., 2008).
It was shown that various organic compounds (e.g., sodium glutamate, glycine, serine, disodium EDTA) stabilized solutions of thiamine in seawater and it was suggested that they function by chelating trace metals that are promoting the degradation (Gold, 1968). Some compounds (e.g., thioglycolic acid, thio‐malic acid, methionine, thiourea, and asparagine) have been patented as stabilizers for their ability to inhibit or delay the formation of an unidentified precipitate in solutions of pure thiamine (pH 2–3.5) and in solutions of thiamine with other B vitamins (pH 3–5) (Kuijvenhoven & Westerterp, 1952). Some noteworthy thiol‐containing compounds that have been patented for stabilizing thiamine solutions are thioglycerol, thiosorbitol, and thioglucose (Bray, 1948). As there are several functional groups in these compounds, they may be achieving the stabilization effect via oxygen‐scavenging, disulfide formation, metal chelation, or any combination of the above.
Regarding encapsulation methodologies, the interaction of thiamine HCl with cyclodextrins leads to the formation of 1:1 complexes but the complexes proved to be chemically unstable and less soluble (Im, Lee, Park, & Kim, 1983).
6. VITAMIN B6
6.1. Function
Vitamin B6 comprises a group of six‐related compounds: pyridoxine (48), pyridoxal (49), pyridoxamine (50) (Figure 7), and their respective 5′‐phosphate derivatives (Urbanski, 1949). At least one form is essentially required in CCM (Eagle, 1955; Vijayasankaran et al., 2010). These six compounds are all referred to as vitamers of vitamin B6, that is, they all have very different chemical functionalities but the same physiological activity as they are interconvertible in cellular systems. Redox‐active enzymes effect the conversion between the amine‐alcohol‐aldehyde functionalities and kinases phosphorylate these to the enzymatically active phosphate forms (Depeint, Bruce, Shangari, Mehta, & O'Brien, 2006; Snell, 1958). Vitamin B6 participates in a plethora of biochemical reactions including fatty acid and folate metabolism, coenzyme Q synthesis, gluconeogenesis, and heme biosynthesis, as well as AA racemization, transamination, and elimination. The mode of action for all of these biochemical reactions is based on the formation of a Schiff base between the vitamin aldehyde functionality and a primary amine substrate, in which AAs are the most common reactions partners (Metzler, Ikawa, & Snell, 1954; Snell, 1963). In CCM, the most common variant of B6 used is pyridoxine HCl because of its comparatively good stability to external factors, low chemical reactivity, and high solubility compared with the other B6 variants (Bühler, 1988).
Figure 7.
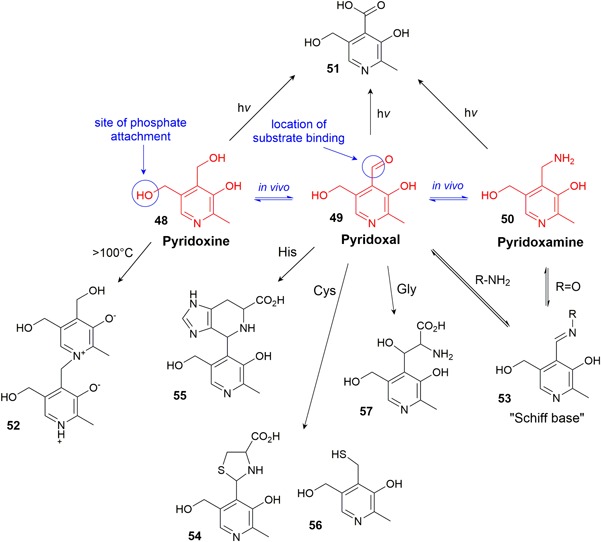
Vitamin B6 forms and their known degradation and interaction products [Color figure can be viewed at wileyonlinelibrary.com]
6.2. Stability, reactivity, degradation products
6.2.1. Impact of external factors
All forms of vitamin B6 are sensitive towards light (Hochberg, Melnick, & Oser, 1944; Shephard & Labadarios, 1986) where 4‐pyridoxic acid (51) (Figure 7) is identified as the main photoproduct (Reiber, 1972). High heat initiates the dimerization of pyridoxine to 52 and subsequent loss of activity, and prolonged heating could induce polymerization in the same way that dimerization occurs (Harris, 1941; Metzler & Snell, 1955). The phosphate forms are not more stable as the phosphate group is easily cleaved by hydrolysis (Shephard & Labadarios, 1986).
6.2.2. Interaction with other CCM components
Pyridoxal is widely known for its reactivity with primary amines, such as AAs. It readily forms a Schiff base (53) and in this form catalyzes the degradation of the amine, itself reforming pyridoxal or being converted to pyridoxamine following further reactions. The reversible Schiff base formation is enhanced by heat, some metal ions (e.g., Fe3+, Cu2+, and Al3+), and phosphate ions (Heyl, Harris, & Folkers, 1948; Metzler, Ikawa, et al., 1954; Snell, 1963). The formation of the Schiff base is more efficient in pyridoxal phosphate than pyridoxal (Heyl et al., 1948). The most common reactions following Schiff base formation are: (a) transaminations, converting AAs to α‐ketoacids and the vitamin to pyridoxamine; (b) racemizations at the α‐carbon; and (c) eliminations, such as deamination, decarboxylation, or removal of heteroatoms in side chains (such as serine or cysteine) (Metzler, Ikawa, et al., 1954; Metzler, Longenecker, & Snell, 1954; Snell, 1963).
Pyridoxal also reacts reversibly to form cyclic structures, such as thiazolidine 54 from cysteine or the nitrogen‐containing bicyclic 55 from histidine (Heyl et al., 1948; Schonbeck, Skalski, & Shafer, 1975) (Figure 7). At higher temperatures, an irreversible reaction between pyridoxal and cysteine can form mercaptopyridoxine derivative 56, which can, like most thiols, form a disulfide dimer (Wendt & Bernhart, 1960).
Glycine reaction with two pyridoxal molecules, catalyzed by reaction with Fe3+ or Al3+ at 100°C forms a chelate that can dissociate into β‐pyridoxylserine (57) and pyridoxal upon boiling with a strong acid (Metzler, Longenecker, et al., 1954).
6.2.3. Stabilization
The first approach to stabilize vitamin B6 was initiated with the predominant application of the stable pyridoxine instead of the reactive pyridoxal form in early CCM, such as Dulbecco’s modified Eagle’s medium (Dulbecco, Vogt, & February, 1954). Another benefit of pyridoxine includes its antioxidative properties that may contribute in stabilizing other critical CCM components (Depeint et al., 2006; Jain & Lim, 2001).
As most unwanted vitamin B6 reactions are enhanced by metal ions (Metzler, Ikawa, et al., 1954; Metzler, Longenecker, et al., 1954; Snell, 1963), the efficient chelation of metals in CCM would be a strategy to inhibit degradation of both the vitamin and CCM compounds in general.
7. STABLE VITAMINS: BIOTIN, PANTOTHENIC ACID, and NICOTINAMIDE
7.1. Function
Biotin (vitamin B7) functions directly as a coenzyme, whereas pantothenic acid (vitamin B5) and nicotinamide/nicotinic acid (vitamin B9) are coenzyme precursors.
Biotin (58) (Figure 8) is utilized primarily in bicarbonate‐dependent carboxylation reactions, thus playing a key role in metabolic processes, such as the citric acid cycle, fatty acid synthesis, and the catabolism of branched‐chain AAs (Medicine, 1998). As a coenzyme, biotin is covalently linked via an amide bond to the primary amine in lysine residues in the enzyme (Combs, 2012).
Figure 8.
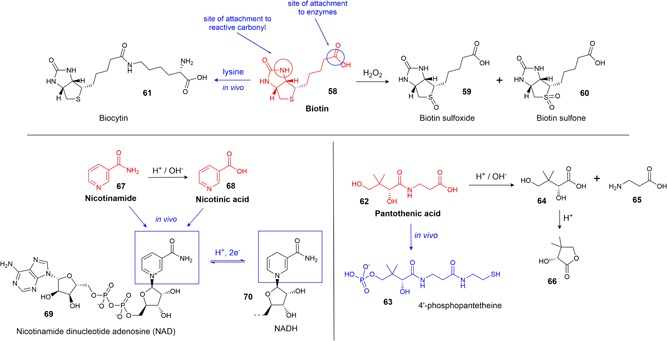
Some in vivo transformations and degradation products of biotin, pantothenic acid, and nicotinamide [Color figure can be viewed at wileyonlinelibrary.com]
Pantothenic acid (62) (Figure 8) is a constituent of 4′‐phosphopantetheine (63) and coenzyme A, which both act as carriers of acyl groups and activators of carbonyl groups in many metabolic processes. Both of these compounds function via the formation of a thioester bond between the terminal thiol group and carboxylic acids in the metabolic substrate, which then activates the carboxyl group for further reactions (Combs, 2012; Medicine, 1998).
Nicotinamide (67) and nicotinic acid (aka. niacin) (68) (Figure 8) are vitamers of B9 (i.e., intracellularly interconvertible, but containing completely different chemical functional groups), and both forms are converted in vivo into the coenzymes nicotinamide adenine dinucleotide (NAD) (69) and nicotinamide adenine dinucleotide phosphate (NADP). These coenzymes participate in important redox reactions, where the transfer of a hydride converts them between these oxidized forms and their reduced forms, NADH (70) and NADPH. The NAD/NADH redox pair are found in many reductases and dehydrogenases, which are central parts of many enzymatic processes. Among these processes are the important energy producing cycles, such as glycolysis, the electron transport chain and the citric acid cycle, and anabolic reactions, such as in fatty acid and steroid syntheses (Aguilera‐Méndez, Fernández‐Lainez, Ibarra‐González, & Fernandez‐Mejia, 2012; Combs, 2012).
7.2. Stability, reactivity, degradation products
7.2.1. Impact of external factors
Biotin and nicotinamide have good stability and are unlikely to be degraded in the conditions that CCM is usually exposed to. Biotin can be inactivated in very strong acid or alkaline conditions at high temperatures and upon exposure to ultraviolet radiation, although the mechanism of degradation and subsequent degradation products have not been determined (Brown & Du Vigneaud, 1941; Bühler, 1988; Charlton & Ewing, 2007). The only known degradation products are from oxidation by hydrogen peroxide or peroxide radicals in which the sulfur atom of biotin may be oxidized to produce biotin sulfoxide (59) or biotin sulfone (60) (Hofmann, Melville, & du Vigneaud, 1941; Melville, 1954). Biotin is stable to oxygen (Brown & Du Vigneaud, 1941; Combs, 2012; O’Neil, 2006).
Nicotinamide will be hydrolyzed in acidic or basic solutions (pH < 4, pH > 8) to the B9 vitamer, nicotinic acid, but as nicotinic acid is equally soluble and does not have any further reactivity, this has no effect on the bioavailability of the vitamin (Bühler, 1988). Both amide and acid forms are stable to oxygen and light but have been reported as being unstable at high temperatures, especially in alkaline conditions and in the presence of metal ions (Charlton & Ewing, 2007; Muhamad, Yusoff, & Gimbun, 2015). However, as with biotin, there are no reports on the mechanism of this degradation or subsequent degradation products. In pharmaceutical formulations, nicotinamide is often preferred, as nicotinic acid has a side effect of producing cutaneous flushing (Bodor & Offermanns, 2008) though the forms should be equivalent as CCM additives.
Pure pantothenic acid is a viscous, hygroscopic, chemically‐unstable oil, so in CCM the vitamin is commonly added as calcium pantothenate, a soluble and stable solid. The calcium salt is preferred to the sodium salt, as solid forms of the sodium salt are much more hygroscopic (O’Neil, 2006). Calcium pantothenate is stable to light and oxygen but is unstable to both acid and alkali (Caşcaval, Kloetzer, Blaga & Galaction, 2017; Charlton & Ewing, 2007). Few conclusive studies have been done on the heat stability of solutions of calcium pantothenate at neutral pH. Frost showed that a solution at pH 5.5–7 was stable for 20 days at 60°C (D. Frost, 1943) and that solid calcium pantothenate degrades upon heating only when some water is present. Heat degradation was avoided when the solid was mixed with a desiccant (D. Frost, 1943).
While stable at neutral pH, at either acidic or alkaline pH values, calcium pantothenate is hydrolyzed to pantoic acid (64) and β‐alanine (65). In acidic solutions, pantoic acid can react further, losing water to form pantolactone (66) (D. Frost, 1943). In general, pantothenic acid is more stable under slightly alkaline conditions when compared with acidic conditions (Coelho, 2002) and increasing temperature in the presence of acid or base will accelerate the degradation (D. Frost, 1943; D. V. Frost & McIntire, 1944).
7.2.2. Interaction with other CCM components
Owing to their general stability and minimal functional groups, biotin, pantothenate, and nicotinamide have not as yet been shown to display any significant interactions with other CCM components. Although biotin reacts with carbonyl compounds in enzymatic reactions, these reactions do not occur between compounds in solution (Brown & Du Vigneaud, 1941).
Phosphate buffers (and electrolytes in general) enhance the rate of destruction of pantothenate at all pH values but the effect is more pronounced in pH values closer to where pantothenate would otherwise be most stable (pH 5–7) (D. Frost, 1943). Nicotinamide was shown to stabilize pantothenate at pH 7–9 but to enhance degradation at pH 4–5 (D. Frost, 1943).
7.3. Stabilization
As mentioned earlier, these compounds are already highly stable and to our knowledge, no specific stabilization methods have been developed for these three vitamins.
A biotin analog, biocytin (61) (Figure 8), is a naturally occurring compound formed from the interaction of biotin and lysine. It is a primary source of this essential vitamin for mammals and can be used as a precursor via cleavage by the enzyme biotinidase (Hymes & Wolf, 1996).
8. CONCLUSION
In recent years chemically defined CCM have superseded undefined media containing serum or hydrolysates to a substantial extent, thus contributing significantly to increased lot‐to‐lot consistency. However, with the removal of stabilizing serum proteins, unstable vitamins are more prone to react and degrade within the CCM. Key factors that promote vitamin degradation include basic or acidic pH, oxidation, (UV) light, heat or reactivity in the presence of selected reaction partners (see a summary of these interactions in Table 1).
Mixtures of the B vitamins are considerably more stable in the solid state and as such it is preferable to buy CCM in powdered form and solubilize immediately before using. However, some CCM formulations may not be available in powder form, and on a manufacturing scale solubilizing immediately before each addition timepoint may be unfeasible for an efficient workflow. In liquid form, considerable degradation can be mitigated by storing CCM at neutral pH, in the dark, and at 2–8°C. These conditions minimize the most common degradation pathways brought about by acid/base‐catalyzed reactions and photooxidation/UV light‐initiated decomposition reactions and thermal degradation, respectively, and is indeed already common practice in most laboratories. For bulk manufacturing, these storage conditions are not always practical and enhanced mitigation strategies may need to be implemented to inhibit those reactions that result in inconsistencies in the biomanufacturing of therapeutic antibodies.
More specific degradation avoidance strategies, which can easily be implemented, include the use of more stable forms of particular vitamins (e.g., pyridoxine in place of pyridoxal and calcium pantothenate for B5) and strategies to limit the reactivity of negative interaction partners in the solution. Most notably, efficient metal chelation could significantly reduce the degradation of many B vitamins induced by the catalytic activity of the transition metals. For vitamins prone to oxidative decomposition (e.g., folic acid and pyridoxine), new antioxidative molecules, such as thiazolidines, can tremendously inhibit these degradation pathways (Kuschelewski, Schnellbaecher, Pering, Wehsling, & Zimmer, 2017). For highly unstable vitamins, encapsulation approaches arise as suitable strategies as they can protect the vitamin from the reactive CCM milieu for some time. Here, especially the encapsulation of vitamins such as riboflavin within the hydrophobic cavity of cyclodextrin molecules or the application of nanoemulsions could prove valuable.
While this review has outlined stabilization strategies pertaining to the B vitamins individually, applying such strategies is not necessarily straightforward. For CCM, as in many pharmaceutical preparations, the B vitamins are usually all present together, and strategies that stabilize one vitamin might cause problems for another. Finding a set of conditions that minimizes reactions across the entire panel of CCM components is a careful balancing act. Such an example of a balancing act was shown for determining an optimal pH for storing a mixture of thiamine and pantothenate—the former decreasing in stability with increasing pH and the latter exhibiting the reverse trend. The study determined an optimal balancing point at pH 4.6, which resulted in an equal degradation of ~75% for both vitamins (D. V. Frost & McIntire, 1944). Gambier & Rahn (1957) examined the optimal pH and vitamin ratios to obtain maximal stability for several mixtures of B vitamins. Important considerations that studies like this bring to light are the chain effects that can occur when multiple compounds are combined in a single set of conditions, aspects that are not necessarily clearly conveyed when looking at data from more simple mixtures. For example, thiamine/cyanocobalamin mixtures are stable at lower pH values, however, when riboflavin or nicotinamide are included, this degrades thiamine, whose degradation products now subsequently degrade cyanocobalamin (Gambier & Rahn, 1957). These chain reactions can be minimized by choice of component ratios in CCM. Although many researchers have investigated optimizing formulation pH to minimize degradation, this strategy applies more to food, drinks, and pharmaceuticals than it does to biotechnology. The storage pH for liquid CCM is dictated more by practicalities than by a desire to enhance formulation stability—CCM is stored at neutral pH so that it can be used in culture immediately as needed. This same balancing act could potentially be applied to choosing other parameters, for example, buffer, transition metal chelator, wavelength of light for storage and cell culture environments, or any change to the redox potential of the solution.
The inclusion of any new compound to stabilize a particular CCM component is fraught with the danger of creating new reactivities, so the addition of complexation agents, chelation agents, or antioxidants would need to be approached with an eye to side reactivities. In addition, the biochemical reactivity of each CCM component needs to be maintained, so complexing agents or chelators cannot be so good at stabilizing that as they reduce the bioavailability of the biologically necessary CCM component, and any analog needs to be assessed for its ability to be transported into the cell and converted back to its physiologically active form.
Improved knowledge and ongoing research into vitamin instabilities and reactivities in CCM will surely contribute to increasing lot‐to‐lot consistencies or shelf lives and will thus further improve chemically defined CCM in the future.
CONFLICTS OF INTEREST
All authors are employees of Merck KGaA, Darmstadt, Germany.
Schnellbaecher A, Binder D, Bellmaine S, Zimmer A. Vitamins in cell culture media: Stability and stabilization strategies. Biotechnology and Bioengineering. 2019;116:1537–1555. 10.1002/bit.26942
References
REFERENCES
- Aguilera‐Méndez, A. , Fernández‐Lainez, C. , Ibarra‐González, I. , & Fernandez‐Mejia, C. (2012). Chapter 7: The chemistry and biochemistry of niacin (B3), B vitamins and folate: chemistry, analysis, function and effects (108–126). Cambridge: The Royal Society of Chemistry. [Google Scholar]
- Ahmad, I. , Ahmed, S. , Sheraz, M. A. , Aminuddin, M. , & Vaid, F. H. M. (2009). Effect of caffeine complexation on the photoylsis of riboflavin in aqueous solution: A kinetic study. Chemical and Pharmaceutical Bulletin, 57(12), 1363–1370. 10.1248/cpb.57.1363 [DOI] [PubMed] [Google Scholar]
- Ahmad, I. , Ahmed, S. , Sheraz, M. A. , Vaid, F. H. M. , & Ansari, I. A. (2010). Effect of divalent anions on photodegradation kinetics and pathways of riboflavin in aqueous solution. International Journal of Pharmaceutics, 390(2), 174–182. 10.1016/j.ijpharm.2010.01.042 [DOI] [PubMed] [Google Scholar]
- Ahmad, I. , Ansari, I. A. , & Ismail, T. (2003). Effect of nicotinamide on the photolysis of cyanocobalamin in aqueous solution. Journal of Pharmaceutical and Biomedical Analysis, 31(2), 369–374. 10.1016/S0731-7085(02)00337-0 [DOI] [PubMed] [Google Scholar]
- Ahmad, I. , Anwar, Z. , Ahmed, S. , Sheraz, M. A. , & Khattak, S. R. (2017). Metal ion mediated photolysis reactions of riboflavin: A kinetic study. Journal of Photochemistry and Photobiology, B: Biology, 173, 231–239. 10.1016/j.jphotobiol.2017.05.033 [DOI] [PubMed] [Google Scholar]
- Ahmad, I. , Anwar, Z. , Ali, S. A. , Hasan, K. A. , Sheraz, M. A. , & Ahmed, S. (2016). Ionic strength effects on the photodegradation reactions of riboflavin in aqueous solution. Journal of Photochemistry and Photobiology, B: Biology, 157, 113–119. 10.1016/j.jphotobiol.2016.02.010 [DOI] [PubMed] [Google Scholar]
- Ahmad, I. , Hafeez, A. , Akther, N. , Vaid, F. H. M. , & Qadeer, K. (2012). Effect of riboflavin on the photolysis of cyanocobalamin in aqueous solution. The Open Analytical Chemistry Journal, 6, 22–27. 10.2174/1874065001206010022 [DOI] [Google Scholar]
- Ahmad, I. , Qadeer, K. , Zahid, S. , Sheraz, M. A. , Ismail, T. , Hussain, W. , & Ansari, I. A. (2014). Effect of ascorbic acid on the degradation of cyanocobalamin and hydroxocobalamin in aqueous solution: A kinetic study. AAPS PharmSciTech, 15(5), 1324–1333. 10.1208/s12249-014-0160-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akberova, S. I. (2002). New biological properties of p‐aminobenzoic acid. Biology Bulletin of the Russian Academy of Sciences, 29(4), 390–393. 10.1023/a:1016871219882 [DOI] [Google Scholar]
- Akhtar, M. J. , Khan, M. A. , & Ahmad, I. (1999). Photodegradation of folic acid in aqueous solution. Journal of Pharmaceutical and Biomedical Analysis, 19(3‐4), 269–275. 10.1016/S0731-7085(98)00038-7 [DOI] [PubMed] [Google Scholar]
- Akhtar, M. J. , Khan, M. A. , & Ahmad, I. (2000). Effect of riboflavin on the photolysis of folic acid in aqueous solution. Journal of Pharmaceutical and Biomedical Analysis, 23(6), 1039–1044. 10.1016/s0731-7085(00)00383-6 [DOI] [PubMed] [Google Scholar]
- Allen, L. H. , & Prentice, A. (2012). Encyclopedia of human nutrition. San Diego: Academic Press. [Google Scholar]
- Al‐Rashood, K. A. M. , Al‐Shammary, F. J. , & Mian, N. A. A. (1990). Analytical profile of thiamine hydrochloride In Florey K., Al‐Badr A. A., Forcier G. A., Brittain H. G., & Grady L. T. (Eds.), Analytical profiles of drug substances (18, pp. 413–458). San Diego: Academic Press. [Google Scholar]
- Ansari, I. A. , Vaid, F. H. , & Ahmad, I. (2004). Chromatographic study of photolysis of aqueous cyanocobalamin solution in presence of vitamins B and C. Pakistan Journal of Pharmaceutical Sciences, 17(1), 19–24. https://doi.org/ncbi.nlm.nih.gov/pubmed/16414582 [PubMed] [Google Scholar]
- Asker, A. F. , & Habib, M. J. (2008). Effect of certain stabilizers on photobleacing of riboflavin solutions. Drug Development and Industrial Pharmacy, 16(1), 149–156. 10.3109/03639049009115991 [DOI] [Google Scholar]
- Baier, J. , Maisch, T. , Maier, M. , Engel, E. , Landthaler, M. , & Bäumler, W. (2006). Singlet oxygen generation by UVA light exposure of endogenous photosensitizers. Biophysical Journal, 91(4), 1452–1459. 10.1529/biophysj.106.082388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, L. B. , & Gregory, J. F., 3rd (1999). Folate metabolism and requirements. The Journal of Nutrition, 129(4), 779–782. 10.1093/jn/129.4.779 [DOI] [PubMed] [Google Scholar]
- Banerjee, R. , & Ragsdale, S. W. (2003). The many faces of vitamin B12: Catalysis by cobalamin‐dependent enzymes. Annual Review of Biochemistry, 72(1), 209–247. 10.1146/annurev.biochem.72.121801.161828 [DOI] [PubMed] [Google Scholar]
- Bartilucci, A. , & Foss, N. E. (1954). Cyanocobalamin (Vitamin B12) I. A study of the stability of cyanocobalamin and ascorbic acid in liquid formulations. Journal of the American Pharmaceutical Association, 43(3), 159–162. 10.1002/jps.3030430311 [DOI] [PubMed] [Google Scholar]
- Beadle, B. W. , Greenwood, D. A. , & Kraybill, H. R. (1943). Stability of thiamine to heat: I. Effect of pH and buffer salts in aqueous solutions. Journal of Biological Chemistry, 149(2), 339–347. https://doi.org/jbc.org/content/149/2/339.citation [Google Scholar]
- Begley, T. P. (1996). The biosynthesis and degradation of thiamin (vitamin B1). Natural Product Reports, 13(3), 177–185. 10.1039/NP9961300177 [DOI] [PubMed] [Google Scholar]
- Bender, D. A. (2003a). Do we really know vitamin and mineral requirements for infants and children?. Journal of the Royal Society for the Promotion of Health, 123(3), 154–158. https://doi.org/ncbi.nlm.nih.gov/pubmed/14526752 [DOI] [PubMed] [Google Scholar]
- Bender, D. A. (2003b). Vitamin B2—Riboflavin In Bender D. A. (Ed.), Nutritional biochemistry of the vitamins (2 ed, pp. 172–199). Cambridge: Cambridge University Press. [Google Scholar]
- Biamonte, A. R. , & Schneller, G. H. (1951). A study of folic acid stability in solutions of the B complex vitamins. Journal of the American Pharmaceutical Association, 40(7), 313–320. https://doi.org/ncbi.nlm.nih.gov/pubmed/14850354 [PubMed] [Google Scholar]
- Bieganowski, R. , & Friedrich, W. (1981). Über die Eisenanaloga von Cobalamin und Cobyrsäure [About the iron analogues of cobalamin and cobyric acid]. Zeitschrift für Naturforschung C, 36, 9–15. [Google Scholar]
- Blakley, R. L. (1969). The biochemistry of folic acid and related pteridines. Amsterdam: North‐Holland Publishing Company. [Google Scholar]
- Blitz, M. , Eigen, E. , & Gunsberg, E. (1956). Studies relating to the stability of vitamin B12 in B‐complex injectable solutions. Journal of the American Pharmaceutical Association (Scientific ed.), 45(12), 803–806. 10.1002/jps.3030451210 [DOI] [PubMed] [Google Scholar]
- Bodor, E. T. , & Offermanns, S. (2008). Nicotinic acid: An old drug with a promising future. British Journal of Pharmacology, 153(Suppl 1), S68–S75. 10.1038/sj.bjp.0707528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnett, R. (1963). The chemistry of the vitamin B12 group. Chemical Reviews, 63(6), 573–605. 10.1021/cr60226a002 [DOI] [Google Scholar]
- Bonnett, R. , Cannon, J. R. , Clark, V. M. , Johnson, A. W. , Parker, L. F. J. , Smith, E. L. , & Todd, A. (1957). Chemistry of the vitamin B12 group. Part V. The structure of the chromophoric grouping. Journal of the Chemical Society (Resumed), (0), 1158–1168. 10.1039/JR9570001158 [DOI] [Google Scholar]
- Booth, R. G. (1943). The thermal decomposition of aneurin and cocarboxylase at varying hydrogen ion concentrations. Biochemical Journal, 37(4), 518–522. 10.1042/bj0370518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, M. D. (1948). United States of America Patent No.
- Brown, G. B. , & Du Vigneaud, V. (1941). The effect of certain reagents on the activity of biotin. Journal of Biological Chemistry, 141, 85–89. [Google Scholar]
- Bühler, V. (1988). Vandemecum for vitamin formulations. Stuttgart: Wissenschaftliche Verlagsgesellschaft. [Google Scholar]
- Cardoso, D. R. , Libardi, S. H. , & Skibsted, L. H. (2012). Riboflavin as a photosensitizer. Effects on human health and food quality. Food & Function, 3(5), 487–502. 10.1039/c2fo10246c [DOI] [PubMed] [Google Scholar]
- Carr, D. O. (1960). Mechanism of riboflavin‐catalyzed oxidations. Iowa: Iowa State University. [Google Scholar]
- Casirola, D. , Kasai, S. , Gastaldi, G. , Ferrari, G. , & Matsui, K. (1994). Specificity of riboflavin molecular groups for riboflavin binding to rat small intestinal brush border membrane. Journal of Nutritional Science and Vitaminology (Tokyo), 40(4), 289–301. https://doi.org/ncbi.nlm.nih.gov/pubmed/7844636 [DOI] [PubMed] [Google Scholar]
- Caşcaval, D. , P., M. , Kloetzer, L. , Blaga, A.C. , Galaction, A.I. (2017). Chemical Stability of Vitamin B5. Paper presented at the International Conference on Advancements of Medicine and Health Care through Technology, Romania.
- Charlton, S. J. , & Ewing, W. N. (2007). The vitamins directory. Leicestershire: Context. [Google Scholar]
- Choe, E. , Huang, R. , & Min, D. B. (2005). Chemical reactions and stability of riboflavin in foods. Journal of Food Science, 70(1), R28–R36. 10.1111/j.1365-2621.2005.tb09055.x [DOI] [Google Scholar]
- Coelho, M. (2002). Vitamin stability in premixes and feeds: A practical approach in ruminant diets. Paper presented at the 13th Annual Florida Ruminant Nutrition Symposium, Florida, USA.
- Combs, G. F. (2012). The vitamins: Fundamental aspects in nutrition and health. San Diego: Elsevier Academic Press. [Google Scholar]
- De Brouwer, V. , Zhang, G. F. , Storozhenko, S. , Van der straeten, D. , & Lambert, W. E. (2007). pH stability of individual folates during critical sample preparation steps in prevision of the analysis of plant folates. Phytochemical Analysis, 18(6), 496–508. 10.1002/pca.1006 [DOI] [PubMed] [Google Scholar]
- Depeint, F. , Bruce, W. R. , Shangari, N. , Mehta, R. , & O’Brien, P. J. (2006). Mitochondrial function and toxicity: Role of the B vitamin family on mitochondrial energy metabolism. Chemico‐Biological Interactions, 163(1‐2), 94–112. 10.1016/j.cbi.2006.04.014 [DOI] [PubMed] [Google Scholar]
- DeRitter, E. (1982). Vitamins in pharmaceutical formulations. Journal of Pharmaceutical Sciences, 71(10), 1073–1096. 10.1002/jps.2600711003 [DOI] [PubMed] [Google Scholar]
- Du, C. , Martin, R. , Huang, Y. , Borwankar, A. , Tan, Z. , West, J. , … Li, Z. J. (2018). Vitamin B12 association with mAbs: Mechanism and potential mitigation strategies. Biotechnology and Bioengineering, 115(4), 900–909. 10.1002/bit.26511 [DOI] [PubMed] [Google Scholar]
- Dulbecco, R. , Vogt, M. , & February, P. (1954). Poliomyelitis viruses preparation of tissue cultures. The Journal of Experimental Medicine, 99, 167–182. 10.1084/jem.99.2.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta, N. K. , Mehta, C. J. , & Narayanan, K. G. A. (1952). Study on the stability of vitamins in oral preparations: Part I. Effects of salts of iron ·and copper on vitamin B1. Indian Journal of Pharmacy, 14, 53. [Google Scholar]
- Dwivedi, B. K. , & Arnold, R. G. (1973). Chemistry of thiamine degradation in food products and model systems: A review. Journal of Agriculture and Food Chemistry, 21(1), 54–60. 10.1021/jf60185a004 [DOI] [PubMed] [Google Scholar]
- Dántola, M. L. , Denofrio, M. P. , Zurbano, B. , Gimenez, C. S. , Ogilby, P. R. , Lorente, C. , & Thomas, A. H. (2010). Mechanism of photooxidation of folic acid sensitized by unconjugated pterins. Photochemical and Photobiological Sciences, 9(12), 1604–1612. 10.1039/c0pp00210k [DOI] [PubMed] [Google Scholar]
- Eagle, H. (1955). Nutrition needs of mammalian cells in tissue culture. Science, 122, 501–504. 10.1126/science.122.3168.501 [DOI] [PubMed] [Google Scholar]
- Farrer, K. T. (1941). The influence of pH on the destruction of aneurin (vitamin B1) at 100°C. Australian Chemical Institute Journal and Proceedings, 8, 113–118. [Google Scholar]
- Farrer, K. T. H. (1947). The thermal destruction of vitamin B1: 3. The influence of copper on the rate of destruction of aneurin in buffer solutions at 100°C. Biochemical Journal, 41(2), 167–169. 10.1042/bj0410167 [DOI] [PubMed] [Google Scholar]
- Farrer, K. T. H. (1955). The thermal destruction of vitamin B1 in foods In Mrak E. M., & Stewart G. F. (Eds.), Advances in food research (6, pp. 257–311). New York: Academic Press. [Google Scholar]
- Feller, B. A. , & Macek, T. J. (1955). Effect of thiamine hydrochloride on the stability of solutions of crystalline vitamin B12. Journal of the American Pharmaceutical Association, 44(11), 662–665. 10.1002/jps.3030441106 [DOI] [PubMed] [Google Scholar]
- Fife, D. J. , & Moore, W. M. (1979). The reduction and quenching of photoexcited flavins by EDTA. Photochemistry and Photobiology, 29(1), 43–47. 10.1111/j.1751-1097.1979.tb09257.x [DOI] [Google Scholar]
- Frost, D. (1943). Pantothenic acid optical rotation as a measure of stability. Industrial & Engineering Chemistry Analytical Edition, 15(5), 306–310. 10.1021/i560117a002 [DOI] [Google Scholar]
- Frost, D. V. , & McIntire, F. C. (1944). The hydrolysis of pantothenate: A first order reaction.1 Relation to thiamin stability. Journal of the American Chemical Society, 66(3), 425–427. 10.1021/ja01231a036 [DOI] [Google Scholar]
- Fujiwara, M. , & Watanabe, H. (1952). Allithiamine, a newly found compound of vitamin B1. Proceedings of the Japan Academy, 28(3), 156–158. 10.2183/pjab1945.28.156 [DOI] [Google Scholar]
- Gakenheimer, W. C. , & Feller, B. A. (1949). A note on a preliminary observation of the incompatibility of vitamin b12 and ascorbic acid. Journal of the American Pharmaceutical Association, 38(12), 660–660. 10.1002/jps.3030381213 [DOI] [PubMed] [Google Scholar]
- Gambier, A. S. , & Rahn, E. P. G. (1957). The combination of B‐complex vitamins and ascorbic acid in aqueous solutions. Journal of the American Pharmaceutical Association (Scientific ed.), 46(2), 134–140. 10.1002/jps.3030460216 [DOI] [PubMed] [Google Scholar]
- Gasparro, F. P. (1985). UV‐induced photoproducts of para‐aminobenzoic acid. Photodermatology, 2(3), 151–157. https://doi.org/ncbi.nlm.nih.gov/pubmed/3875085 [PubMed] [Google Scholar]
- Gazzali, A. M. , Lobry, M. , Colombeau, L. , Acherar, S. , Azaïs, H. , Mordon, S. , … Frochot, C. (2016). Stability of folic acid under several parameters. European Journal of Pharmaceutical Sciences, 93, 419–430. 10.1016/j.ejps.2016.08.045 [DOI] [PubMed] [Google Scholar]
- Gold, K. (1968). Some factors affecting the stability of thiamine. Limnology and Oceanography, 13(1), 185–188. 10.4319/lo.1968.13.1.0185 [DOI] [Google Scholar]
- Gregory, J. F., 3rd (1997). Bioavailability of folate. European Journal of Clinical Nutrition, 51(Suppl 1), S54–S59. https://doi.org/ncbi.nlm.nih.gov/pubmed/9023482 [PubMed] [Google Scholar]
- Hannibal, L. , Bunge, S. D. , van Eldik, R. , Jacobsen, D. W. , Kratky, C. , Gruber, K. , & Brasch, N. E. (2007). X‐ray structural characterization of imidazolylcobalamin and histidinylcobalamin: Cobalamin models for aquacobalamin bound to the B12 transporter protein transcobalamin. Inorganic Chemistry, 46(9), 3613–3618. 10.1021/ic070022n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, S. A. (1941). Chemistry of vitamin B6. IV. Reactions in solutions at elevated temperatures. Journal of the American Chemical Society, 63, 3363–3367. 10.1021/ja01857a038 [DOI] [Google Scholar]
- Heyl, D. , Harris, S. A. , & Folkers, K. (1948). The chemistry of vitamin B6. VI. Pyridoxylamino acids. Journal of the American Chemical Society, 70, 3429–3431. 10.1021/ja01190a061 [DOI] [PubMed] [Google Scholar]
- Hochberg, M. , Melnick, D. , & Oser, B. L. (1944). Chemical determination of pyridoxine. Reactions in pure systems. Journal of Biological Chemistry, 155(1), 109–117. https://doi.org/jbc.org/content/155/1/109.short [Google Scholar]
- Hofmann, K. , Melville, D. B. , & du Vigneaud, V. (1941). Characterization of the functional groups of biotin. Journal of Biological Chemistry, 141(1), 207–214. https://doi.org/jbc.org/content/141/1/207.short [Google Scholar]
- Hogenkamp, H. P. C. , Barker, H. A. , & Mason, H. S. (1963). An electron‐spin resonance study of coenzyme B12. Archives of Biochemistry and Biophysics, 100(3), 353–359. doi:. 10.1016/0003-9861(63)90097-3 [DOI] [PubMed] [Google Scholar]
- Hu, M.‐L. , Chen, Y.‐K. , Chen, L.‐C. , & Sano, M. (1995). Para‐aminobenzoic acid scavenges reactive oxygen species and protects DNA against UV and free radical damage. The Journal of Nutritional Biochemistry, 6(9), 504–508. doi:. 10.1016/0955-2863(95)00082-b [DOI] [Google Scholar]
- Huvaere, K. , Andersen, M. L. , Storme, M. , Van Bocxlaer, J. , Skibsted, L. H. , & De Keukeleire, D. (2006). Flavin‐induced photodecomposition of sulfur‐containing amino acids is decisive in the formation of beer lightstruck flavor. Photochemical & Photobiological Sciences, 5(10), 961–969. 10.1039/b609337j [DOI] [PubMed] [Google Scholar]
- Hymes, J. , & Wolf, B. (1996). Biotinidase and its roles in biotin metabolism. Clinica Chimica Acta, 255(1), 1–11. [DOI] [PubMed] [Google Scholar]
- Im, S. , Lee, W. K. , Park, M. K. , & Kim, B.‐K. (1983). Studies on the interaction of thiamines and cyclodextrins. Archives of Pharmacal Research, 6(1), 35–44. 10.1007/bf02855700 [DOI] [Google Scholar]
- Insińska‐Rak, M. , & Sikorski, M. (2014). Riboflavin interactions with oxygen—A survey from the photochemical perspective. Chemistry, 20(47), 15280–15291. 10.1002/chem.201403895 [DOI] [PubMed] [Google Scholar]
- Jain, S. K. , & Lim, G. (2001). Pyridoxine and pyridoxamine inhibits superoxide radicals and prevents lipid peroxidation, protein glycosylation, and (Na++K+)‐ATPase activity reduction in high glucose‐treated human erythrocytes. Free Radical Biology and Medicine, 30, 232–237. 10.1016/S0891-5849(00)00462-7 [DOI] [PubMed] [Google Scholar]
- Juzeniene, A. , & Nizauskaite, Z. (2013). Photodegradation of cobalamins in aqueous solutions and in human blood. Journal of Photochemistry and Photobiology, B: Biology, 122, 7–14. 10.1016/j.jphotobiol.2013.03.001 [DOI] [PubMed] [Google Scholar]
- Jägerstad, M. , & Arkbåge, K. (2003). Cobalamins|properties and determination In Caballero B., Trugo L., & Finglas P. (Eds.), Encyclopedia of Food Sciences and Nutrition (pp. 1419–1427). Amsterdam: Elsevier Science. [Google Scholar]
- Kanner, J. D. , & Fennema, O. (1987). Photooxidation of tryptophan in the presence of riboflavin. Journal of Agricultural and Food Chemistry, 35, 71–76. [Google Scholar]
- Kawasaki, C. (1964). Modified thiamine compounds In Harris R. S., Wool I. G., & Loraine J. A. (Eds.), Vitamins & Hormones (21, pp. 69–111). San Diego: Academic Press. [PubMed] [Google Scholar]
- Kearsley, M. W. , & Rodriguez, N. (2007). The stability and use of natural colours in foods: Anthocyanin, β‐carotene and riboflavin. International Journal of Food Science & Technology, 16(4), 421–431. 10.1111/j.1365-2621.1981.tb01833.x [DOI] [Google Scholar]
- Kinnersley, H. W. , O’Brien, J. R. , & Peters, R. A. (1935). The properties of blue fluorescent substances formed by oxidation of vitamin B1 (quinochromes). Biochemical Journal, 29(10), 2369–2384. https://doi.org/biochemj.org/content/29/10/2369.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschbaum, J. (1981). Analytical profiles of drug substances. Cyanocobalamin, 10, 183–288. 10.1016/s0099-5428(08)60642-5 [DOI] [Google Scholar]
- Koft, B. , & Sevag, M. G. (1949). The liberation of diazotizable amine from pteroylglutamic acid. Journal of the American Chemical Society, 71(9), 3245–3245. 10.1021/ja01177a513 [DOI] [Google Scholar]
- Kuijvenhoven, L. M., & Westerterp, C. (1952). US2734902A, Washington DC: United States Patent and Trademark Office.
- Kuschelewski, J. , Schnellbaecher, A. , Pering, S. , Wehsling, M. , & Zimmer, A. (2017). Antioxidant effect of thiazolidine molecules in cell culture media improves stability and performance. Biotechnology Progress, 33(3), 759–770. 10.1002/btpr.2458 [DOI] [PubMed] [Google Scholar]
- Landauer, K. (2014). Designing media for animal cell culture: CHO cells, the industrial standard. Methods in Molecular Biology, 1104, 89–103. 10.1007/978-1-62703-733-4_7 [DOI] [PubMed] [Google Scholar]
- Larson, R. A. , Stackhouse, P. L. , & Crowley, T. O. (1992). Riboflavin tetraacetate: A potentially useful photosensitizing agent for the treatment of contaminated waters. Environmental Science & Technology, 26(9), 1792–1798. [Google Scholar]
- Le Huerou, Y. , Gunawardana, I. , Thomas, A. A. , Boyd, S. A. , de Meese, J. , Dewolf, W. , … Lin, J. (2008). Prodrug thiamine analogs as inhibitors of the enzyme transketolase. Bioorganic and Medicinal Chemistry Letters, 18(2), 505–508. 10.1016/j.bmcl.2007.11.100 [DOI] [PubMed] [Google Scholar]
- Lee, E. S. (1988). The influence of pH, water activity, and reducing sugars on kinetics of thermal thiamin breakdown in model systems and ground pork (8825413 Ames, Iowa: Iowa State University. [Google Scholar]
- Liang, X. S. , Zhao, F. Q. , & Hao, L. X. (2013). Research on stability of synthetic folic acid. Advanced Materials Research, 781‐784, 1215–1218. 10.4028/www.scientific.net/AMR.781-784.1215 [DOI] [Google Scholar]
- Lonsdale, D. (2006). A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives. Evidence‐Based Complementary and Alternative Medicine, 3(1), 49–59. 10.1093/ecam/nek009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry, O. H. , Bessey, O. A. , & Crawford, E. J. (1949). Photolytic and enzymatic transformations of pteroylglutamic acid. The Journal of Biological Chemistry, 180(1), 389–398. https://doi.org/ncbi.nlm.nih.gov/pubmed/18133404 [PubMed] [Google Scholar]
- Macek, T. J. (1960). Stability problems with some vitamins in pharmaceuticals. American Journal of Pharmacy and the Sciences Supporting Public Health, 132, 433–455. https://doi.org/ncbi.nlm.nih.gov/pubmed/13764760 [PubMed] [Google Scholar]
- Maier, G. D. , & Metzler, D. E. (1957). Structures of thiamine in basic solution 1. Journal of the American Chemical Society, 79(16), 4386–4391. 10.1021/ja01573a040 [DOI] [Google Scholar]
- Matherly, L. H. , & Hou, Z. (2008). Chapter 5 structure and function of the reduced folate carrier: A paradigm of a major facilitator superfamily mammalian nutrient transporter In Litwack G. (Ed.), Vitamins & hormones (79, pp. 145–184). San Diego: Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa, T. , Iwatsu, T. , & Yurugi, S. (1951). Studies on vitamin B1 and its related compounds. XIV. Behaviour of vitamin B1 in water. Journal of the Pharmaceutical Society of Japan, 71(5), 369–371. 10.1248/yakushi1947.71.5_369 [DOI] [Google Scholar]
- Matsukawa, T. , Yurugi, S. , & Oka, Y. (1962). The synthesis of S‐acyl thiamine derivatives and their stability. Annals of the New York Academy of Sciences, 98(2), 430–444. 10.1111/j.1749-6632.1962.tb30564.x [DOI] [PubMed] [Google Scholar]
- McIntire, F. C. , & Frost, D. V. (1944). Thiamin stability. Effect of amino acids and related compounds and of thiamin concentration. Journal of the American Chemical Society, 66(8), 1317–1318. 10.1021/ja01236a033 [DOI] [Google Scholar]
- Medicine, I. O. (1998). Dietary reference intakes for thiamin, riboflavin, niacin, vitamin b6, folate, vitamin B12, pantothenic acid, biotin, and choline. Washington, DC: The National Academies Press. [PubMed] [Google Scholar]
- Melville, D. B. (1954). Biotin sulfoxide. Journal of Biological Chemistry, 208, 495–502. [PubMed] [Google Scholar]
- Metzler, D. E. , & Cairns, W. L. (1971). Photochemical degradation of flavins. VI. A new photoproduct and its use in studying the photolytic mechanism. Journal of the American Chemical Society, 93(11), 2772–2777. https://doi.org/ncbi.nlm.nih.gov/pubmed/5573725 [DOI] [PubMed] [Google Scholar]
- Metzler, D. E. , Ikawa, M. , & Snell, E. E. (1954). A general mechanism for vitamin B6‐catalyzed reactions. Journal of the American Chemical Society, 76, 648–652. 10.1021/ja01632a004 [DOI] [Google Scholar]
- Metzler, D. E. , Longenecker, J. B. , & Snell, E. E. (1954). The reversible catalytic cleavage of hydroxyamino acids by pyridoxal and metal salts. Journal of the American Chemical Society, 76, 639–644. 10.1021/ja01632a002 [DOI] [Google Scholar]
- Metzler, D. E. , & Snell, E. E. (1955). Spectra and ionization constants of the vitamin B6 group and related 3‐hydroxypyridine derivatives. Journal of the American Chemical Society, 77, 2431–2437. 10.1021/ja01614a022 [DOI] [Google Scholar]
- Miller, C. S. , Sprague, J. M. , & Krampitz, L. O. (1962). The reaction of thiamine with carbonyl compounds. Annals of the New York Academy of Sciences, 98(2), 401–411. 10.1111/j.1749-6632.1962.tb30562.x [DOI] [PubMed] [Google Scholar]
- Mizuhara, S. , & Handler, P. (1954). Mechanism of thiamine‐catalyzed reactions 1. Journal of the American Chemical Society, 76(2), 571–573. 10.1021/ja01631a071 [DOI] [Google Scholar]
- Mohberg, J. , & Johnson, M. J. (1963). Stability of vitamins in a chemically defined medium for 929‐L fibroblasts. Journal of the National Cancer Institute, 31, 603–610. https://doi.org/ncbi.nlm.nih.gov/pubmed/14059005 [PubMed] [Google Scholar]
- Monajjemzadeh, F. , Ebrahimi, F. , Zakeri‐Milani, P. , & Valizadeh, H. (2014). Effects of formulation variables and storage conditions on light protected vitamin B12 mixed parenteral formulations. Advanced Pharmaceutical Bulletin, 4(4), 329–338. 10.5681/apb.2014.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhamad, N. , Yusoff, M. M. , & Gimbun, J. (2015). Thermal degradation kinetics of nicotinic acid, pantothenic acid and catechin derived from Averrhoa bilimbi fruits. RSC Advances, 5(90), 74132–74137. 10.1039/C5RA11950B [DOI] [Google Scholar]
- Mulley, E. A. , Stumbo, C. R. , & Hunting, W. M. (1975). Effect of pH and form of the vitamin on its rate of destruction. Journal of Food Science, 40(5), 989–992. 10.1111/j.1365-2621.1975.tb02250.x [DOI] [Google Scholar]
- Newmark, H. L. (1958). U.S. Patent No. 2,823,167, https://doi.org/Washington DC: US Patent and Trademark Office [Google Scholar]
- Obata, Y. , & Tanaka, H. (2014). Studies on the photolysis of l‐cysteine and l‐cystine. Agricultural and Biological Chemistry, 29(3), 191–195. 10.1080/00021369.1965.10858375 [DOI] [Google Scholar]
- Off, M. K. , Steindal, A. E. , Porojnicu, A. C. , Juzeniene, A. , Vorobey, A. , Johnsson, A. , & Moan, J. (2005). Ultraviolet photodegradation of folic acid. The Journal of Photochemistry and Photobiology B: Biology, 80(1), 47–55. 10.1016/j.jphotobiol.2005.03.001 [DOI] [PubMed] [Google Scholar]
- Ottaway, P. B. (1993). The technology of vitamins in food. Stability of Vitamins in Food, 90–113. 10.1007/978-1-4615-2131-0_5 [DOI] [Google Scholar]
- Pan, Y.‐L. , Pinnick, R. G. , Hill, S. C. , Niles, S. , Holler, S. , Bottiger, J. R. , … Chang, R. K. (2001). Dynamics of photon‐induced degradation and fluorescence in riboflavin microparticles. Applied Physics B, 72(4), 449–454. 10.1007/s003400100532 [DOI] [Google Scholar]
- Pezacka, E. , Green, R. , & Jacobsen, D. W. (1990). Glutathionylcobalamin as an intermediate in the formation of cobalamin coenzymes. Biochemical and Biophysical Research Communications, 169(2), 443–450. https://doi.org/ncbi.nlm.nih.gov/pubmed/2357215 [DOI] [PubMed] [Google Scholar]
- Pratt, J. M. (1964). The chemistry of vitamin B12. Part II. Photochemical reactions. Journal of the Chemical Society (Resumed), 5154–5160. 10.1039/jr9640005154 [DOI] [Google Scholar]
- Reddy, K. H. (1999). Coordination compounds in biology. Resonance, 4(6), 67–77. 10.1007/bf02834637 [DOI] [Google Scholar]
- Reiber, H. (1972). Photochemical reactions of vitamin B6 compounds, isolation and properties of products. Biochimica et Biophysica Acta, 279, 310–315. 10.1016/0304-4165(72)90148-1 [DOI] [PubMed] [Google Scholar]
- Rosenberg, A. J. (1956). The copper‐promoted decomposition of vitamin B12 in solutions of ascorbic acid. Journal of Biological Chemistry, 219(2), 951–956. Retrieved from.http://www.jbc.org/content/219/2/951.short [PubMed] [Google Scholar]
- Ruiz‐Rico, M. , Pérez‐Esteve, É. , Lerma‐García, M. J. , Marcos, M. D. , Martínez‐Máñez, R. , & Barat, J. M. (2017). Protection of folic acid through encapsulation in mesoporous silica particles included in fruit juices. Food Chemistry, 218, 471–478. 10.1016/j.foodchem.2016.09.097 [DOI] [PubMed] [Google Scholar]
- Sarkar, B. , Das, U. , Bhattacharyya, S. , & Bose, S. K. (1997). Studies on the aerobic photooxidation of cysteine using riboflavin as a sensitizer: Evidence for the photogeneration of a superoxide anion and hydrogen peroxide. Biological & Pharmaceutical Bulletin, 20(8), 910–912. 10.1248/bpb.20.910 [DOI] [PubMed] [Google Scholar]
- Sato, Y. , Yokoo, M. , Takahashi, S. , & Takahashi, T. (1982). Biphasic photolysis of riboflavine with a low‐intensity light source. Chemical & Pharmaceutical Bulletin, 30(5), 1803–1810. 10.1248/cpb.30.1803 [DOI] [Google Scholar]
- Scheindlin, S. , & Griffith, I. (1951). The action of ascorbic acid on folic acid. American Journal of Pharmacy and the Sciences Supporting Public Health, 123(3), 78–86. Retrieved from.http://www.ncbi.nlm.nih.gov/pubmed/14829563 [PubMed] [Google Scholar]
- Scheindlin, S. , Lee, A. , & Griffith, I. (1952). The action of riboflavin on folic acid. Journal of the American Pharmaceutical Association, 41(8), 420–427. 10.1002/jps.3030410806 [DOI] [PubMed] [Google Scholar]
- Schneider, M. , Klotzsche, M. , Werzinger, C. , Hegele, J. , Waibel, R. , & Pischetsrieder, M. (2002). Reaction of folic acid with reducing sugars and sugar degradation products. Journal of Agricultural and Food Chemistry, 50(6), 1647–1651. 10.1021/jf011042p [DOI] [PubMed] [Google Scholar]
- Schneider, Z. , & Stroiński, A. (1987). Comprehensive B12: Chemistry, biochemistry, nutrition, ecology, medicine. New York: De Gruyter. [Google Scholar]
- Schonbeck, N. D. , Skalski, M. , & Shafer, J. A. (1975). Reactions of pyridoxal 5′‐phosphate, 6‐aminocaproic acid, cysteine, and penicilamine. Models for reactions of Schiff base linkages in pyridoxal 5′‐phosphate‐requiring enzymes. Journal of Biological Chemistry, 250(14), 5343–5351. https://doi.org/jbc.org/content/250/14/5343 [PubMed] [Google Scholar]
- Scurachio, R. S. , Skibsted, L. H. , Metzker, G. , & Cardoso, D. R. (2011). Photodegradation of folate sensitized by riboflavin. Photochemistry and Photobiology, 87(4), 840–845. 10.1111/j.1751-1097.2011.00916.x [DOI] [PubMed] [Google Scholar]
- Shephard, G. S. , & Labadarios, D. (1986). Degradation of vitamin B6 standard solutions. Clinica Chimica Acta, 160, 307–311. 10.1016/0009-8981(86)90198-1 [DOI] [PubMed] [Google Scholar]
- Sheraz, M. A. , Kazi, S. H. , Ahmed, S. , Anwar, Z. , & Ahmad, I. (2014). Photo, thermal and chemical degradation of riboflavin. Beilstein Journal of Organic Chemistry, 10, 1999–2012. 10.3762/bjoc.10.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, E. , & Godoy, J. (1994). Riboflavin sensitized photooxidation of tyrosine. International Journal for Vitamin and Nutrition Research, 64(4), 253–256. Retrieved from.http://www.ncbi.nlm.nih.gov/pubmed/7883461 [PubMed] [Google Scholar]
- Skeggs, H. R. (1952). United States of America Patent No.
- Snell, E. E. (1958). Chemical structure in relation to biological activities of vitamin B6. Vitamins and Hormones, 16, 77–125. 10.1016/S0083-6729(08)60314-3 [DOI] [PubMed] [Google Scholar]
- Snell, E. E. (1963). Comprehensive biochemistry. Vitamin B6 Comprehensive Biochemistry, 11, 48–58. [Google Scholar]
- Steindal, A. H. , Porojnicu, A. C. , & Moan, J. (2007). Is the seasonal variation in cancer prognosis caused by sun‐induced folate degradation?. Medical Hypotheses, 69(1), 182–185. 10.1016/j.mehy.2006.07.063 [DOI] [PubMed] [Google Scholar]
- Stokstad, E. L. , Fordham, D. , & De Grunigen, A. (1947). The inactivation of pteroylglutamic acid (liver Lactobacillus casei factor) by light. The Journal of Biological Chemistry, 167(3), 877 https://doi.org/ncbi.nlm.nih.gov/pubmed/20287925 [PubMed] [Google Scholar]
- Surrey, A. R. , & Nachod, F. C. (1951). Alkaline hydrolysis of riboflavin. Journal of the American Chemical Society, 73(5), 2336–2338. 10.1021/ja01149a128 [DOI] [Google Scholar]
- Tansey, R. P. , & Schneller, G. H. (1955). Studies in the stabilization of folic acid in liquid pharmaceutical preparations. Journal of the American Pharmaceutical Association, 44(1), 34–37. https://doi.org/ncbi.nlm.nih.gov/pubmed/13233093 [DOI] [PubMed] [Google Scholar]
- Taub, A. , & Lieberman, H. (1953). Stability of vitamin B12; folic acid parenteral solutions. Journal of the American Pharmaceutical Association, 42(4), 183–186. https://doi.org/ncbi.nlm.nih.gov/pubmed/13052522 [DOI] [PubMed] [Google Scholar]
- Terekhova, I. V. , Koźbiał, M. , Kumeev, R. S. , & Alper, G. A. (2011). Inclusion complex formation between modified cyclodextrins and riboflvin and alloxazine in aqueous solution. Journal of Solution Chemistry, 40(8), 1435–1446. 10.1007/s10953-011-9724-0 [DOI] [Google Scholar]
- Terekhova, I. V. , Tikhova, M. N. , Volkova, T. V. , Kumeev, R. S. , & Perlovich, G. L. (2010). Inclusion complex formation of α‐ and β‐cyclodextrins with riboflavin and alloxazine in aqueous solution: Thermodynamic study. Journal of Inclusion Phenomena and Macrocyclic Chemistry, 69(1‐2), 167–172. 10.1007/s10847-010-9827-z [DOI] [Google Scholar]
- O’Neil, M. J. (2006). The merck index: An encyclopedia of chemicals, drugs, and biologicals (14th ed [Google Scholar]
- Thomas, A. H. , Lorente, C. , Capparelli, A. L. , Martínez, C. G. , Braun, A. M. , & Oliveros, E. (2003). Singlet oxygen (1Δg) production by pterin derivatives in aqueous solutions. Photochemical & Photobiological Sciences, 2(3), 245–250. 10.1039/B209993D [DOI] [PubMed] [Google Scholar]
- Tofzikovskaya, Z. , O’Connor, C. , & McNamara, M. (2012). Synthesis, characterisation and photo‐stability of a folate‐modified β‐cyclodextrin as a functional food additive. Journal of Inclusion Phenomena and Macrocyclic Chemistry, 74(1‐4), 437–445. 10.1007/s10847-012-0139-3 [DOI] [Google Scholar]
- Toohey, J. I. (1965). A vitamin B12 compound containing no cobalt. Proceedings of the National Academy of Sciences, 54(3), 934–942. 10.1073/pnas.54.3.934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenner, N. R. , Buhs, R. P. , Bacher, F. A. , & Gakenheimer, W. C. (1950). A note concerning the incompatibility of vitamin B12 and ascorbic acid. Journal of the American Pharmaceutical Association, 39(6), 361–361. 10.1002/jps.3030390619 [DOI] [PubMed] [Google Scholar]
- Tzeng, D. D. , Lee, M.‐H. , Chung, K.‐R. , & DeVay, J. E. (1990). Products in light‐mediated reactions of free methionine‐riboflavin mixtures that are biocidal to microorganisms. Canadian Journal of Microbiology, 36(7), 500–506. 10.1139/m90-087 [DOI] [Google Scholar]
- Uchida, T. , Suguri, T. , & Harjinder, S. (2002). US6500472B2, https://doi.org/Washington DC: US Patent and Trademark Office [Google Scholar]
- Uprety, M. C. , R., S. , & Rao, V. K. M. (1961). Stabilization of vitamins in pharmaceutical preparations. 4. Influence of amino acids on the stability of vitamins A, B1, & C. Journal of Scientific and Industrial Research, 20D, 124–126. [Google Scholar]
- Urbanski, T. (1949). Vitamin B6; pyridoxine, pyridoxal and pyridoxamine. Research, 2(11), 507–512. https://doi.org/ncbi.nlm.nih.gov/pubmed/15407094 [PubMed] [Google Scholar]
- Vaid, F. H. (1997). Photolysis and interaction of thiamin hydrochloride with riboflavin in aqueous solution, Karachi: University of Karachi. https://doi.org/(PhD Thesis) [Google Scholar]
- Vijayasankaran, N. , Li, J. , Shawley, R. , Chen, A. , Shiratori, M. , Gawlitzek, M. … Amanullah, A. (2010). Animal cell culture media In Flickinger M. C. (Ed.), Encyclopedia of industrial biotechnology: Bioprocess, bioseparation, and cell technology. New York: John Wiley & Sons. [Google Scholar]
- Wang, X. (2003). A comparison of folic acid and 5‐methyltetrahydrofolate for prevention of DNA damage and cell death in human lymphocytes in vitro. Mutagenesis, 18(1), 81–86. https://doi.org/ncbi.nlm.nih.gov/pubmed/12473740 [DOI] [PubMed] [Google Scholar]
- Wang, X.‐M. , & Chen, H.‐Y. (1996). A spectroelectrochemical study of the interaction of riboflavin with β‐cyclodextrin. Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy, 52(5), 599–605. 10.1016/0584-8539(95)01561-2 [DOI] [Google Scholar]
- Watanabe, A. (1939). Ueber die hydrolyoe von vitamin B1 in waesoeriger Loesung. Yakugaku Zasshi, 59(7), 500–502. 10.1248/yakushi1881.59.7_500 [DOI] [Google Scholar]
- Weidenheimer, J. F. , & Carstensen, J. T. (1954). US 2695860 A, https://doi.org/Washington DC: US Patent and Trademark Office [Google Scholar]
- Wendt, G. , & Bernhart, F. W. (1960). The structure of a sulfur‐containing compound with vitamin B6 activity. Archives of Biochemistry and Biophysics, 88, 270–272. 10.1016/0003-9861(60)90234-4 [DOI] [PubMed] [Google Scholar]
- Windheuser, J. J. , & Higuchi, T. (1962). Kinetics of thiamine hydrolysis. Journal of Pharmaceutical Sciences, 51(4), 354–364. 10.1002/jps.2600510415 [DOI] [PubMed] [Google Scholar]
- Windheuser, J. J. , & Higuchi, T. (1963). Effect of various compounds on the rate of thiamine hydrolysis. Journal of Pharmaceutical Sciences, 52, 557–559. https://doi.org/ncbi.nlm.nih.gov/pubmed/14001136 [DOI] [PubMed] [Google Scholar]
- Wu, Z , Li, X. , Hou, C. , … Qian, Y. (2010). Solubility of folic acid in water at pH values between 0 and 7 at temperatures (298.15, 303.15, and 313.15) K. Journal of Chemical & Engineering Data, 55(9), 3958–3961. 10.1021/je1000268 [DOI] [Google Scholar]
- Yang, S. F. , Ku, H. S. , & Pratt, H. K. (1967). Photochemical production of ethylene from methionine and its analogues in the presence of flavin mononucleotide. The Journal of Biological Chemistry, 242(22), 5274–5280. https://doi.org/jbc.org/content/242/22/5274 [PubMed] [Google Scholar]
- Yoshimura, A. , & Ohno, T. (1988). Lumiflavin‐sensitized photooxygenation of indole. Photochemistry and Photobiology, 48(5), 561–565. 10.1111/j.1751-1097.1988.tb02864.x [DOI] [PubMed] [Google Scholar]
- Zima, O. , & Williams, R. R. (1940). Über ein antineuritisch wirksames Oxydationsprodukt des Aneurins. Berichte der Deutschen Chemischen Gesellschaft (A and B Series), 73(9), 941–949. 10.1002/cber.19400730904 [DOI] [Google Scholar]
- Zuck, D. A. , & Conine, J. W. (1963). Stabilization of vitamin B12 I. Complex cyanides. Journal of Pharmaceutical Sciences, 52(1), 59–63. 10.1002/jps.2600520112 [DOI] [PubMed] [Google Scholar]